2.1. Protein Characterization
To verify that the Trp to phenylalanine (Phe) mutations did not adversely affect the stability and function of midW mutant DHFR enzymes, the melting temperature and activity of the enzymes were examined. The effects of the Trp to Phe mutations on enzyme catalysis were determined by comparing wt-DHFR and midW mutant activity by monitoring nicotinamide adenine dinucleotide phosphate (NADPH) absorbance at 340 nm. As shown in
Table S1, the midW22 mutant retained 90% activity compared to the wild-type enzyme. The high activity of midW22 compared to wt-DHFR demonstrates that W30, W47, W74, and W133 have little importance in the catalytic activity of DHFR, since the midW22 mutant has all of the Trp residues, except W22 is replaced with Phe. The other four midW mutants have reduced activity (20–33%), which can be explained by disrupting the hydrogen bond network between W22, D27, T113, and the substrate DHF [
19].
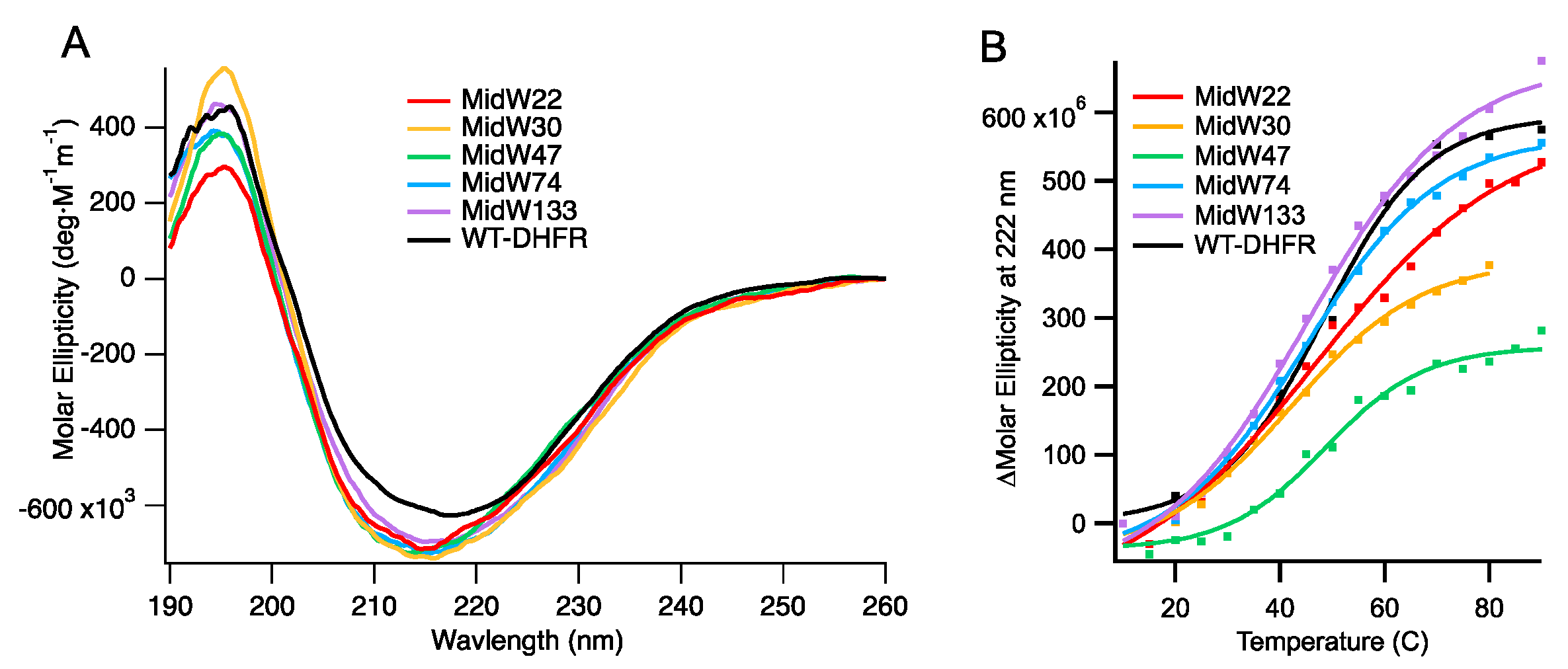
Circular dichroism (CD) spectroscopy was used to examine the fold and the stability of the midW mutants.
