
Screening of Heteroaromatic Scaffolds against Cystathionine Beta-Synthase Enables Identification of Substituted Pyrazolo[3,4-c]Pyridines as Potent and Selective Orthosteric Inhibitors

 , , , ,
, , , ,  ,
,  and
and 
Abstract
1. Introduction
2. Results and Discussion
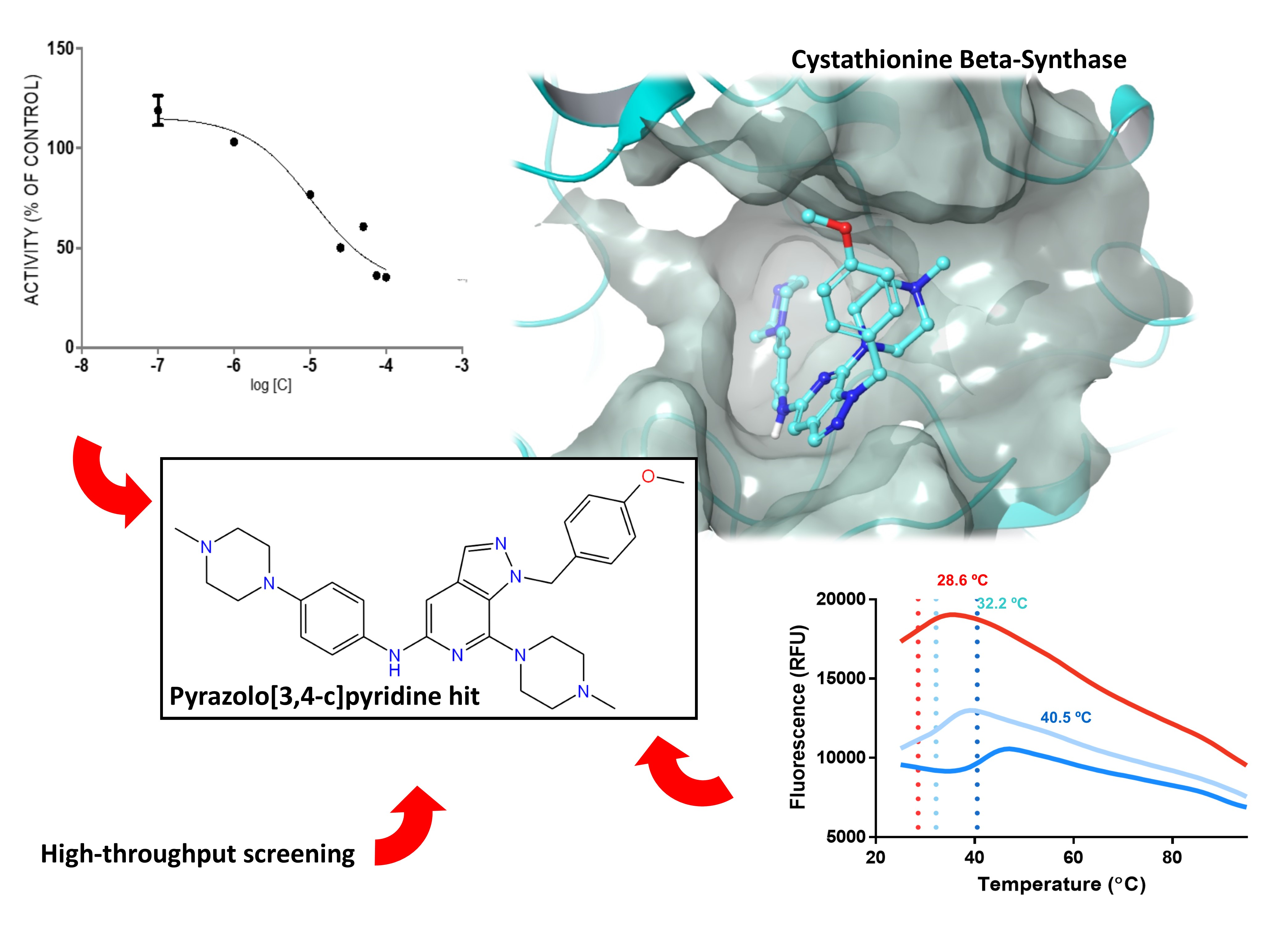
2.1. Rationale and Compound Selection
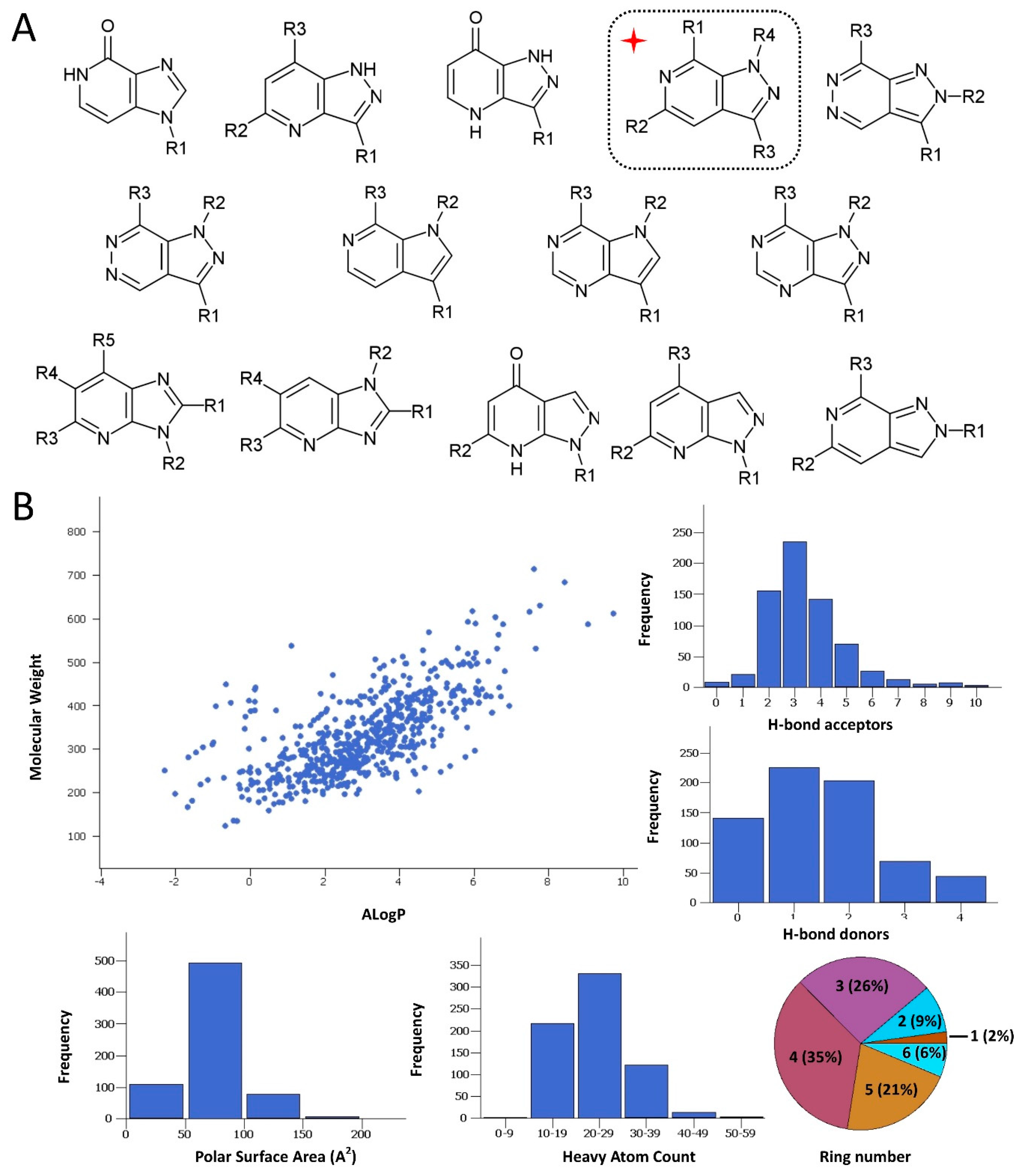
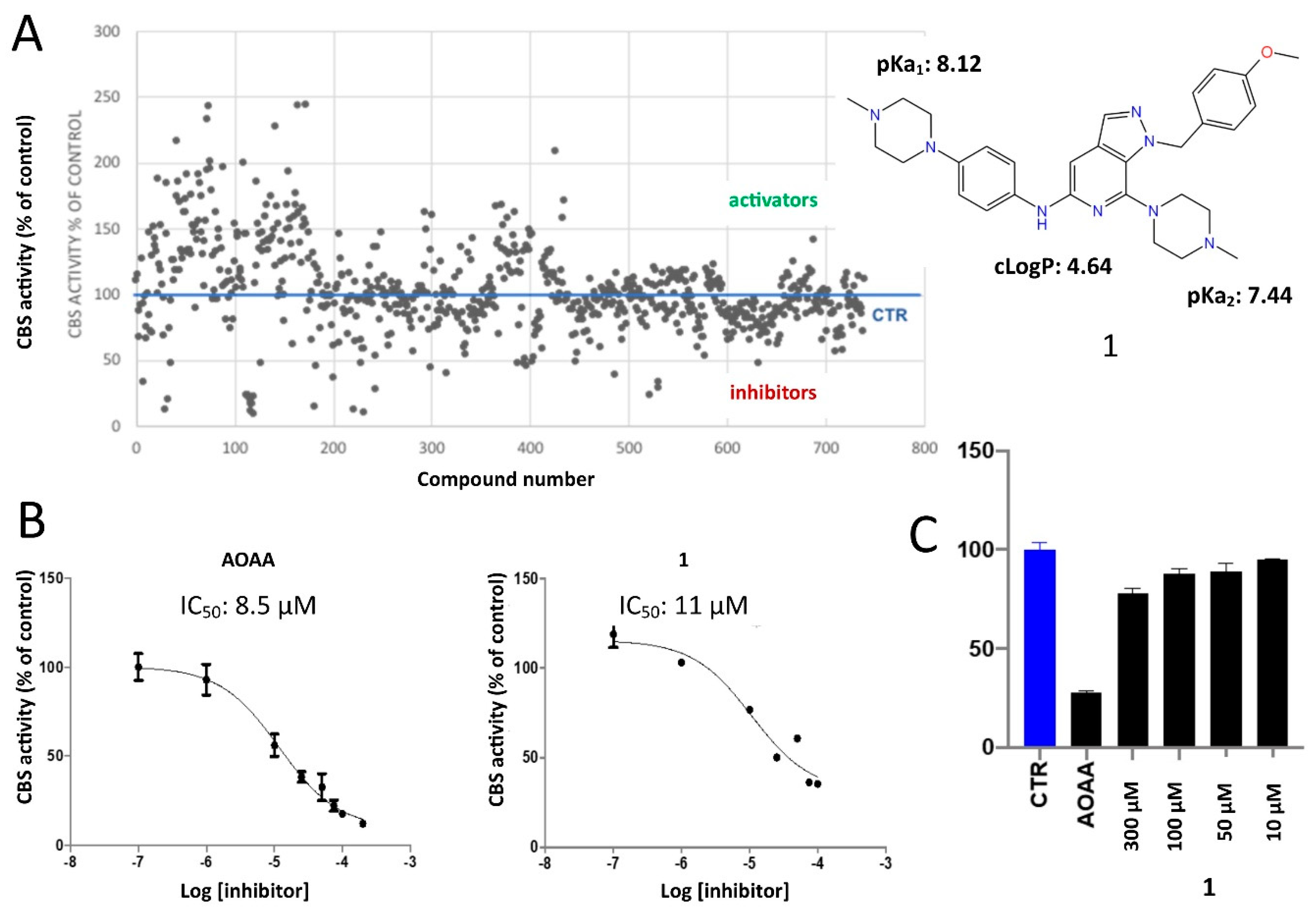
2.2. Protein Expression and In Vitro Evaluations
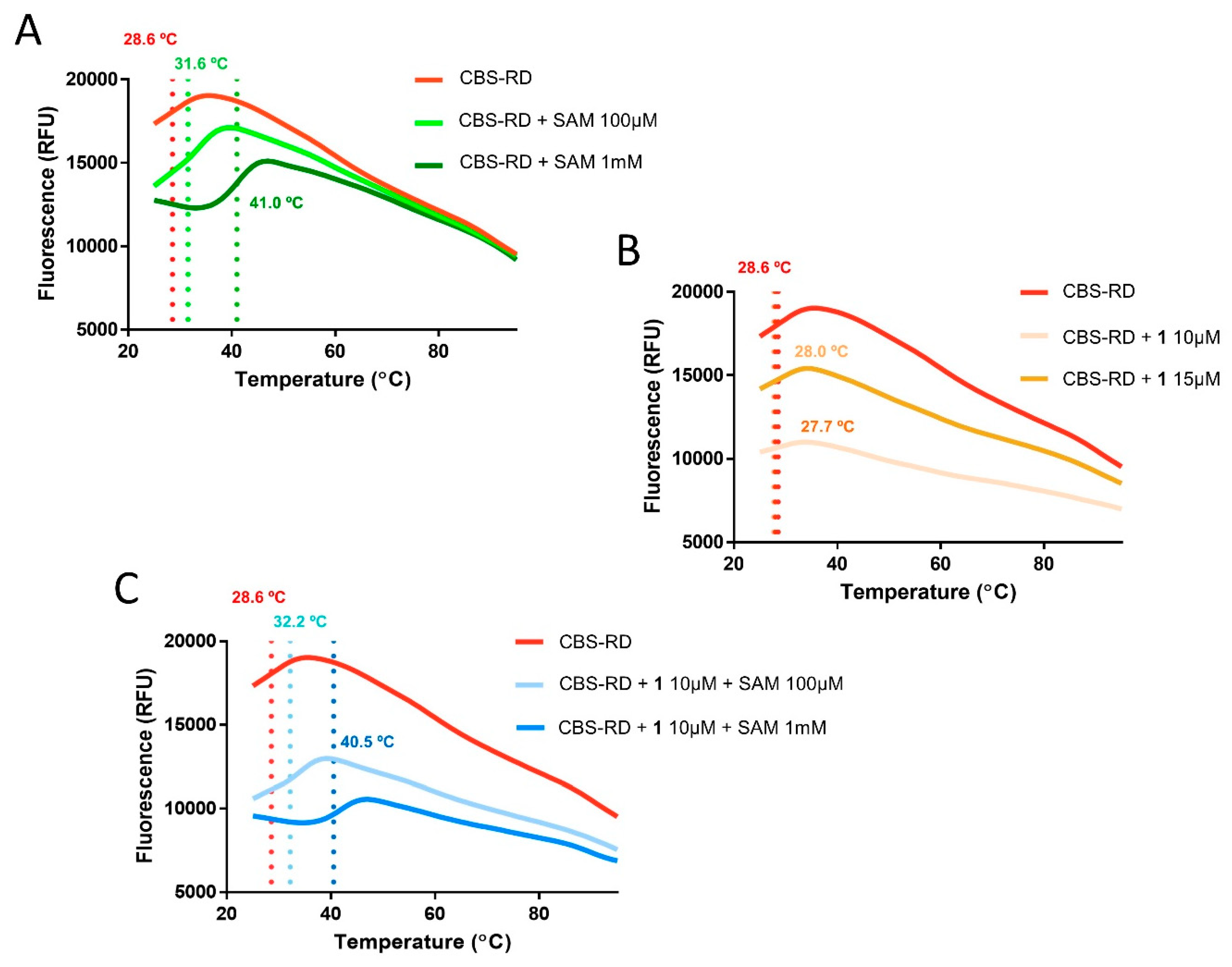
2.3. Differential Scanning Fluorimetry
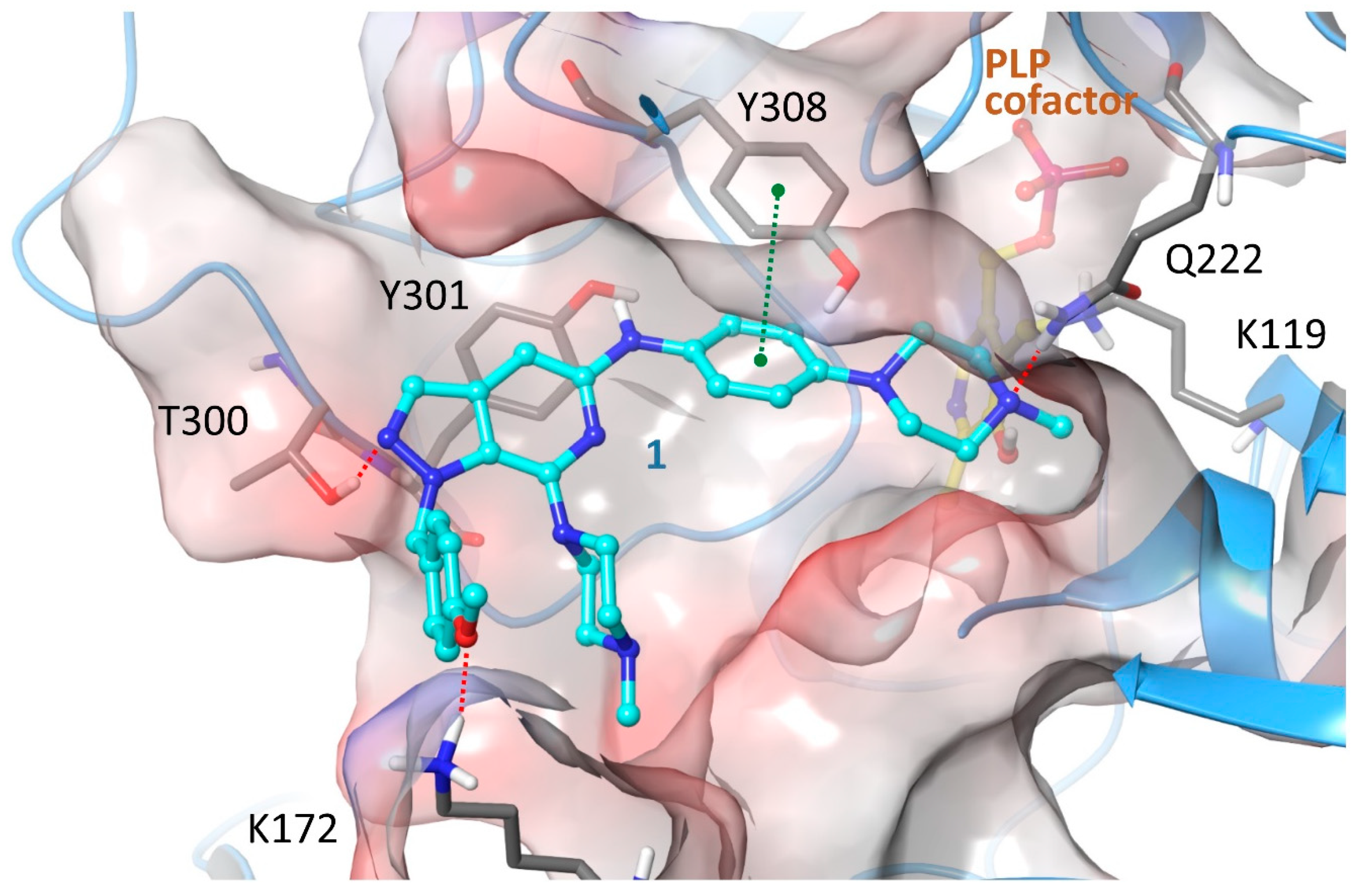
2.4. Structure-Activity Relationships, Neural Networks Modeling and Theoretical Simulations
3. Materials and Methods
3.1. Expression and Purification of Full Length GST-CBS and His Tag-CBS Regulatory Domain
3.2. Sample Preparation and Library Administration
3.3. H2S Detection Using the Methylene Blue Assay
3.4. H2S Detection Using the 7-Azido-4-Methylcoumarin Assay
3.5. Differential Scanning Fluorimetry
3.6. Deep Neural Networks
3.7. Molecular Simulations
3.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wang, R. Physiological implications of hydrogen sulfide: A whiff exploration that blossomed. Physiol. Rev. 2012, 92, 791–896. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C.; Papapetropoulos, A. International Union of Basic and Clinical Pharmacology. CII: Pharmacological Modulation of H2S Levels: H2S Donors and H2S Biosynthesis Inhibitors. Pharmacol. Rev. 2017, 69, 497–564. