
Putative SARS-CoV-2 Mpro Inhibitors from an In-House Library of Natural and Nature-Inspired Products: A Virtual Screening and Molecular Docking Study



Abstract
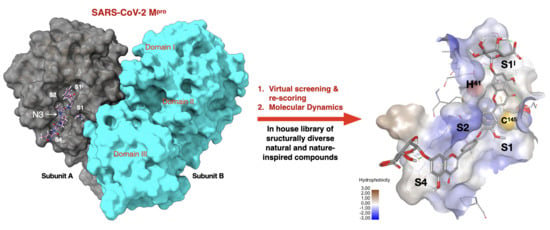
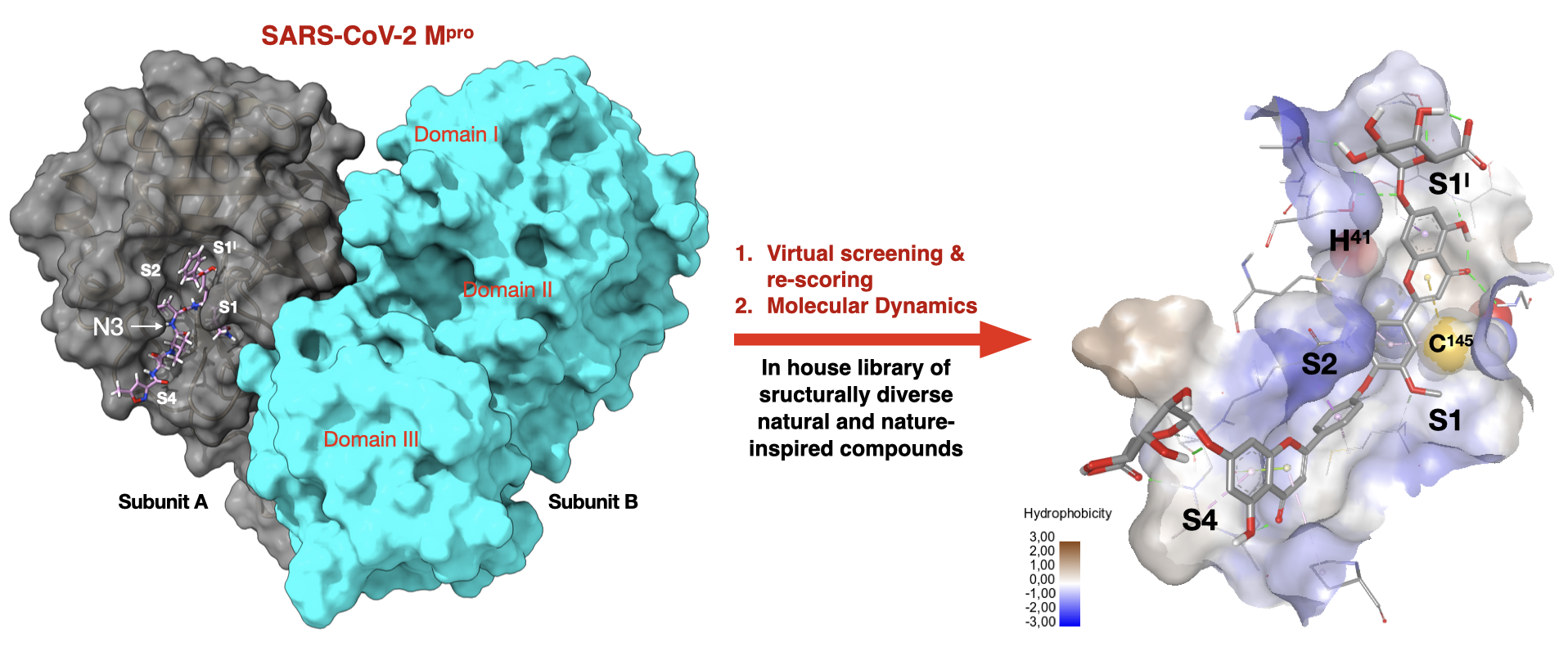
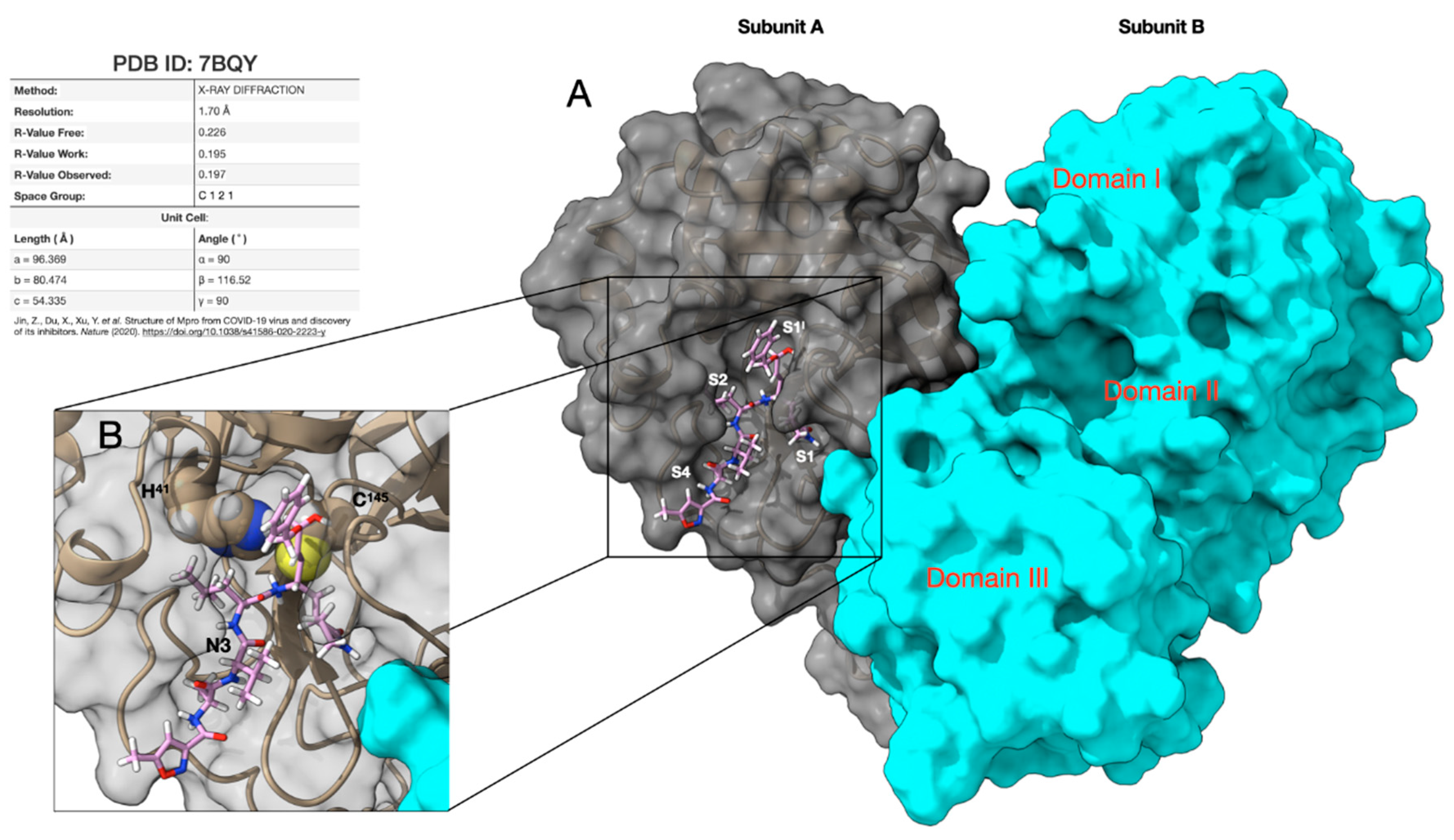
1. Introduction
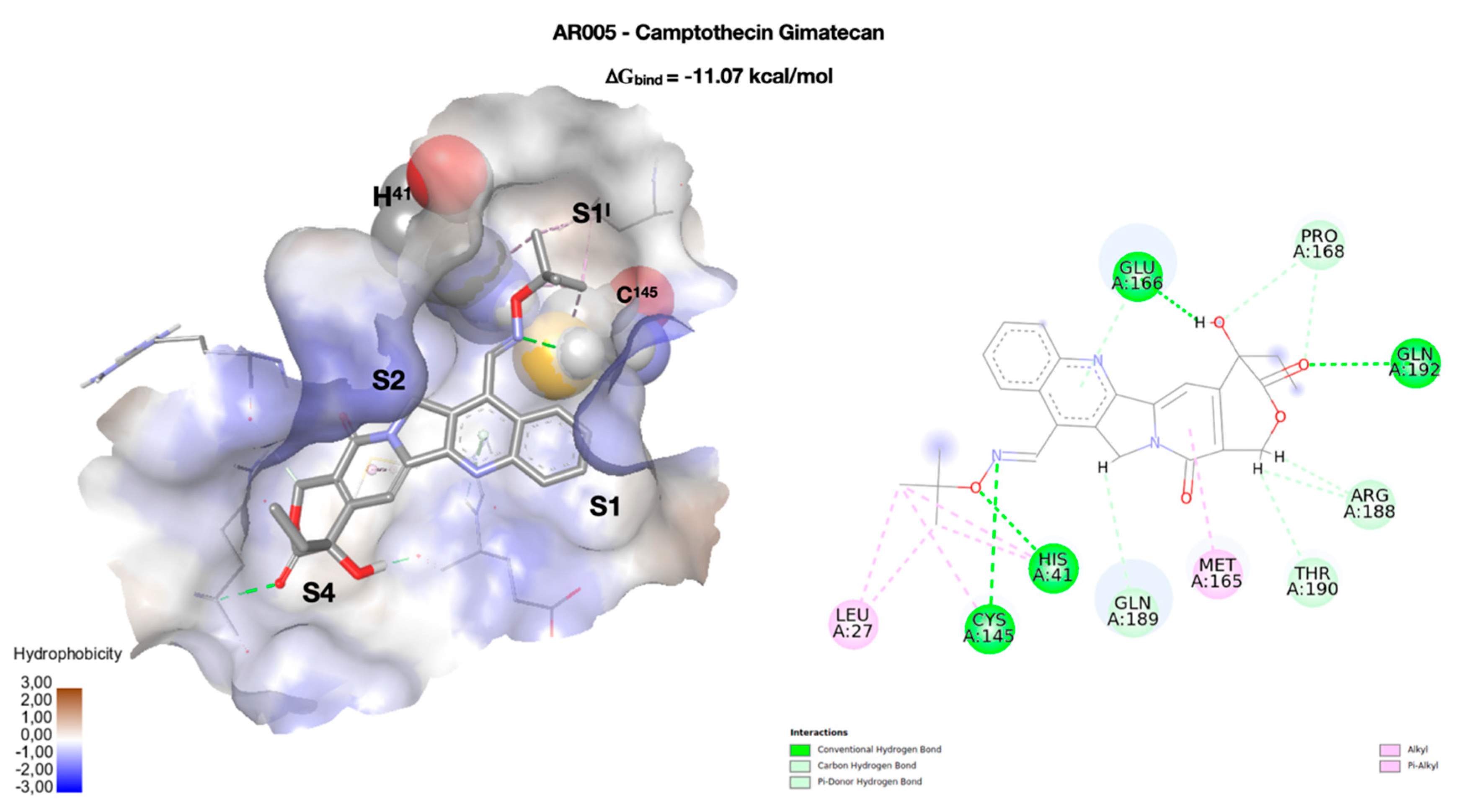
2. Results
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hui, D.S.; Azhar, E.I.; Madani, T.A.; Ntoumi, F.; Kock, R.; Dar, O.; Ippolito, G.; McHugh, T.D.; Memish, Z.A.; Drosten, C.; et al. The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health-The latest 2019 novel coronavirus outbreak in Wuhan, China. Int. J. Infect. Dis. 2020, 91, 264–266. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- WHO Director-General’s Opening Remarks at the Media Briefing on COVID-19–11th March 2020. Available online: https://www.who.int/dg/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19 (accessed on 15 March 2020).