Figure 2A shows the CD spectra. Notably, the molar ellipticity of the midW mutants is lower compared to the wild-type enzyme. In the midW mutants, all but one of the five native Trp residues have been mutated to Phe. Previously, single Trp to Phe, valine, or leucine mutations have been shown to impact the CD spectra from 190 to 250 nm [
20]. The mutations caused a loss of ellipticity, gain of ellipticity, or a mixture of the two in different regions of the CD spectra depending on the location of the Trp mutation and which amino acid was chosen to replace the Trp residue. In particular, exciton coupling between W47 and W74 has a large impact on the CD spectra of DHFR. In each of the midW mutants, W47, W74, or both have been removed, making a direct comparison to the wt-DHFR spectra difficult to interpret. However, the CD spectra does show that the midW mutants have a minimum band in molar ellipticity around 215 nm and a maximum around 195 nm, which is characteristic for folded proteins with both alpha helices and beta sheets. The melting temperatures of the midW mutants were determined by monitoring the loss of ellipticity at 222 nm with increasing temperatures via CD spectroscopy (
Figure 2B). We determined the melting temperature of wt-DHFR to be 48 ± 1 °C, which is similar to the previously determined melting temperature of 49.3 °C [
21]. The melting temperatures of the midW mutants range from 42 to 49 °C (
Table 1). The melting temperature is an important consideration, since the proteins are transiently heated to 36 °C during the T-jump experiments. For midW22, midW47, midW74, and midW133, the final temperature is 9 °C or more below the melting temperature, so unfolding is not likely to play a large role. MidW30 has a lower melting temperature of 42 °C, so unfolding effects may be relevant to the observed dynamics.
2.2. Equilibrium Fluorescence