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Banerjee, R. PLP-dependent H2S biogenesis. Biochim. Biophys. Acta 2011, 1814, 1518–1527. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.; Zou, C.G. Redox regulation and reaction mechanism of human cystathionine-beta-synthase: A PLP-dependent hemesensor protein. Arch. Biochem. Biophys. 2005, 433, 144–156. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Ding, Y.P.; Wang, Z.; Kong, Y.; Gao, R.; Chen, G. Hydrogen sulfide therapy in brain diseases: From bench to bedside. Med. Gas Res. 2017, 7, 113–119. [Google Scholar] [CrossRef]
- Keller, R.; Chrastina, P.; Pavlíková, M.; Gouveia, S.; Ribes, A.; Kölker, S.; Blom, H.J.; Baumgartner, M.R.; Bártl, J.; Dionisi-Vici, C.; et al. Newborn screening for homocystinurias: Recent recommendations versus current practice. J. Inherit. Metab. Dis. 2019, 42, 128–139. [Google Scholar] [CrossRef]
- Wen, Y.D.; Wang, H.; Zhu, Y.Z. The drug developments of hydrogen sulfide on cardiovascular disease. Oxidative Med. Cell. Longev. 2018, 2018, 4010395. [Google Scholar] [CrossRef]
- Wu, D.; Si, W.; Wang, M.; Lv, S.; Ji, A.; Li, Y. Hydrogen sulfide in cancer: Friend or foe? Nitric Oxide Biol. Chem. 2015, 50, 38–45. [Google Scholar] [CrossRef]
