
Effects of N-Substituents on the Functional Activities of Naltrindole Derivatives for the δ Opioid Receptor: Synthesis and Evaluation of Sulfonamide Derivatives

 ,
, 
Abstract
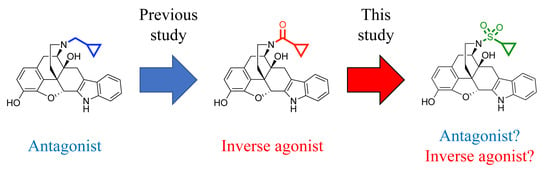
1. Introduction
2. Results and Discussion
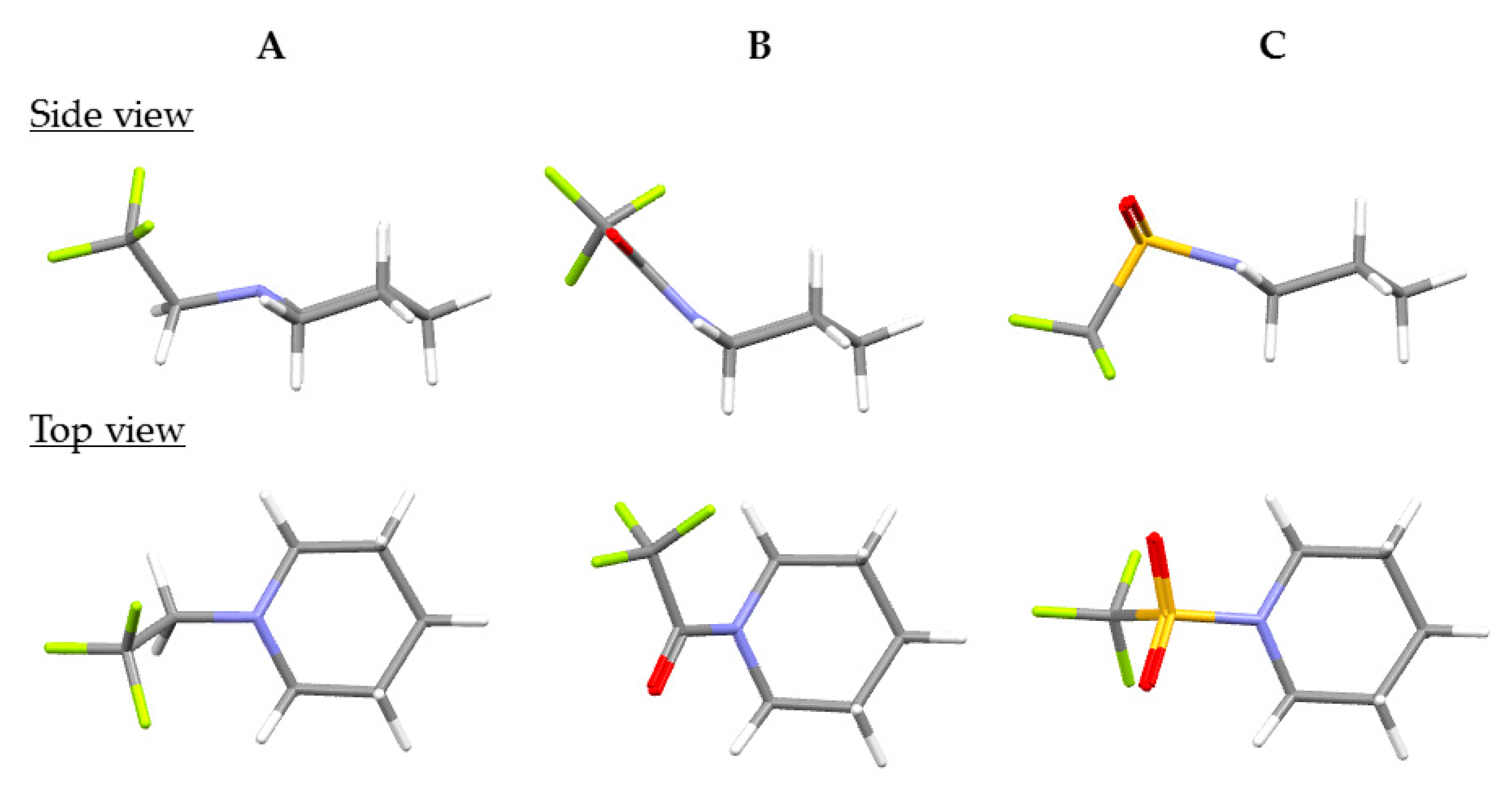
2.1. The Most Stable Conformations of the Corresponding Alkyl-, Amide-, and Sulfonamide-Type Compounds and the Comparison of the Basicities of Their Nitrogen Atoms
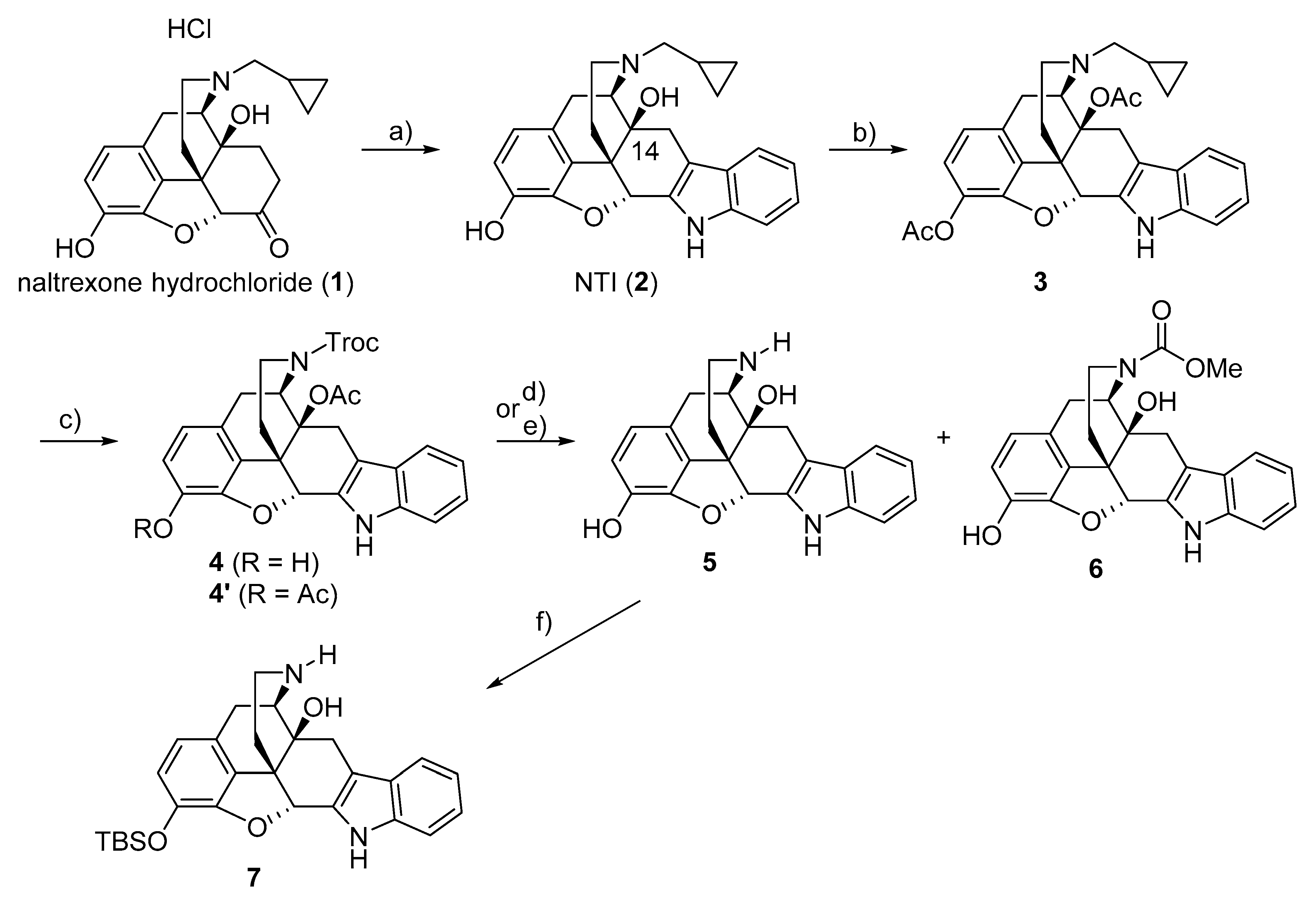
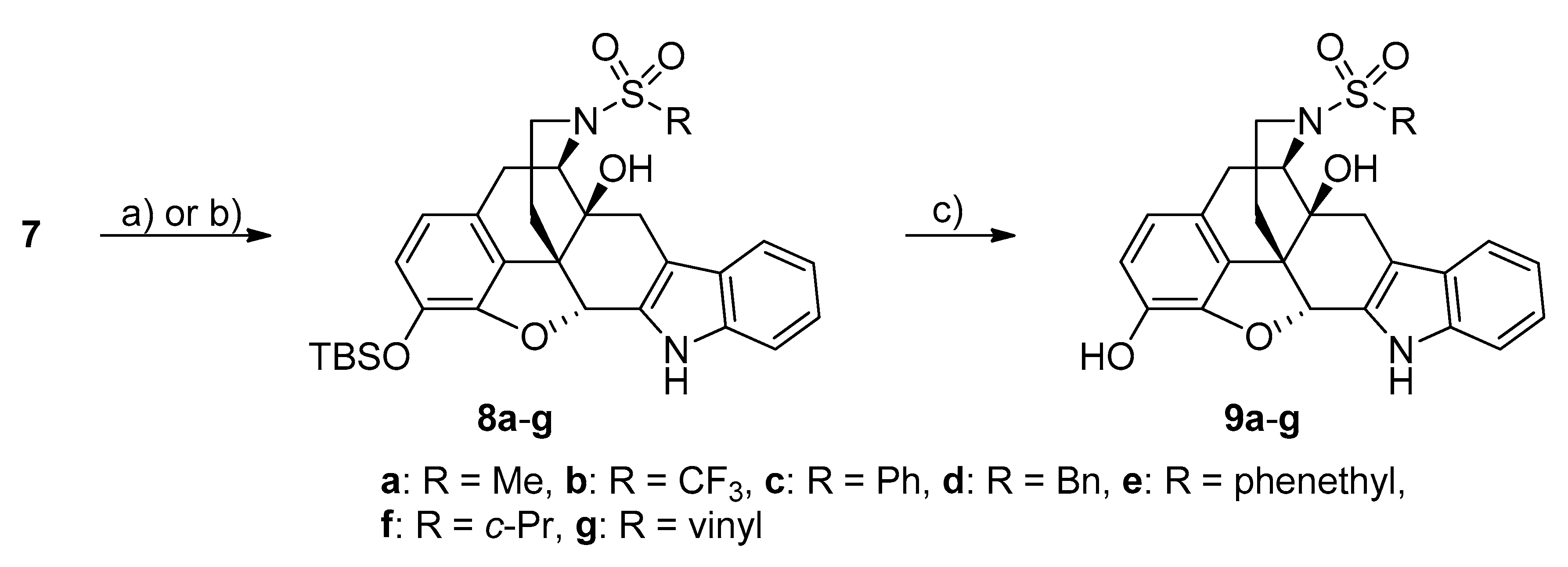
2.2. Chemical Synthesis
2.3. Binding Affinity and Functional Activity
3. Materials and Methods
3.1. General Information
3.2. Procedures for the Synthesis All the New Compounds and Their Spectroscopic Data
3.2.1. 17-Cyclopropyl-6,7-didehydro-4,5α-epoxyindolo[2′,3′:6,7]morphinan-3,14β-diyl Diacetate (3)
3.2.2. 6,7-Didehydro-4,5α-epoxyindolo[2′,3′:6,7]morphinan-3,14β-diol (nor-NTI) (5)
3.2.3. Methyl 6,7-Didehydro-4,5α-epoxy-3,14β-dihydorxyindolo[2′,3′:6,7]morphinan-17-ylcarboxylate (6)
3.2.4. General Synthesis of Sulfonamides 8
3-((tert-Butyldimethylsilyl)oxy)-6,7-didehydro-4,5α-epoxy-17-(methylsulfonyl)indolo[2′,3′:6,7]morphinan-14β-ol (8a)
3-((tert-Butyldimethylsilyl)oxy)-6,7-didehydro-4,5α-epoxy-17-(1,1,1-trifluoromethylsulfonyl)indolo[2′,3′:6,7]morphinan-14β-ol (8b)
3-((tert-Butyldimethylsilyl)oxy)-6,7-didehydro-4,5α-epoxy-17-(phenylsulfonyl)indolo[2′,3′:6,7]morphinan-14β-ol (8c)
17-(Benzylsulfonyl)-3-((tert-butyldimethylsilyl)oxy)-6,7-didehydro-4,5α-epoxyindolo[2′,3′:6,7]morphinan-14β-ol (8d)
3-((tert-Butyldimethylsilyl)oxy)-6,7-didehydro-4,5α-epoxy-17-((2-phenyethyl)sulfonyl)indolo[2′,3′:6,7]morphinan-14β-ol (8e)
3-((tert-Butyldimethylsilyl)oxy)-17-(cyclopropylsulfonyl)-6,7-didehydro-4,5α-epoxyindolo[2′,3′:6,7]morphinan-14β-ol (8f)
3.2.5. 3-((tert-Butyldimethylsilyl)oxy)-6,7-didehydro-4,5α-epoxy-17-(ethenylsulfonyl)indolo[2′,3′:6,7]morphinan-14β-ol (8g)
3.2.6. General Synthesis of Test Compounds 9
6,7-Didehydro-4,5α-epoxy-17-(methylsulfonyl)indolo[2′,3′:6,7]morphinan-3,14β-diol (9a)
6,7-Didehydro-4,5α-epoxy-17-(1,1,1-trifluoromethylsulfonyl)indolo[2′,3′:6,7]morphinan-3,14β-diol (9b)
6,7-Didehydro-4,5α-epoxy-17-(phenylsulfonyl)indolo[2′,3′:6,7]morphinan-3,14β-diol (9c)
17-(Benzylsulfonyl)-6,7-didehydro-4,5α-epoxyindolo[2′,3′:6,7]morphinan-3,14β-diol (9d)
6,7-Didehydro-4,5α-epoxy-17-((2-phenylethyl)sulfonyl)indolo[2′,3′:6,7]morphinan-3,14β-diol (9e)
17-(Cyclopropylsulfonyl)-6,7-didehydro-4,5α-epoxyindolo[2′,3′:6,7]morphinan-3,14β-diol (9f)
6,7-Didehydro-4,5α-epoxy-17-(ethenylsulfonyl)indolo[2′,3′:6,7]morphinan-3,14β-diol (9g)