The tryptophan fluorescence spectra of all five midW mutants were measured for the apoenzyme and three different enzyme·ligand complexes: E·Folate, E·NADP
+, and E·NADP
+·Folate. Each of these enzyme–ligand complexes are model systems for the different enzyme states in the catalytic cycle: the binary product complex, the holoenzyme, and the Michaelis complex, respectively. The fluorescence intensity of each midW mutant was much less than the wild-type enzyme (
Figure S2), as expected, since the midW mutants each contain only one tryptophan, whereas wt-DHFR contains 5 tryptophans. As shown in
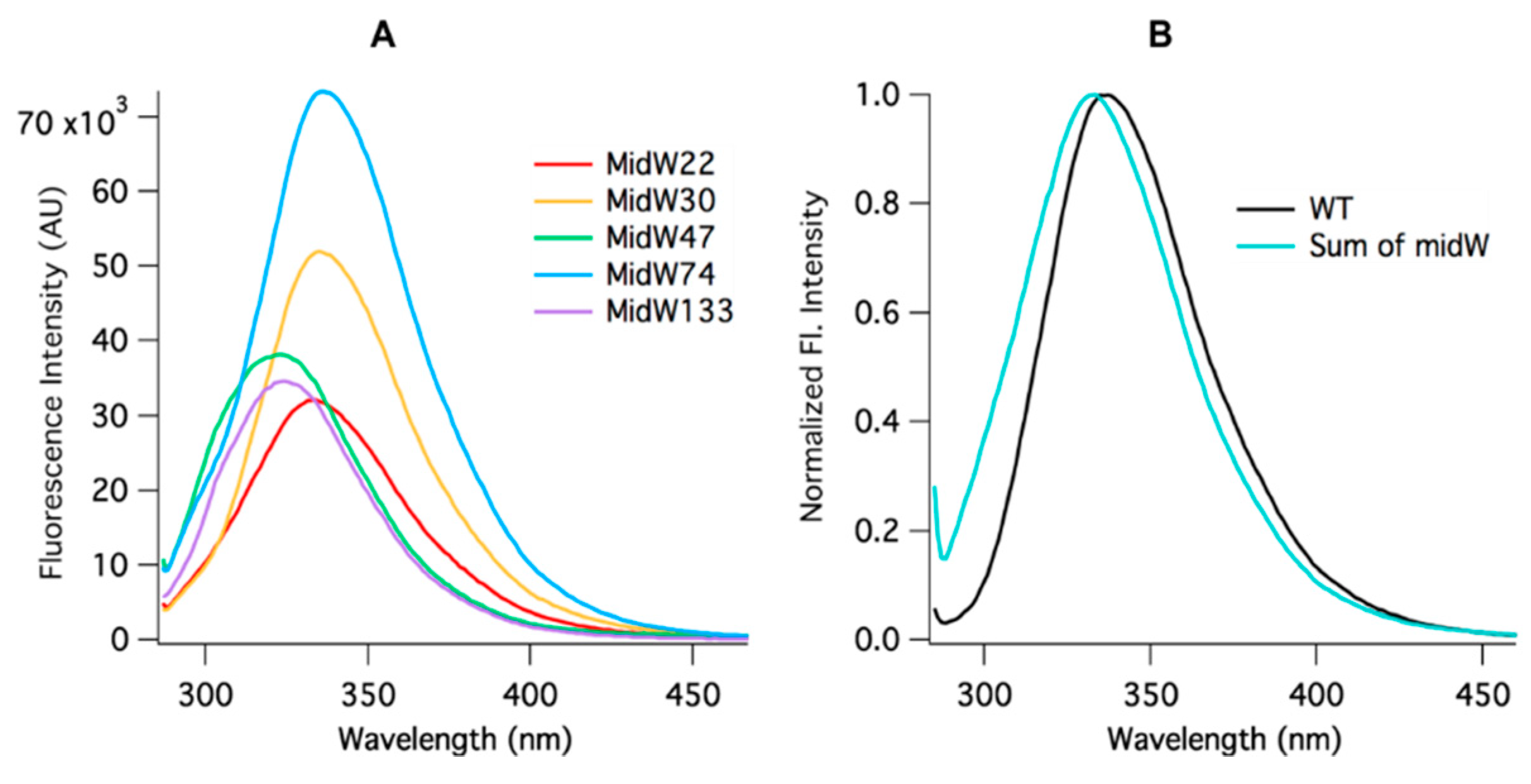
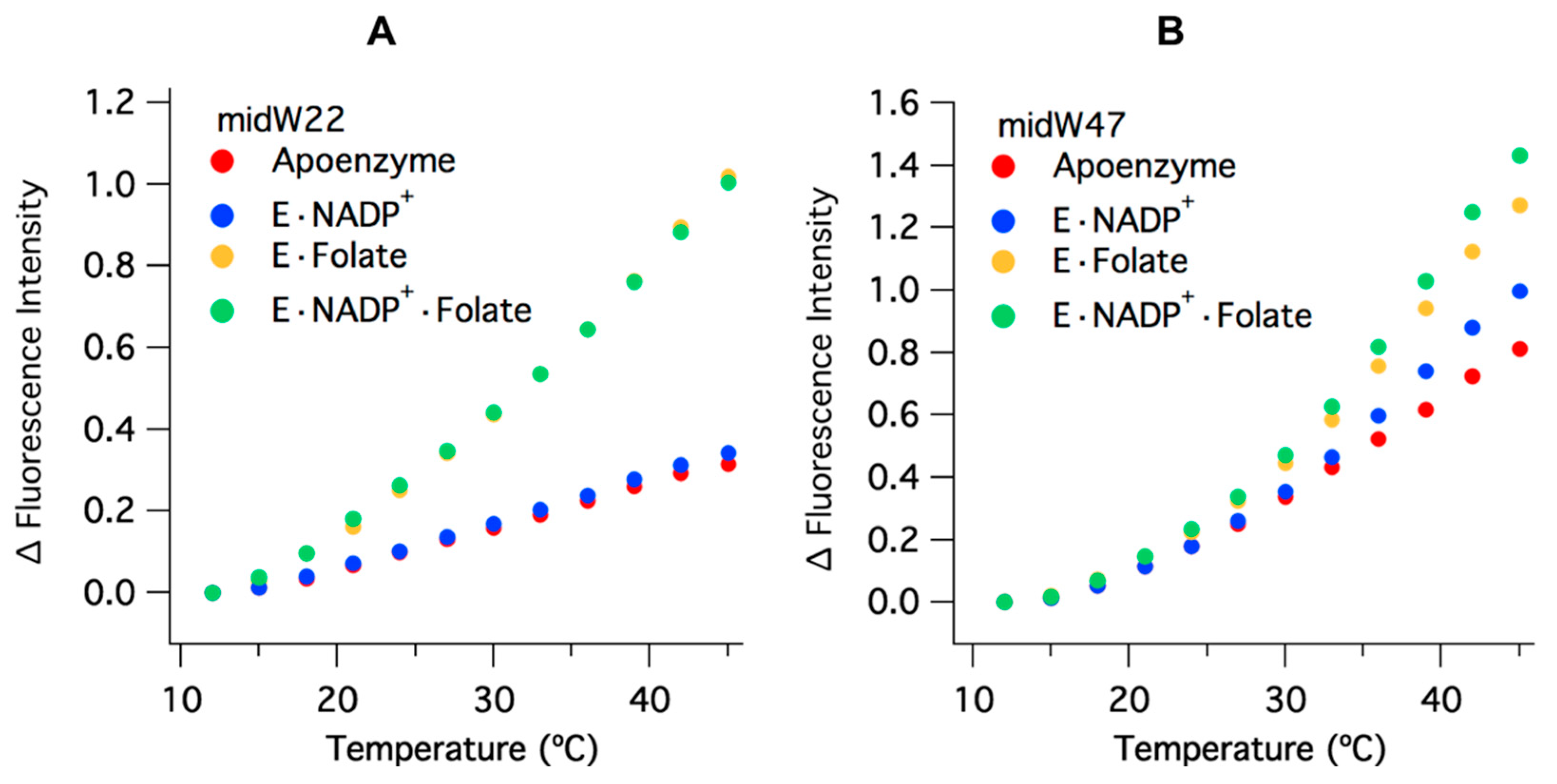
Figure 3A, the intensity of the Trp fluorescence of each midW mutant differs, with midW74 exhibiting the greatest fluorescence intensity, followed by midW30, midW47, midW133, and midW22 with the lowest fluorescence intensity. Additionally, the shift in the Trp fluorescence maximum between midW mutants shows that the Trp in each mutant is in a different environment. Typically, a blue shift indicates a more hydrophobic environment and tends to be concomitant with an increase in fluorescence intensity. A red shift indicates a more hydrophilic environment, which usually corresponds with a decrease in fluorescence intensity, due to quenching by water. Interestingly, the Trp fluorescence of the midW mutants does not necessarily follow these trends. For example, midW47 and midW133 are blue shifted compared to midW74 and midW30, yet midW47 and midW133 display lower fluorescence intensities. This indicates that there must be some quenching interactions with W47 and W133 in the apoenzyme. Additionally, there are likely quenching interactions at position 22, given that midW22 exhibits a similar shift compared to midW74 and midW30. The similar shift suggests a similar environment; however, midW22 Trp fluorescence is drastically reduced.