- Asimakopoulou, A.; Panopoulos, P.; Chasapis, C.T.; Coletta, C.; Zhou, Z.; Cirino, G.; Giannis, A.; Szabo, C.; Spyroulias, G.A.; Papapetropoulos, A. Selectivity of commonly used pharmacological inhibitors for cystathionine beta synthase (CBS) and cystathionine gamma lyase (CSE). Br. J. Pharmacol. 2013, 169, 922–932. [Google Scholar] [CrossRef]
- Druzhyna, N.; Szczesny, B.; Olah, G.; Módis, K.; Asimakopoulou, A.; Pavlidou, A.; Szoleczky, P.; Gerö, D.; Yanagi, K.; Törö, G.; et al. Screening of a composite library of clinically used drugs and well-characterized pharmacological compounds for cystathionine β-synthase inhibition identifies benserazide as a drug potentially suitable for repurposing for the experimental therapy of colon cancer. Pharmacol. Res. 2016, 113, 18–37. [Google Scholar] [CrossRef]
- Hellmich, M.R.; Coletta, C.; Chao, C.; Szabo, C. The therapeutic potential of cystathionine beta-synthetase/hydrogen sulfide inhibition in cancer. Antioxid. Redox Signal. 2015, 22, 424–448. [Google Scholar] [CrossRef] [PubMed]
- Panagaki, T.; Randi, E.B.; Augsburger, F.; Szabo, C. Overproduction of H(2)S, generated by CBS, inhibits mitochondrial Complex IV and suppresses oxidative phosphorylation in Down syndrome. Proc. Natl. Acad. Sci. USA 2019, 116, 18769–18771. [Google Scholar] [CrossRef] [PubMed]
- Marechal, D.; Brault, V.; Leon, A.; Martin, D.; Lopes Pereira, P.; Loaëc, N.; Birling, M.-C.; Friocourt, G.; Blondel, M.; Herault, Y. Cbs overdosage is necessary and sufficient to induce cognitive phenotypes in mouse models of Down syndrome and interacts genetically with Dyrk1α. Hum. Mol. Genet. 