3.3. Calculation of the Most Stable Conformations of Alkyl-, Amide-, and Sulfonamide-Type Piperidines
3.4. Bioassays
3.4.1. Membrane Preparation
3.4.2. Competitive Binding Assays
3.4.3. [35S]GTPγS Binding Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chalmers, D.T.; Behan, D.P. The use of constitutively active gpcrs in drug discovery and functional genomics. Nat. Rev. Drug Discov. 2002, 1, 599–608. [Google Scholar] [CrossRef]
- Costa, T.; Cotecchia, S. Historical review: Negative efficacy and the constitutive activity of G-protein coupled receptors. Trends Pharmacol. Sci. 2005, 26, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Bond, R.A.; IJzerman, A.P. Recent developments in constitutive receptor activity and inverse agonism, and their potential for GPCR drug discovery. Trends Pharmacol. Sci. 2006, 27, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, S.; Fujii, H. δ Opioid Receptor Inverse Agonists and their In Vivo Pharmacological Effects. Curr. Top. Med. Chem. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Recent examples (from ref. 5 to ref. 20), Cherney, R.J.; Cornelius, L.A.M.; Srivastava, A.; Weigelt, C.A.; Marcoux, D.; Duan, J.J.-W.; Shi, Q.; Batt, D.G.; Liu, Q.; Yip, S.; et al. Discovery of BMS-986251: A Clinically Viable, Potent, and Selective RORγt Inverse Agonist. ACS Med. Chem. Lett. 2020, 11, 1221–1227. [Google Scholar] [CrossRef]
- Gege, C.; Albers, M.; Kinzel, O.; Kleymann, G.; Schlütera, T.; Steeneck, C.; Hoffmann, T.; Xue, X.; Cummingsc, M.D.; Spurlinoc, J.; et al. Optimization and biological evaluation of thiazole-bis-amide inverse agonists of RORγt. Bioorg. Med. Chem. Lett. 2020, 30, 127205. [Google Scholar] [CrossRef]
- Nakajima, R.; Oono, H.; Sugiyama, S.; Matsueda, Y.; Ida, T.; Kakuda, S.; Hirata, J.; Baba, A.; Makino, A.; Matsuyama, R.; et al. Discovery of [1,2,4]Triazolo[1,5-a]pyridine Derivatives as Potent and Orally Bioavailable RORγt Inverse Agonists. ACS Med. Chem. Lett. 2020, 11, 528–534. [Google Scholar] [CrossRef]
- Sun, N.; Ma, X.; Zhou, K.; Zhu, C.; Cao, Z.; Wang, Y.; Xu, J.; Fu, W. Discovery of novel N-sulfonamide- tetrahydroquinolines as potent retinoic acid receptor-related orphan receptor γt inverse agonists for the treatment of autoimmune diseases. Eur. J. Med. Chem. 2020, 187, 111984. [Google Scholar] [CrossRef]
- Shrader, S.H.; Song, Z. Discovery of endogenous inverse agonists for G protein-coupled receptor 6. Biochem. Biophys. Res. Commun. 2020, 522, 1041–1045. [Google Scholar] [CrossRef]