Figure 3B shows the comparison of the wt-DHFR Trp fluorescence spectrum to the sum of the Trp fluorescence spectra of the midW mutants. The intensities have been normalized to 1 to better demonstrate the difference in lambda max between the two spectra. The difference in the spectra shows that the wt-DHFR Trp fluorescence is not a simple linear combination of the fluorescence of the individual tryptophans. This has been observed previously, albeit in a less direct manner by Ohmae et al., where a single tryptophan was mutated to various hydrophobic residues [
20]. They observed that the Trp fluorescence decreased in varying amounts depending on which tryptophan was mutated. In this way, they came to the same conclusion as stated above. Furthermore, W47 and W74 are known to form an excimer pair [
20]. Typically, the emission of the excimer is red shifted compared to the emission of the fluorescent monomers, which may contribute to the observed red shift of the wild-type DHFR fluorescence compared to the sum of the fluorescence of the individual midW mutants.
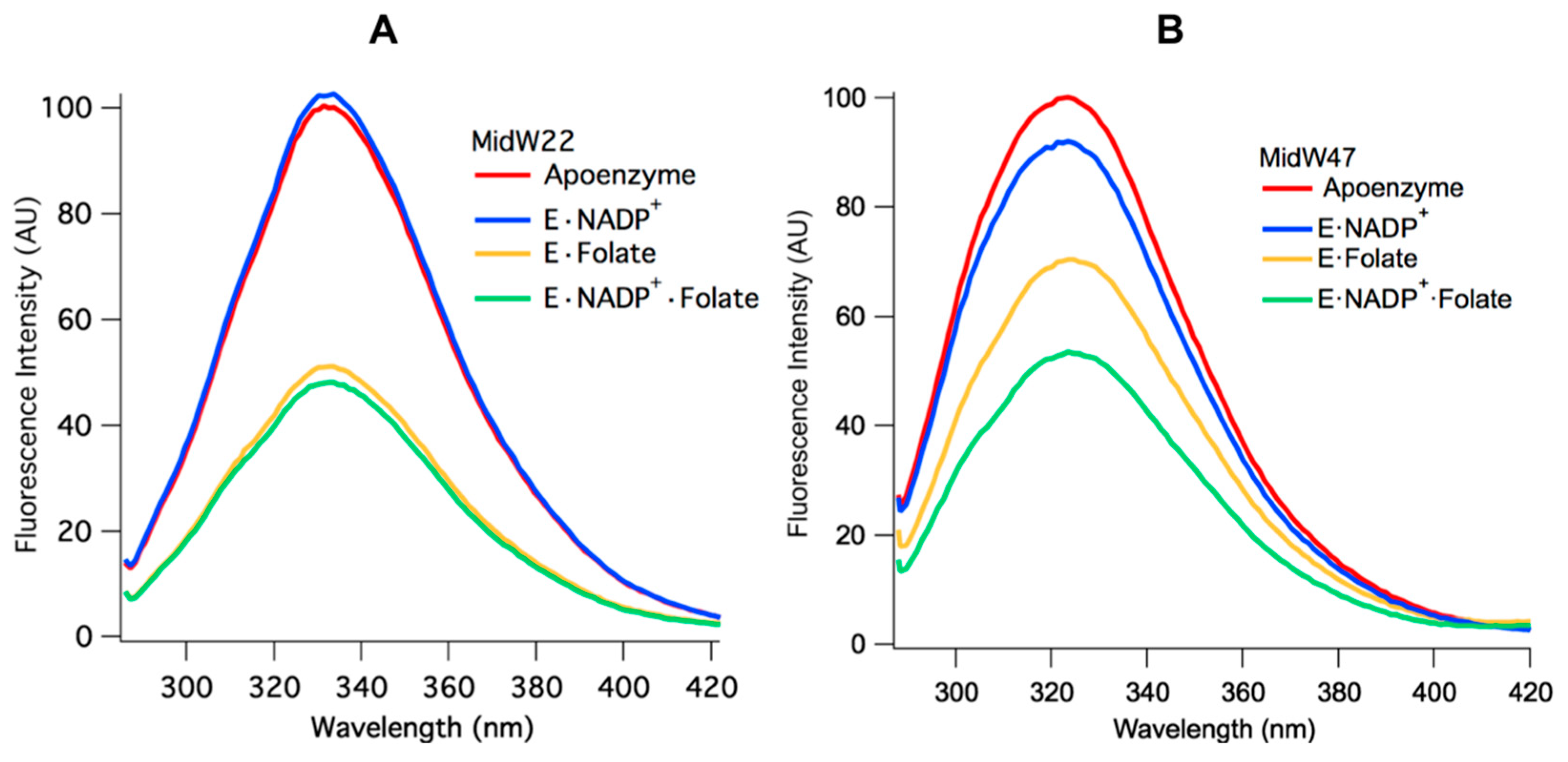
Two patterns emerge when the Trp fluorescence of the midW mutant ligand complexes are compared.