2019, 28, 1561–1577. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, K.; Davisson, M. The sequence of human chromosome 21 and implications for research into Down syndrome. Genome Biol. 2000, 1, reviews0002.1. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Skovby, F.; Krassikoff, N.; Francke, U. Assignment of the gene for cystathionine β-synthase to human chromosome 21 in somatic cell hybrids. Hum. Genet. 1984, 65, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Kamoun, P.P. Mental retardation in Down syndrome: Two ways to treat. Med. Hypotheses 2019, 131, 109289. [Google Scholar] [CrossRef]
- Szabo, C. The re-emerging pathophysiological role of the cystathionine-beta-synthase-hydrogen sulfide system in Down syndrome. FEBS J. 2020. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Saha, S.; Giri, K.; Lanza, I.R.; Nair, K.S.; Jennings, N.B.; Rodriguez-Aguayo, C.; Lopez-Berestein, G.; Basal, E.; Weaver, A.L.; et al. Cystathionine beta-synthase (CBS) contributes to advanced ovarian cancer progression and drug resistance. PLoS ONE 2013, 8, e79167. [Google Scholar] [CrossRef]
- Kashfi, K. The dichotomous role of H2S in cancer cell biology? Déjà vu all over again. Biochem. Pharmacol. 2018, 149, 205–223. [Google Scholar] [CrossRef]
- Untereiner, A.A.; Pavlidou, A.; Druzhyna, N.; Papapetropoulos, A.; Hellmich, M.R.; Szabo, C. Drug resistance induces the upregulation of H2S-producing enzymes in HCT116 colon cancer cells. Biochem. Pharmacol. 2018, 149, 174–185. [Google Scholar] [CrossRef]