- Poli, G.; Dimmito, M.P.; Mollica, A.; Zengin, G.; Benyhe, S.; Zador, F.; Stefanucci, A. Discovery of Novel μ-Opioid Receptor Inverse Agonist from a Combinatorial Library of Tetrapeptides through Structure-Based Virtual Screening. Molecules 2019, 24, 3872. [Google Scholar] [CrossRef]
- Sato, A.; Fukase, Y.; Kono, M.; Ochida, A.; Oda, T.; Sasaki, Y.; Ishii, N.; Tomata, Y.; Fukumoto, S.; Imai, Y.N. Design and Synthesis of Conformationally Constrained RORγt Inverse Agonists. ChemMedChem 2019, 14, 1917–1932. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, N.S.; Iyer, J.P.; Munot, Y.S.; Mukhopadhyay, P.P.; Raje, A.A.; Nagaraj, R.; Jamdar, V.; Gavhane, R.; Lohote, M.; Sherkar, P.; et al. Discovery and pharmacological evaluation of indole derivatives as potent and selective RORγt inverse agonist for multiple autoimmune conditions. Bioorg. Med. Chem. Lett. 2019, 29, 2208–2217. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Duan, J.J.-W.; Xiao, H.; Neels, J.; Wu, D.; Weigelt, C.A.; Sack, J.S.; Khan, J.; Ruzanov, M.; An, Y.; et al. Identification of potent, selective and orally bioavailable phenyl ((R)-3-phenylpyrrolidin-3-yl)sulfone analogues as RORγt inverse agonists. Bioorg. Med. Chem. Lett. 2019, 29, 2265–2269. [Google Scholar] [CrossRef] [PubMed]
- Amato, G.; Manke, A.; Wiethe, R.; Vasukuttan, V.; Snyder, R.; Yueh, Y.L.; Decker, A.; Runyon, S.; Maitra, R. Functionalized 6-(Piperidin-1-yl)-8,9-Diphenyl Purines as Peripherally Restricted Inverse Agonists of the CB1 Receptor. J. Med. Chem. 2019, 62, 6330–6345. [Google Scholar]
- Amaudrut, J.; Argiriadi, M.A.; Barth, M.; Breinlinger, E.C.; Bressac, D.; Broqua, P.; Calderwood, D.J.; Chatar, M.; Cusac, K.P.; Gauld, S.B.; et al. Discovery of novel quinoline sulphonamide derivatives as potent, selective and orally active RORγ inverse agonists. Bioorg. Med. Chem. Lett. 2019, 29, 1799–1806. [Google Scholar] [CrossRef]
- Tanis, V.M.; Venkatesan, H.; Cummings, M.D.; Albers, M.; Barbay, J.K.; Herman, K.; Kummer, D.A.; Milligan, C.; Nelen, M.I.; Nishimura, R.; et al. 3-Substituted Quinolines as RORγt Inverse Agonists. Bioorg. Med. Chem. Lett. 2019, 29, 1463–1470. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, X.; Xue, X.; Li, C.; Wang, J.; Wang, R.; Zhang, C.; Wang, C.; Shi, Y.; Zou, L.; et al. Discovery and Characterization of XY101, a Potent, Selective, and Orally Bioavailable RORγ Inverse Agonist for Treatment of Castration-Resistant Prostate Cancer. J. Med. Chem. 2019, 62, 4716–4730. [Google Scholar]
- Troxler, T.; Feuerbach, D.; Zhang, X.; Yang, C.R.; Lagu, B.; Perrone, M.; Wang, T.-L.; Briner, K.; Bock, M.G.; Auberson, Y.P. The Discovery of LML134, a Histamine H3 Receptor Inverse Agonist for the Clinical Treatment of Excessive Sleep Disorders. ChemMedChem 2019, 14, 1238–1247. [Google Scholar] [CrossRef]