Figure 4A shows the midW22 mutant complexes, which exhibit the same trend as all the other mutants except for midW47, which is shown in panel B.
Table 2 shows the integrated fluorescence intensity of each complex as a percentage of the apoenzyme fluorescence intensity. MidW22 and the other three midW mutants (midW30, midW74, and midW133) show very little change upon binding NADP
+ compared to the apoenzyme and show a significant change compared to apoenzyme for the binary complex with folate and the tertiary complex. Importantly, there is only a small difference in the Trp fluorescence intensity between the E·Folate and E·NADP
+·Folate complexes, which exist in the occluded and closed conformations, respectively. This indicates that W22, W30, W74, and W133 are not particularly sensitive to the closed versus occluded conformational states of the Met20 loop and are instead primarily sensitive to the presence of folate. Conversely, the Trp fluorescence of midW47 (
Figure 4B) decreases by different amounts based on which ligands are bound. There is a small decrease upon binding NADP
+, a larger decrease upon binding folate, and a further decrease in the tertiary complex. However, the combined decrease in fluorescence intensity of individually binding folate and NADP
+ is less than the total decrease observed for the tertiary complex. This means that the decrease in fluorescence intensity observed in the tertiary complex is not simply due to quenching of the Trp fluorescence from the presence of ligands; there is also a contribution from the conformation of the Met20 loop. Thus, midW47 is more sensitive to changes in the enzyme conformation compared to the other midW mutants.
The temperature-dependent Trp fluorescence of the midW mutants is shown in
Figure 5. The values have been corrected for the intrinsic temperature dependence of Trp fluorescence, which decreases with increasing temperature (
Figure S3). This is opposite of the trend observed for the enzyme complexes. All of the midW mutants follow the same general trend as the wild-type enzyme: an increase in fluorescence intensity with increasing temperature, due primarily to ligand dissociation and accompanying loop conformation changes. Each of the midW mutants follow the trend as expected based on their change in Trp fluorescence with different ligands bound. An additional contribution to the increased Trp fluorescence is likely caused by loosening of the enzyme structure at elevated temperatures, which in turn reduces quenching interactions within the protein. This explains the increase in Trp fluorescence for the apoenzyme where no ligands are present to dissociate.
Figure 5 shows the change in fluorescence intensity for midW22 and midW47 as representative data.
2.3. Temperature Jump Enzyme Dynamics
The T-jump transients for all five midW mutants with folate share a few key characteristics. The transients fit to double exponentials with two distinct phases (
Tables S3 and S4). To determine the origin of the observed relaxation rates, we can examine the correlation between the sum of the concentrations of the free enzyme and free ligand. Since T-jump spectroscopy is a relaxation method that provides information about both the forward and reverse reactions, if the observed rate is concentration dependent, then there must be a bimolecular step—in this case, ligand binding.