- Modis, K.; Coletta, C.; Asimakopoulou, A.; Szczesny, B.; Chao, C.; Papapetropoulos, A.; Hellmich, M.R.; Szabo, C. Effect of S-adenosyl-l-methionine (SAM), an allosteric activator of cystathionine-beta-synthase (CBS) on colorectal cancer cell proliferation and bioenergetics in vitro. Nitric Oxide Biol. Chem. 2014, 41, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Giuffrè, A.; Tomé, C.S.; Fernandes, D.G.F.; Zuhra, K.; Vicente, J.B. Hydrogen sulfide metabolism and signaling in the tumor microenvironment. Adv. Exp. Med. Biol. 2020, 1219, 335–353. [Google Scholar] [CrossRef] [PubMed]
- Zuhra, K.; Augsburger, F.; Majtan, T.; Szabo, C. Cystathionine-β-synthase: Molecular regulation and pharmacological inhibition. Biomolecules 2020, 10, 697. [Google Scholar] [CrossRef] [PubMed]
- Meier, M.; Janosik, M.; Kery, V.; Kraus, J.P.; Burkhard, P. Structure of human cystathionine beta-synthase: A unique pyridoxal 5’-phosphate-dependent heme protein. EMBO J. 2001, 20, 3910–3916. [Google Scholar] [CrossRef] [PubMed]
- Ereno-Orbea, J.; Majtan, T.; Oyenarte, I.; Kraus, J.P.; Martinez-Cruz, L.A. Structural insight into the molecular mechanism of allosteric activation of human cystathionine beta-synthase by S-adenosylmethionine. Proc. Natl. Acad. Sci. USA 2014, 111, E3845–E3852. [Google Scholar] [CrossRef]