- Kim, J.; Song, J.; Ji, H.D.; Yoo, E.K.; Lee, J.-E.; Lee, S.B.; Oh, J.M.; Lee, S.; Hwang, J.S.; Yoon, H.; et al. Discovery of Potent, Selective, and Orally Bioavailable Estrogen-Related Receptor-γ Inverse Agonists To Restore the Sodium Iodide Symporter Function in Anaplastic Thyroid Cancer. J. Med. Chem. 2019, 62, 1837–1858. [Google Scholar]
- Nirogi, R.; Shinde, A.; Mohammed, A.R.; Badange, R.K.; Reballi, V.; Bandyala, T.R.; Saraf, S.K.; Bojja, K.; Manchineella, S.; Achanta, P.K.; et al. Discovery and Development of N-[4-(1-Cyclobutylpiperidin-4-yloxy)phenyl]-2-(morpholin-4-yl)acetamide Dihydrochloride (SUVNG3031): A Novel, Potent, Selective, and Orally Active Histamine H3 Receptor Inverse Agonist with Robust Wake-Promoting Activity. J. Med. Chem. 2019, 62, 1203–1217. [Google Scholar]
- Costa, T.; Herz, A. Antagonists with negative intrinsic activity at δ opioid receptors coupled to GTP-binding proteins. Proc. Natl. Acad. Sci. USA 1989, 86, 7321–7325. [Google Scholar] [CrossRef] [PubMed]
- Shaw, W.N. Long-term treatment of obese Zucker rats with LY255582 and other appetite suppressants. Pharmacol. Biochem. Behav. 1993, 46, 653–659. [Google Scholar] [CrossRef]
- Emmerson, P.J.; McKinzie, J.H.; Surface, P.L.; Suter, T.M.; Mitch, C.H.; Statnick, M.A. Na+ modulation, inverse agonism, and anorectic potency of 4-phenylpiperidine opioid antagonists. Eur. J. Pharmacol. 2004, 494, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, S.; Iwai, T.; Higashi, E.; Nakamura, M.; Iwamatsu, C.; Itoh, K.; Nemoto, T.; Tanabe, M.; Fujii, H. Discovery of δ opioid receptor full inverse agonists and their effects on restraint stress induced cognitive impairment in mice. ACS Chem. Neurosci. 2019, 10, 2237–2242. [Google Scholar] [CrossRef] [PubMed]
- Higashi, E.; Hirayama, S.; Nikaido, J.; Shibasaki, M.; Kono, T.; Honjo, A.; Ikeda, H.; Kamei, J.; Fujii, H. Development of novel δ opioid receptor inverse agonists without a basic nitrogen atom and their antitussive effects in mice. ACS Chem. Neurosci. 2019, 10, 3939–3945. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, T.; Iihara, Y.; Hirayama, S.; Iwai, T.; Higashi, E.; Fujii, H.; Nagase, H. Naltrindole derivatives with fluorinated ethyl substituents on the 17-nitrogen as δ opioid receptor inverse agonists. Bioorg. Med. Chem. Lett. 2015, 25, 2927–2930. [Google Scholar] [CrossRef]
- Portoghese, P.S.; Sultana, M.; Nagase, H.; Takemori, A.E. Application of the message-address concept in the design of highly potent and selective non-peptide δ opioid receptor antagonists. J. Med. Chem. 1988, 31, 281–282. [Google Scholar] [CrossRef]