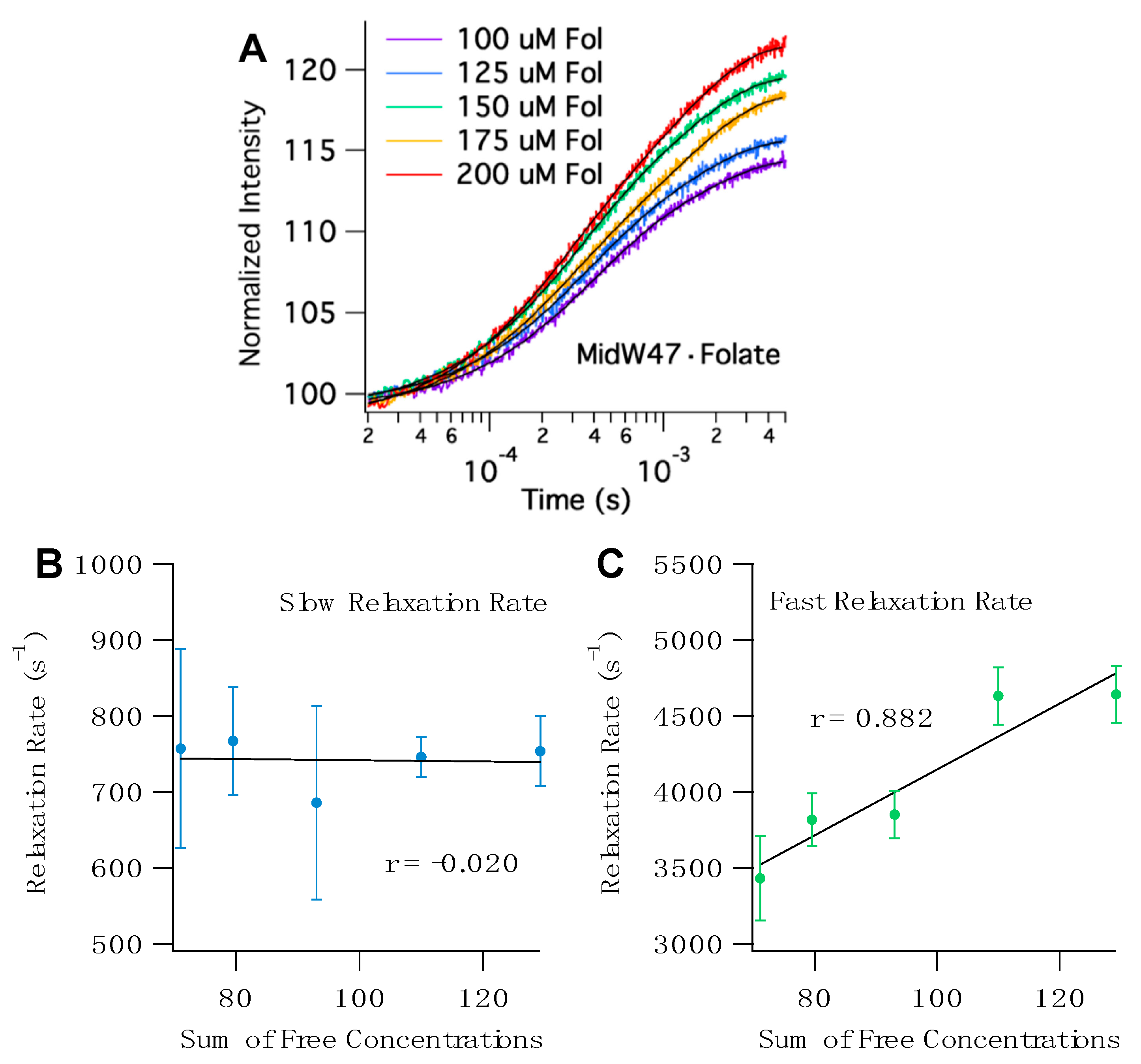
Figure 6 shows representative T-jump transients of midW47 and the concentration-dependent plots. The sum of free concentrations at 36 °C, the temperature after the laser pulse, was calculated using the K
d and ∆H of folate binding determined by isothermal titration calorimetry (ITC). The ITC results are summarized in
Table S2. The concentration dependence of the fast and slow relaxation rates for the midW mutants are shown in
Figure 6 and
Figure S4. The fast relaxation rate of the midW mutants are positively correlated with the sum of the concentrations of the free enzyme and free ligand. The strength of the correlation can be quantitatively described by the linear correlation coefficient, r. The r values range from 0.867 to 0.954 for midW30, midW47, midW74, and midW133. The threshold for significance at a 99% confidence level is 0.505 (DF = 23). [
22] Thus, all of the midW mutants except for midW22 are significantly correlated with concentration and can be qualitatively described as strongly concentration dependent. MidW22 exhibits a weak concentration dependence with an r value of 0.366, which is higher than the cutoff for significance at the 90% confidence level (0.337, degrees of freedom (DF) = 23). The concentration dependence of the fast relaxation rate indicates that the fast relaxation phase is convolved with ligand binding/unbinding. Conversely, the correlation coefficients of the slow relaxation rate vary from −0.659 to 0.124. A negative correlation with regard to concentration is nonsensical. Therefore, we can conclude that the slow relaxation rate of midW22, midW30, midW47, and midW133 is not concentration dependent. MidW74 has an r value of 0.124, which is a very low correlation coefficient. Even at an 80% confidence level, the midW74 correlation coefficient is lower than the threshold value (0.265, DF = 23), indicating that there is no significant correlation between the slow relaxation rate and the sum of the free concentrations. The concentration dependence of the midW mutant relaxation rates follows the same trend as the wild-type relaxation profile. A fast concentration-dependent relaxation rate and a slow concentration-independent relaxation rate were also observed for wt-DHFR [
14].