- Maier, H.; Bickerton, E.; Britton, P. Coronaviruses. Methods and Protocols. Methods in Molecular Biology; Springer: Berlin, Germany, 2015; p. 1282. [Google Scholar]
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef]
- Sunil, K.; Lal, S.K. Molecular Biology of the SARS-Coronavirus; Springer: Berlin, Germany, 2010. [Google Scholar]
- Satatij, N.; Lal, S.K. The Molecular Biology of SARS Coronavirus. Ann. N. Y. Acad. Sci. 2007, 1102, 26–38. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-2. PNAS 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, X.; et al. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Yang, H.; Xie, W.; Xue, X.; Yang, K.; Ma, J.; Liang, W.; Zhao, Q.; Zhou, Z.; Pei, D.; Ziebuhr, J.; et al. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005, 3, e324. [Google Scholar] [CrossRef]
- Coronavirus (COVID-19) Update: FDA Issues Emergency Use Authorization for Potential COVID-19 Treatment. Available online: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-issues-emergency-use-authorization-potential-covid-19-treatment. (accessed on 1 May 2020).
- Treatments and Vaccines for COVID-1. Available online: https://www.ema.europa.eu/en/human-regulatory/overview/public-health-threats/coronavirus-disease-covid-19/treatments-vaccines-covid-19 (accessed on 5 July 2020).
- De Wit, E.; Feldmann, F.; Cronin, J.; Jordan, R.; Okumura, A.; Thomas, T.; Scott, D.; Cihlar, T.; Deldmann, H. Prophylactic and therapeutic remdesivir (GS-5734) treatment in the rhesus macaque model of MERS-CoV infection. Proc. Natl. Acad. Sci. USA 2020, 117, 6771–6776. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, X.; Lu, Y.; Chen, F.; Zhang, W. Clinical characteristics and therapeutic procedure for four cases with 2019 novel coronavirus pneumonia receiving combined Chinese and Western medicine treatment. Biosci. Trends 2020, 14, 64–68. [Google Scholar] [CrossRef]
- Arabi, Y.M.; Fowler, R.; Hayden, F.G. Critical care management of adults with community-acquired severe respiratory viral infection. Intensive Care Med. 2020, 46, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Wax, R.S.; Christian, M.D. Practical recommendations for critical care and anesthesiology teams caring for novel coronavirus (2019-nCoV) patients. Directives concretes a l’intention des equipes de soins intensifs et d’anesthesiologie prenant soin de patients atteints du coronavirus 2019-nCoV. Can. J. Anaesth. 2020, 67, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef]