- Portoghese, P.S.; Sultana, M.; Takemori, A.E. Design of peptidomimetic δ opioid receptor antagonists using the message-address concept. J. Med. Chem. 1990, 33, 1714–1720. [Google Scholar] [CrossRef]
- Fujii, H.; Uchida, Y.; Shibasaki, M.; Nishida, M.; Yoshioka, T.; Kobayashi, R.; Honjo, A.; Itoh, K.; Yamada, D.; Hirayama, S.; et al. Discovery of δ opioid receptor full agonists lacking a basic nitrogen atom and their antidepressant-like effects. Bioorg. Med. Chem. Lett. 2020, 30, 127176. [Google Scholar] [CrossRef]
- Casy, A.F.; Parfitt, R.T. Opioid Analgesics: Chemistry and Receptors; Plenum Press: New York, NY, USA, 1986; pp. 9–104. [Google Scholar]
- Che, T.; Majumdar, S.; Zaidi, S.A.; Ondachi, P.; McCorvy, J.D.; Wang, S.; Mosier, P.D.; Uprety, R.; Vardy, E.; Krumm, B.E.; et al. Structure of the Nanobody-Stabilized Active State of the Kappa Opioid Receptor. Cell 2018, 172, 55–67. [Google Scholar] [CrossRef]
- Huang, W.; Manglik, A.; Venkatakrishnan, A.J.; Laeremans, T.; Feinberg, E.N.; Sanborn, A.L.; Kato, H.E.; Livingston, K.E.; Thorsen, T.S.; Kling, R.C.; et al. Structural insights into μ-opioid receptor activation. Nature 2015, 524, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Fenalti, G.; Giguere, P.M.; Katritch, V.; Huang, X.-P.; Thompson, A.A.; Cherezov, V.; Roth, B.L.; Stevens, R.C. Molecular control of δ-opioid receptor signaling. Nature 2014, 506, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Granier, S.; Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Weis, W.I.; Kobilka, B.K. Structure of the δ-opioid receptor bound to naltrindole. Nature 2012, 485, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Mafi, A.; Kim, S.-K.; Goddard, W.A., III. The atomistic level structure for the activated human κ-opioid receptor bound to the full Gi protein and the MP1104 agonist. Proc. Natl. Acad. Sci. USA 2020, 117, 5836–5843. [Google Scholar] [CrossRef] [PubMed]
- Koehl, A.; Hu, H.; Maeda, S.; Zhang, Y.; Qu, Q.; Paggi, J.M.; Latorraca, N.R.; Hilger, D.; Dawson, R.; Matile, H.; et al. Structure of the μ-opioid receptor–Gi protein complex. Nature 2018, 558, 547–552. [Google Scholar] [CrossRef]
- Roth, B.L.; Baner, K.; Westkaemper, R.; Siebert, D.; Rice, K.C.; Steinberg, S.; Ernsberger, P.; Rothman, R.B. Salvinorin A: A potent naturally occurring non-nitrogenous κ opioid selective agonist. Proc. Natl. Acad. Sci. USA 2002, 99, 11934–11939. [Google Scholar] [CrossRef]
- Yamaotsu, N.; Fujii, H.; Nagase, H.; Hirono, S. Identification of the three-dimensional pharmacophore of κ-opioid receptor agonists. Bioorg. Med. Chem. 2010, 18, 4446–4452. [Google Scholar] [CrossRef]