- Pey, A.L.; Martinez-Cruz, L.A.; Kraus, J.P.; Majtan, T. Oligomeric status of human cystathionine beta-synthase modulates AdoMet binding. FEBS Lett. 2016, 590, 4461–4471. [Google Scholar] [CrossRef]
- Ereno-Orbea, J.; Oyenarte, I.; Martinez-Cruz, L.A. CBS domains: Ligand binding sites and conformational variability. Arch. Biochem. Biophys. 2013, 540, 70–81. [Google Scholar] [CrossRef]
- Catazaro, J.; Caprez, A.; Guru, A.; Swanson, D.; Powers, R. Functional evolution of PLP-dependent enzymes based on active-site structural similarities. Proteins Struct. Funct. Bioinform. 2014, 82, 2597–2608. [Google Scholar] [CrossRef]
- Koutmos, M.; Kabil, O.; Smith, J.L.; Banerjee, R. Structural basis for substrate activation and regulation by cystathionine beta-synthase (CBS) domains in cystathionine {beta}-synthase. Proc. Natl. Acad. Sci. USA 2010, 107, 20958–20963. [Google Scholar] [CrossRef]
- McCorvie, T.J.; Kopec, J.; Hyung, S.J.; Fitzpatrick, F.; Feng, X.; Termine, D.; Strain-Damerell, C.; Vollmar, M.; Fleming, J.; Janz, J.M.; et al. Inter-domain communication of human cystathionine beta-synthase: Structural basis of S-adenosyl-L-methionine activation. J. Biol. Chem. 2014, 289, 36018–36030. [Google Scholar] [CrossRef]
- Hnizda, A.; Spiwok, V.; Jurga, V.; Kozich, V.; Kodicek, M.; Kraus, J.P. Cross-talk between the catalytic core and the regulatory domain in cystathionine beta-synthase: Study by differential covalent labeling and computational modeling. Biochemistry 2010, 49, 10526–10534. [Google Scholar] [CrossRef] [PubMed]