Assuming a simple two-state model where a ligand binds to an enzyme to form an enzyme– ligand complex, a linear fit of the fast relaxation rate as a function of the sum of free concentrations (
Figure 6C and
Figure S4B,D,F,H) provides additional information. The slope of the line is equivalent to k
on and the y-intercept is equivalent to k
off [
23]. The k
on and k
off values for wt-DHFR and the midW mutants are reported in
Table 3. The k
on and k
off are modulated in the midW mutants compared to wt-DHFR. While k
on is similar to what has been reported in the literature for the DHFR–folate complex (57 ± 5 μM
−1s
−1) [
24], the value of k
off is two orders of magnitude larger (35 ± 12 s
−1) [
24]. Furthermore, comparing the K
d values determined by ITC experiments and the K
d values calculated from k
off and k
on reveals a discrepancy. The K
d values calculated from the T-jump experiments are approximately an order of magnitude larger than the K
d values determined from ITC. Taken together, these results indicate that the observed fast relaxation rate is not only due to ligand association/dissociation but is also convolved with associated conformation changes, such as the Met20 or FG loop dynamics. Loop motions within DHFR are critical for enzyme activity. In particular, two mutations have been studied that impact loop flexibility and consequently impact enzyme catalysis. When N23 at the end of the Met20 loop is replaced with a double proline sequence, millisecond fluctuations of the Met20 loop are abrogated [
12]. When G121 on the FG loop is replaced with a valine, the pico-nanosecond and micro-millisecond motions of both the FG and Met20 loops are decreased [
11]. Both of these studies used NMR relaxation experiments and thus were limited to time regimes accessible by NMR. Since T-jump spectroscopy is capable of measuring dynamics on the nanosecond to millisecond timescale, future studies with the N23PP or G121V mutants could use T-jump spectroscopy to fill in the time gap.
Although the T-jump transients of all five mutants show similar behavior with respect to concentration dependence, the relaxation rate of the slow phase varies based on the position of the Trp. The average relaxation rates of the slow phase are reported in
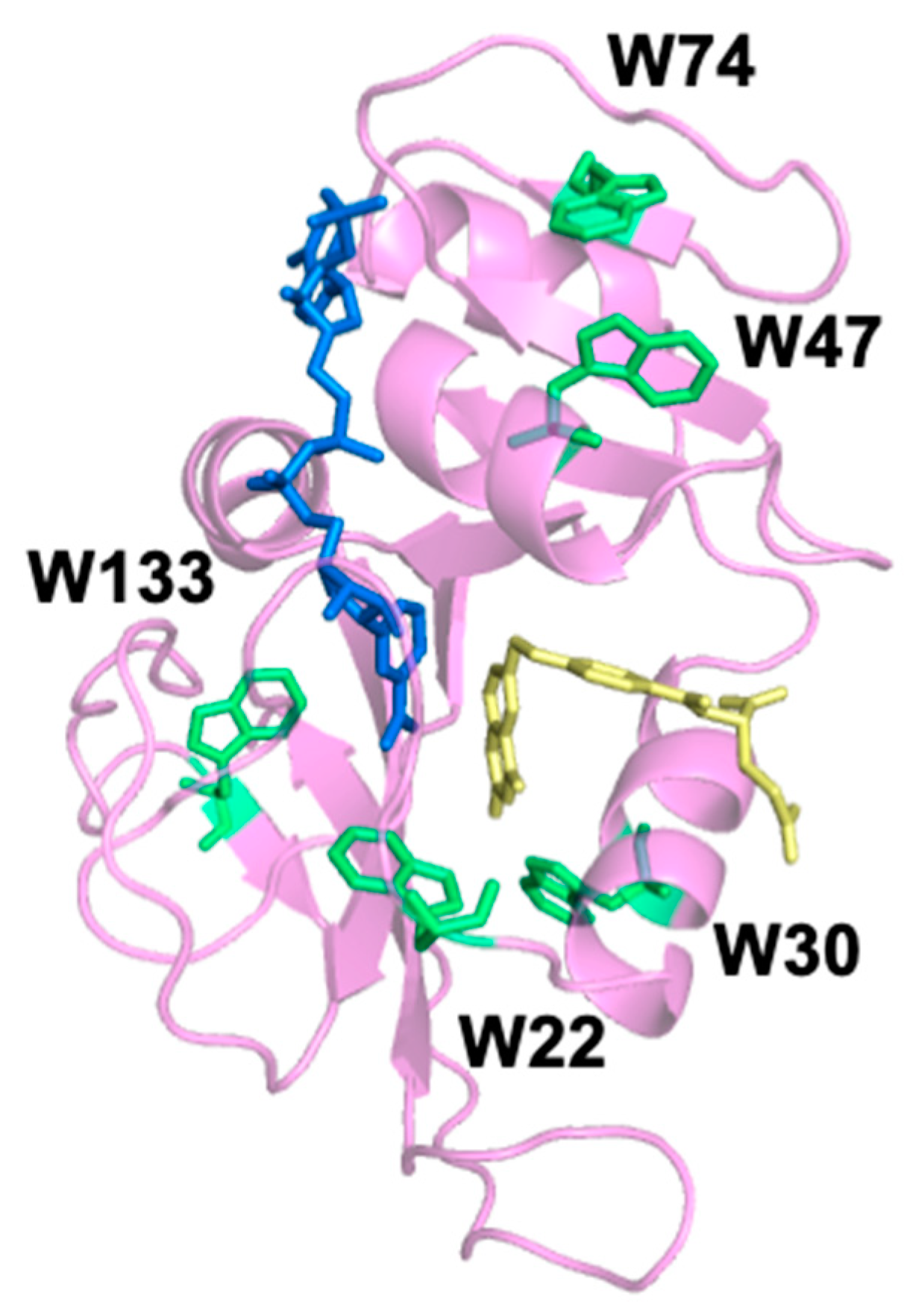
Table 4. Since the slow relaxation rate is concentration independent, the average rate is the combined average of the five replicates at the five ligand concentrations (n = 25). W47 and W74 are spatially close together; they are located at the top of DHFR when orientated as shown in
Figure 1. MidW47 and midW74 have close average slow relaxation rates of approximately 730 s
−1. The slow phase relaxation rates for MidW22 and midW30 are substantially faster, in excess of 1000 s
−1 and are within one standard deviation of one another. Similarly, W22 and W30 are close together spatially; they are located near the substrate binding site. Lastly, W133 is located on a beta sheet near the distal FG loop, and midW133 has the slowest average slow phase relaxation rate of 370 s
−1. To compare, the binary complex of wt-DHFR with folate has a slow concentration-independent relaxation rate of 400 s
−1. This is slower than all but one of the concentration-independent rates observed in the midW mutants. A linear combination of the relaxation rates of midW mutants would not result in the relaxation rate observed in the wild-type enzyme unless the wt-DHFR transients were almost completely dominated by the W133 signal with very little contribution from the other four Trp residues. Our equilibrium fluorescence results show that midW133 has a low fluorescence intensity compared to the other midW mutants. Additionally, Ohmae et al. showed that when W133 is replaced with phenylalanine, the tryptophan fluorescence of DHFR is reduced by less than 15% [