- Olofson, R.A.; Martz, J.T.; Senet, J.P.; Piteau, M.; Malfroot, T. A New Reagent for the Selective, High-Yield N-Dealkylation of Tertiary Amines: Improved Syntheses of Naltrexone and Nalbuphine. J. Org. Chem. 1984, 49, 2081–2082. [Google Scholar] [CrossRef]
- Olofson, R.A.; Schnur, R.C.; Bunes, L.; Pepe, J.P. Selective N-dealkylation of tertiary amines with vinyl chloroformate: An improved synthesis of naloxone. Tetrahedron Lett. 1977, 18, 1567–1570. [Google Scholar] [CrossRef]
- Montzka, T.A.; Matiskella, J.D.; Partyka, R.A. 2,2,2-trichloroethyl chloroformate: A general reagent for demethylation of tertiary methylamines. Tetrahedron Lett. 1974, 15, 1325–1327. [Google Scholar] [CrossRef]
- Fujii, H.; Imaide, S.; Watanabe, A.; Nemoto, T.; Nagase, H. Novel cleavage reaction of the C16–N17 bond in naltrexone derivatives. Tetrahedron Lett. 2008, 49, 6293–6296. [Google Scholar] [CrossRef]
- Wuts, P.G.M. Greene’s Protective Groups in Organic Synthesis, 5th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2014; pp. 921–923. [Google Scholar]
- McLamore, S.; Ullrich, T.; Rothman, R.B.; Xu, H.; Dersch, C.; Coop, A.; Davis, P.; Porreca, F.; Jacobson, A.E.; Rice, K.C. Effect of N-Alkyl and N-Alkenyl Substituents in Noroxymorphindole, 17-Substituted-6,7-dehydro-4,5α-epoxy-3,14-dihydroxy-6,7:2′,3′-indolomorphinans, on Opioid Receptor Affinity, Selectivity, and Efficacy. J. Med. Chem. 2001, 44, 1471–1474. [Google Scholar] [CrossRef] [PubMed]
- Portoghese, P.S.; Larson, D.L.; Sultana, M.; Takemori, A.E. Opioid Agonist and Antagonist Activities of Morphindoles Related to Naltrindole. J. Med. Chem. 1992, 35, 4325–4329. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Karaki, F.; Tayama, K.; Higashi, E.; Hirayama, S.; Itoh, K.; Fujii, H. C-Homomorphinan Derivatives as Lead Compounds to Obtain Safer and More Clinically Useful Analgesics. Chem. Pharm. Bull. 2017, 65, 920–929. [Google Scholar] [CrossRef] [PubMed]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.A.; Cao, Y.; Gunn, J.R.; Day, T.; Gallicchio, E.; Zhou, Z.; Levy, R.; Farid, R. PrimeX and the Schrödinger Computational Chemistry Suite of Programs. Int. Tables Crystallogr. 2012, 534–538. [Google Scholar]