- Bobrowski, T.; Alves, V.M.; Melo-Filho, C.C.; Korn, D.; Auerbach, S.; Schmitt, C.; Muratov, E.N.; Tropsha, A. Computational 1 models identify several FDA approved or experimental drugs as putative agents against SARS-CoV-2. ChemRxiv 2020, 1–20. [Google Scholar] [CrossRef]
- Gul, S.; Ozcan, O.; Asar, S.; Okyar, A.; Baris, I.; Kavakli, I.H. In silico identification of widely used and well tolerated drugs that may inhibit SARS-Cov-2 3C-like protease and viral RNA-dependent RNA polymerase activities, and may have potential to be directly used in clinical trials. ChemRxiv 2020, 1–49. [Google Scholar] [CrossRef]
- Wang, J. Fast identification of possible drug treatment of coronavirus disease-19 (COVID-19) through computational drug repurposing study. J. Chem. Inf. Model 2020, 60, 3277–3286. [Google Scholar] [CrossRef]
- Li, Z.; Li, X.; Huang, Y.Y.; Wu, Y.; Zhou, L.; Liu, R.; Wu, D.; Zhang, L.; Liu, H.; Xu, X.; et al. FEP-based screening prompts drug repositioning against COVID-19. BioRxiv 2020, 1–34. [Google Scholar] [CrossRef]
- Sekiou, O.; Bouziane, I.; Bouslama, Z.; Djemel, A. In-silico identification of potent inhibitors of COVID-19 main protease (Mpro) and angiotensin converting enzyme 2 (ACE2) from natural products: Quercetin, hispidulin, and cirsimaritin exhibited better potential inhibition than hydroxy-chloroquine against COVID-19 main protease active site and ACE2. ChemRxiv 2020, 1–22. [Google Scholar] [CrossRef]
- Singam, E.R.A.; La Merrill, M.A.; Durkin, K.A.; Smith, M.T. Structure-based virtual screening of a natural product database to identify several possible SARS-CoV-2 main protease inhibitors. ChemRxiv 2020, 1–15. [Google Scholar] [CrossRef]
- Umesh, K.D.; Dubey, V.K.; Selvaraj, C.; Singh, S.K. Identification of new anti-nCoV drug chemical compounds from Indian spices exploiting SARS-CoV-2 main protease as target. J. Biomol. Struct. Dyn. 2020, 1–9. [Google Scholar] [CrossRef]