20]. Therefore, it is unlikely that W133 is dominating the fluorescence signal in wt-DHFR. Instead, Trp–Trp interactions as well as the local environment of the Trp residues contribute to the wt-DHFR transients, similar to what we observed with the equilibrium fluorescence. Thus, the motions reported in the multi-Trp system are fundamentally different than the motions reported in the single Trp systems. We conclude that the wt-DHFR signal is dominated by Trp coupling effects due to relative changes in the distance and orientation of the Trp residues, which means that the wt-DHFR transients are reporting on global motions throughout the enzyme. Conversely, the signals from the midW mutants report on local motions that affect the environment of the single tryptophan in the enzyme, as evidenced by the dependence of the slow relaxation rate on the Trp location.
By comparing the relaxation rates of the midW mutants, we can gain information about the environments of the individual Trp residues. Of particular interest are midW22 and midW30 because they both exhibit faster relaxation rates compared to the other midW mutants. The faster relaxation rate indicates that W22 and W30 are sensitive to conformational changes of a local flexible structure. MidW30 has a lower melting temperature compared to wt-DHFR and the other midW mutants. During the T-jump experiments, samples are transiently heated to 36 °C, which is 6 °C lower than the melting temperature of midW30. It is possible that unfolding events may be playing a role. Another possibility is that W22 and W30 could be reporting on the same motion. W30 is located near the active site, and W22 is located at the end of the Met20 loop, which is known to change conformations during the catalytic cycle: opening and closing over the active site as well as protruding into the active site. The conformation of the Met20 loop can be modulated by ligand binding. In the binary complex with folate, the Met20 loop is in the occluded conformation, and in the apoenzyme, the Met20 loop is in the open conformation. Since W22 and W30 are on or near the Met20 loop, it is reasonable that they could be sensitive to the Met20 loop motions. The slow relaxation rates of midW22 and midW30 are approaching that of the fast concentration-dependent relaxation rate. The strong concentration dependence of the faster relaxation rate indicates that it is strongly coupled to ligand association/dissociation. The slow relaxation rate for midW22 and midW30 is not concentration dependent, which means that the slow relaxation rate is not coupled to ligand binding. This suggests that the observed slow relaxation rate is not the open to occluded transition. However, the slow relaxation event sensed by W22 and W30 is still likely related to Met20 loop fluctuations due to the location of the Trp probes and the observed timescale. Loop motions typically occur in tens of nanoseconds to hundreds of microseconds, [
1,
25] and the slow relaxation events of midW22 and midW30 occur at approximately 300–500 microseconds. This is in contrast to what is observed with wt-DHFR and the other midW mutants where the slow relaxation events occur on the order of milliseconds.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}