- Jorgensen, W.L.; Tirado-Rives, J. The OPLS [Optimized Potentials for Liquid Simulations] Potential Functions for Proteins, Energy Minimizations for Crystals of Cyclic Peptides and Crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of Absolute Solvation Free Energies using Molecular Dynamics Free Energy Perturbation and the OPLS Force Field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the Comprehensive, Rapid, and Accurate Prediction of the Favorable Tautomeric States of Drug-Like Molecules in Aqueous Solution. J. Comput.-Aided Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Shelley, J.C.; Cholleti, A.; Frye, L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A Software Program for pKa Prediction and Protonation State Generation for Drug-Like Molecules. J. Comput.-Aided Mol. Design 2007, 21, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford CT, UK, 2009. [Google Scholar]
Sample Availability: Samples of SYK-623, SYK-753, SYK-839, SYK-901, and SYK-903 are available from the authors. |





{kind=link}
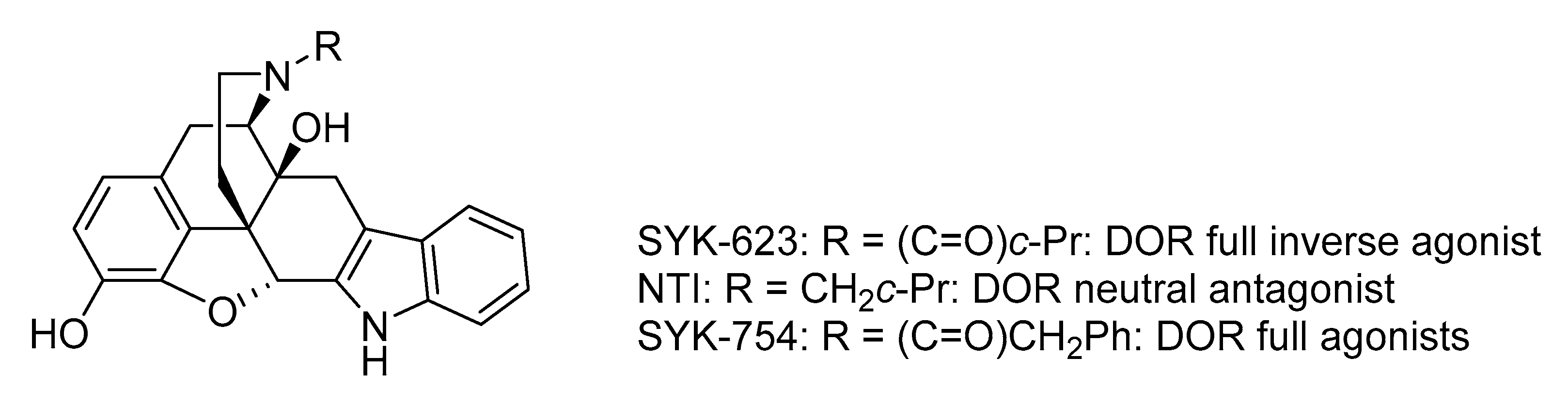{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compd | R | Binding Affinity, nM (95% CI) | Functional Activity for DOR | |||
|---|---|---|---|---|---|---|
| Ki (DOR) | Ki (MOR) | Ki (KOR) | EC50, nM (95% CI) | Emax, % (95% CI) | ||
| DPDPE c | ― | NT d | NT d | NT d | 4.66 (2.08–10.4) | 100 e |
| ICI-174,864 c | ― | 422 (215–829) | NT d | NT d | 114 (67.9–192) | −100 f |
| NTI c | ― | 0.457 (0.192–1.09) | 30.7 (12.5–75.4) | 14.7 (3.16–68.5) | ND g | 7.50 h |
| 9a | Me | 284 (151–535) | 10,100 (1920–16,600) | ND g | 0.468 (0.0816–2.68) | −48.8 (−58.2–−39.5) |
| 9b | CF3 | 365 (193–692) | 11,800 (1740–80,300) | 675 (210–2170) | ND g | −36.1 h |
| 9c | Ph | 49.6 (26.2–93.9) | 20,500 (1560–269,000) | 95.6 (41.4–220) | ND g | ND g |
| 9d | Bn | 734 (326–1660) | 5180 (1930–13,900) | 18,800 (6010–58,800) | 310 (82.1–177) | 47.1 (36.9–57.3) |
| 9e | Phenethyl | 132 (57.0–304) | 1400 (261–7440) | 17,800 (6840–46,300) | 75.8 (39.5–146) | 88.1 (79.2–97.0) |
| 9f | c-Pr | 7.44 (3.53–15.7) | 3900 (1450–10,500) | 13.6 (5.45–33.7) | 1.59 (0.825–3.05) | −80.5 (−87.7–−73.4) |
| 9g | vinyl | 196 (87.5–440) | 3570 (1420–8970) | 59,100 (2190–160,000) | 7.80 (1.58–38.4) | −80.2 (−97.8–−62.6) |

| R | Alkyl-Type, X = CH2 (95% CI) | Amide-Type, X = CO (95% CI) | Sulfonamide-Type, X = SO2 (95% CI) |
|---|---|---|---|
| Me | SYK-323 a Ki = 2.71 nM (1.93–3.82) EC50: ND c Emax: ND c | SYK-747 b Ki = 977 nM (332–2,880) EC50: ND c Emax = −4.86% d | 9a (SYK-884) Ki = 284 nM (151–535) EC50 = 0.468 nM (0.0816–2.68) Emax = −48.8% (−58.2–−39.5) |
| CF3 | SYK-165 a Ki = 134 nM (75.4–239) EC50 = 45.5 nM (17.5–118) Emax = −44.8% (−51.9–−37.7) | SYK-752 b Ki = 7.84 nM (4.30–14.3) EC50: ND c Emax = −1.10% d | 9b (SYK-837) Ki = 365 nM (193–692) EC50: ND c Emax = −36.1% d |
| Ph | SYK-619 e Ki = 15.8 nM (7.23–34.7) EC50 = 16.1 nM (7.31–35.4) Emax = −56.5% (−63.3–−49.8) | SYK-736 b Ki = 290 nM (177–476) EC50 = 322 nM (51.5–2,010) Emax = −92.7% (−122–−62.6) | 9c (SYK-838) Ki = 49.6 nM (26.2–93.9) EC50: ND c Emax: ND c |