- Janošík, M.; Kery, V.; Gaustadnes, M.; Maclean, K.N.; Kraus, J.P. Regulation of human cystathionine β-synthase by S-adenosyl-l-methionine: evidence for two catalytically active conformations involving an autoinhibitory domain in the C-terminal region. Biochemistry 2001, 40, 10625–10633. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Cai, H.; Hu, Y.; Liu, F.; Huang, S.; Zhou, Y.; Yu, J.; Xu, J.; Wu, F. A pharmacological probe identifies cystathionine β-synthase as a new negative regulator for ferroptosis. Cell Death Dis. 2018, 9, 1005. [Google Scholar] [CrossRef] [PubMed]
- Niu, W.; Chen, F.; Wang, J.; Qian, J.; Yan, S. Antitumor effect of sikokianin C, a selective cystathionine β-synthase inhibitor, against human colon cancer in vitro and in vivo. MedChemComm 2018, 9, 113–120. [Google Scholar] [CrossRef]
- Niu, W.; Wu, P.; Chen, F.; Wang, J.; Shang, X.; Xu, C. Discovery of selective cystathionine β-synthase inhibitors by high-throughput screening with a fluorescent thiol probe. MedChemComm 2017, 8, 198–201. [Google Scholar] [CrossRef]
- Zuhra, K.; Sousa, P.M.F.; Paulini, G.; Lemos, A.R.; Kalme, Z.; Bisenieks, I.; Bisenieks, E.; Vigante, B.; Duburs, G.; Bandeiras, T.M.; et al. Screening pyridine derivatives against human hydrogen sulfide-synthesizing enzymes by orthogonal methods. Sci. Rep. 2019, 9, 684. [Google Scholar] [CrossRef]