- Gentile, D.; Patamia, V.; Rescifina, A.; Scala, A.; Sciortino, M.T.; Piperno, A.; Rescifina, A. Putative inhibitors of SARS-CoV-2 main protease from a library of marine natural products: A virtual screening and molecular modeling study. Mar. Drugs 2020, 18, 225. [Google Scholar] [CrossRef]
- Arun, K.; Sharanya, C.S.; Abhithaj, J.; Dileep, F.; Sadasivan, C. Drug repurposing against SARS-CoV-2 using E-pharmacophore based virtual screening and molecular docking with main protease as the target. ChemRxiv 2020, 1–17. [Google Scholar] [CrossRef]
- Ton, A.T.; Gentile, F.; Hsing, M.; Ban, F.; Cherkasov, A. Rapid identification of potential inhibitors of SARS-CoV-2 main protease by deep docking of 1.3 billion compounds. Mol. Inf. 2020, 39, 2000028. [Google Scholar] [CrossRef]
- Tsuji, M. Potential anti-SARS-CoV-2 drug candidates identified through virtual screening of the ChEMBL database for compounds that target the main coronavirus protease. FEBS Open Bio. 2020. [Google Scholar] [CrossRef]
- Sarma, P.; Shekhar, N.; Prajapat, M.; Avti, P.; Kaur, H.; Kumar, S.; Singh, S.; Kumar, H.; Prakash, A.; Prasad Dhibar, D.; et al. In-silico homology assisted identification of inhibitor of RNA binding against 2019-nCoV N-protein (N terminal domain). J. Biomol. Struct. Dyn. 2020, 1–10. [Google Scholar] [CrossRef]
- Wu, C.; Zheng, M.; Li, H.; Liu, Y.; Yang, Y.; Zhang, P.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef]
- Dallavalle, S.; Ferrari, A.; Biasotti, B.; Merlini, L.; Penco, S.; Gallo, G.; Marzi, M.; Tinti, M.O.; Martinelli, R.; Pisano, C.; et al. Novel 7-oxyiminomethyl derivatives of camptothecin with potent in vitro and in vivo antitumor activity. J. Med. Chem. 2001, 44, 3264–3274. [Google Scholar] [CrossRef]
- Dallavalle, S.; Giannini, G.; Alloatti, D.; Casati, A.; Marastoni, E.; Musso, L.; Merlini, L.; Morini, G.; Penco, S.; Pisano, C.; et al. Synthesis and cytotoxic activity of polyamine analogues of camptothecin. J. Med. Chem. 2006, 49, 5177–5186. [Google Scholar] [CrossRef]
- Cincinelli, R.; Musso, L.; Dallavalle, S.; Artali, R.; Tinelli, S.; Colangelo, D.; Zunino, F.; de Cesare, M.; Beretta, G.L.; Zaffaroni, N. Design, modeling, synthesis and biological activity evaluation of camptothecin-linked platinum anticancer agents. Eur. J. Med. Chem. 2013, 63, 387–400. [Google Scholar] [CrossRef]