| Bn | SYK-707 e Ki = 1.94 nM (1.50–2.50) EC50 = 15.1 nM (2.67–85.0) Emax = 40.3% (30.4–50.2) | SYK-754 f Ki = 89.0 nM (66.6–119) EC50 = 127 nM (80.1–201) Emax = 88.5% (81.6–95.3) | 9d (SYK-887) Ki = 734 nM (326–1,660) EC50 = 310 nM (82.1–117) Emax = 47.1% (36.9–57.3) |
| Phenethyl | SYK-903 g Ki = 166 nM (65.5–419) EC50 = 7.37 nM (3.80–14.3) Emax = 105% (96.2–114) | SYK-753 f Ki = 41.5 nM (28.5–60.6) EC50 = 132 nM (51.5–336) Emax = 97.5% (82.6–112) | 9e (SYK-901) Ki = 132 nM (57.0–304) EC50 = 75.8 nM (39.5–146) Emax = 88.1% (79.2–97.0) |
| c-Pr | NTI e Ki = 0.46 nM (0.192–1.09) EC50: ND c Emax = 7.50% d | SYK-623 b Ki = 17.3 nM (10.3–28.9) EC50 = 0.969 nM (0.406–2.93) Emax = −91.2% (−99.8–−82.7) | 9f (SYK-839) Ki = 7.44 nM (3.53–15.7) EC50 = 1.59 nM (0.825–3.05) Emax = −80.5% (−87.7–−73.4) |
| vinyl | SYK-706 e Ki = 0.609 nM (0.407–0.913) EC50 = 101 nM (4.46–2,280) Emax = 16.2% (8.14–24.3) | SYK-836 b Ki = 1.16 nM (0.508–2.63) EC50 = 1.43 nM (0.819–2.59) Emax = −86.2% (−92.2–−80.2) | 9g (SYK-886) Ki = 196 nM (87.5–440) EC50 = 7.80 nM (1.58–38.4) Emax = −80.2% (−97.8–−62.6) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iwamatsu, C.; Hayakawa, D.; Kono, T.; Honjo, A.; Ishizaki, S.; Hirayama, S.; Gouda, H.; Fujii, H. Effects of N-Substituents on the Functional Activities of Naltrindole Derivatives for the δ Opioid Receptor: Synthesis and Evaluation of Sulfonamide Derivatives. Molecules 2020, 25, 3792. https://doi.org/10.3390/molecules25173792
Iwamatsu C, Hayakawa D, Kono T, Honjo A, Ishizaki S, Hirayama S, Gouda H, Fujii H. Effects of N-Substituents on the Functional Activities of Naltrindole Derivatives for the δ Opioid Receptor: Synthesis and Evaluation of Sulfonamide Derivatives. Molecules. 2020; 25(17):3792. https://doi.org/10.3390/molecules25173792
Chicago/Turabian StyleIwamatsu, Chiharu, Daichi Hayakawa, Tomomi Kono, Ayaka Honjo, Saki Ishizaki, Shigeto Hirayama, Hiroaki Gouda, and Hideaki Fujii. 2020. "Effects of N-Substituents on the Functional Activities of Naltrindole Derivatives for the δ Opioid Receptor: Synthesis and Evaluation of Sulfonamide Derivatives" Molecules 25, no. 17: 3792. https://doi.org/10.3390/molecules25173792
APA StyleIwamatsu, C., Hayakawa, D., Kono, T., Honjo, A., Ishizaki, S., Hirayama, S., Gouda, H., & Fujii, H. (2020). Effects of N-Substituents on the Functional Activities of Naltrindole Derivatives for the δ Opioid Receptor: Synthesis and Evaluation of Sulfonamide Derivatives. Molecules, 25(17), 3792. https://doi.org/10.3390/molecules25173792