- Dal Pozzo, A.; Ni, M.H.; Esposito, E.; Dallavalle, S.; Musso, L.; Bargiotti, A.; Pisano, C.; Vesci, L.; Bucci, F.; Castorina, M.; et al. Novel tumor-targeted RGD peptide-camptothecin conjugates: Synthesis and biological evaluation. Bioorg. Med. Chem. 2010, 18, 64–72. [Google Scholar] [CrossRef]
- Cananzi, S.; Merlini, L.; Artali, R.; Beretta, G.L.; Zaffaroni, N.; Dallavalle, S. Synthesis and topoisomerase I inhibitory activity of a novel diazaindeno [2,1-b]phenanthrene analogue of Lamellarin, D. Bioorg. Med. Chem. 2011, 19, 4971–4984. [Google Scholar] [CrossRef]
- Dhavan, A.A.; Kaduskar, R.D.; Musso, L.; Scaglioni, L.; Martino, P.A.; Dallavalle, S. Total synthesis of leopolic acid A, a natural 2, 3-pyrrolidinedione with antimicrobial activity. Beilstein J. Org. Chem. 2016, 12, 1624–1628. [Google Scholar] [CrossRef]
- Cincinelli, R.; Scaglioni, L.; Arnold, N.A.; Dallavalle, S. Structure and absolute configuration of new acidic metabolites from Stachys ehrenbergii. Tetrahedron Lett. 2011, 52, 5972–5975. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Rackers, J.A.; Laury, M.L.; Li, C.; Wang, Z.; LagardeȲre, L.; Piquemal, J.P.; Ren, P.; Jay, W. TINKER 8: A modular software package for molecular design and simulation ponder. J. Chem. Theor. Comp. 2018, 14, 5273–5289. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of parameters for semi-empirical methods i-method. J. Comp. Chem. 1989, 10, 209–220. [Google Scholar] [CrossRef]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar]
- Gasteiger, J.; Marsili, M.M. Iterative partial equalization of orbital electronegativity-a rapid access to atomic charge. Tetrahedron 2008, 36, 3219–3228. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comp. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Huey, R.; Morris, G.M.; Olson, A.J.; Goodsell, D.S.S. A semiempirical free energy force field with charge-based desolvetion. J. Comp. Chem. 2007, 28, 1145–1152. [Google Scholar] [CrossRef]
- Solis, F.J.; Wets, R.J.B. Minimization by random search techniques. Math. Oper. Res. 1981, 6, 19–30. [Google Scholar] [CrossRef]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2009, 31, 671–690. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Nose, S.; Klein, M. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Dassault Systèmes BIOVIA, BIOVIA Discovery Studio Visualize 2019, version 2019; Dassault Systèmes BIOVIA: San Diego, CA, USA, 2019.
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Oprea, Tudor, I. Property distribution of drug-related chemical databases. J. Comp. Aid. Mol. Des. 2000, 14, 251–264. [Google Scholar] [CrossRef]
Sample Availability: Samples of the investigated compounds are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
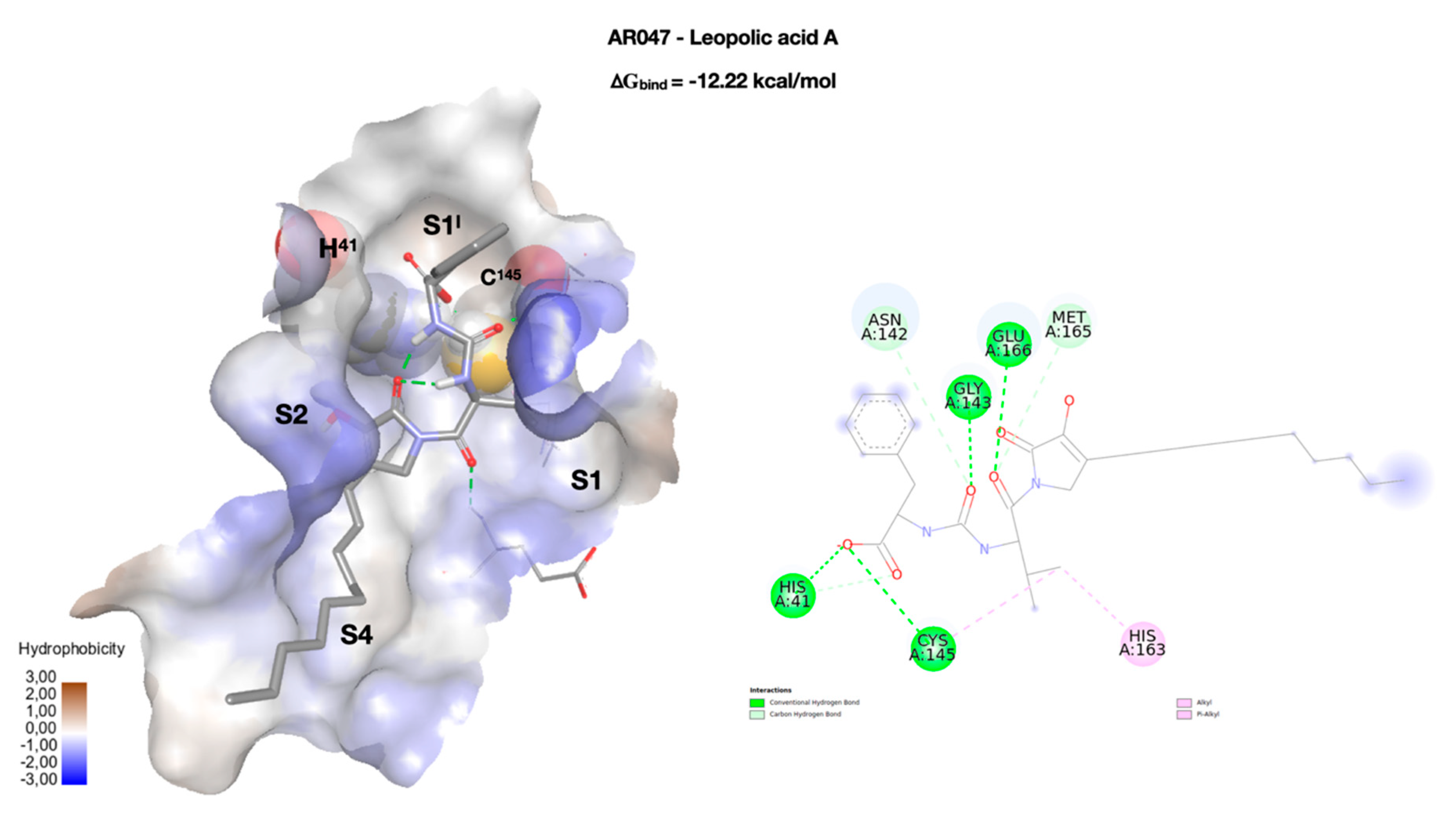{kind=link}
{kind=link}
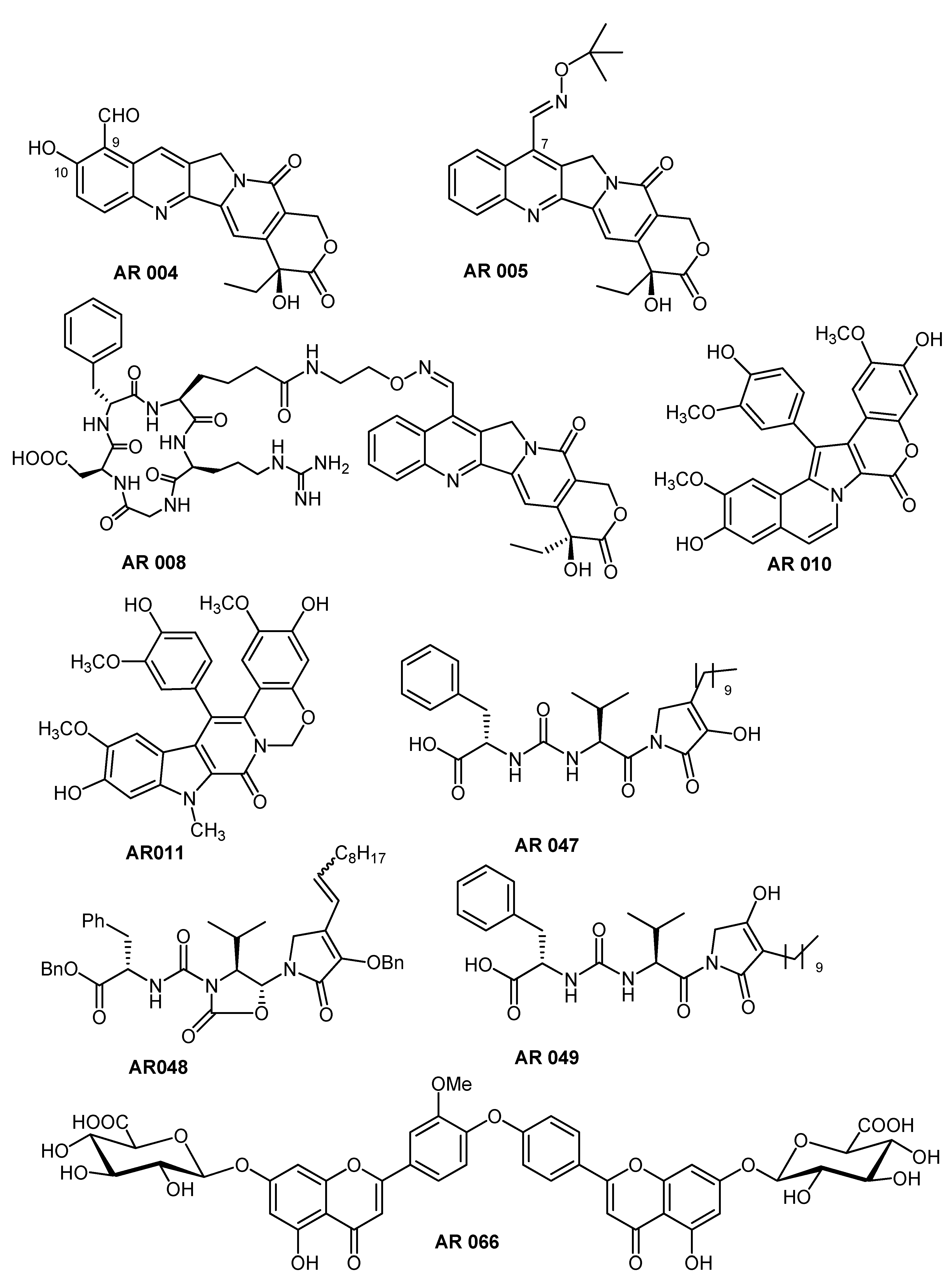{kind=link}
| Cpd | RMSD | RMSF |
|---|---|---|
| AR005 | 0.13 ± 0.02 | 0.07 ± 0.02 |
| AR010 | 0.15 ± 0.02 | 0.08 ± 0.03 |
| AR047 | 0.12 ± 0.02 | 0.06 ± 0.03 |
| AR066 | 0.18 ± 0.02 | 0.09 ± 0.03 |
| Cpd | Box Volume Å3 | TIP3P Molecules | Na + Ions |
|---|---|---|---|
| AR005 | 390.817 | 10977 | 1 |
| AR010 | 391.206 | 10997 | 1 |
| AR047 | 391.835 | 10996 | 1 |
| AR066 | 391.858 | 10897 | 3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazzini, S.; Musso, L.; Dallavalle, S.; Artali, R. Putative SARS-CoV-2 Mpro Inhibitors from an In-House Library of Natural and Nature-Inspired Products: A Virtual Screening and Molecular Docking Study. Molecules 2020, 25, 3745. https://doi.org/10.3390/molecules25163745
Mazzini S, Musso L, Dallavalle S, Artali R. Putative SARS-CoV-2 Mpro Inhibitors from an In-House Library of Natural and Nature-Inspired Products: A Virtual Screening and Molecular Docking Study. Molecules. 2020; 25(16):3745. https://doi.org/10.3390/molecules25163745
Chicago/Turabian StyleMazzini, Stefania, Loana Musso, Sabrina Dallavalle, and Roberto Artali. 2020. "Putative SARS-CoV-2 Mpro Inhibitors from an In-House Library of Natural and Nature-Inspired Products: A Virtual Screening and Molecular Docking Study" Molecules 25, no. 16: 3745. https://doi.org/10.3390/molecules25163745
APA StyleMazzini, S., Musso, L., Dallavalle, S., & Artali, R. (2020). Putative SARS-CoV-2 Mpro Inhibitors from an In-House Library of Natural and Nature-Inspired Products: A Virtual Screening and Molecular Docking Study. Molecules, 25(16), 3745. https://doi.org/10.3390/molecules25163745