- Nakai, T.; Nakagawa, N.; Maoka, N.; Masui, R.; Kuramitsu, S.; Kamiya, N. Structure of P-protein of the glycine cleavage system: Implications for nonketotic hyperglycinemia. EMBO J. 2005, 24, 1523–1536. [Google Scholar] [CrossRef]
- Macarron, R.; Banks, M.N.; Bojanic, D.; Burns, D.J.; Cirovic, D.A.; Garyantes, T.; Green, D.V.; Hertzberg, R.P.; Janzen, W.P.; Paslay, J.W.; et al. Impact of high-throughput screening in biomedical research. Nat. Rev. Drug Discov. 2011, 10, 188–195. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision Glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Dixon, S.L.; Lowrie, J.F.; Sherman, W. Analysis and comparison of 2D fingerprints: Insights into database screening performance using eight fingerprint methods. J. Mol. Graph. Model. 2010, 29, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Sastry, M.; Lowrie, J.F.; Dixon, S.L.; Sherman, W. Large-scale systematic analysis of 2D fingerprint methods and parameters to improve virtual screening enrichments. J. Chem. Inf. Model. 2010, 50, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Bukovska, G.; Kery, V.; Kraus, J.P. Expression of human cystathionine beta-synthase in Escherichia coli: Purification and characterization. Protein Expr. Purif. 1994, 5, 442–448. [Google Scholar] [CrossRef]
- Frank, N.; Kent, J.O.; Meier, M.; Kraus, J.P. Purification and characterization of the wild type and truncated human cystathionine beta-synthase enzymes expressed in E. coli. Arch. Biochem. Biophys. 2008, 470, 64–72. [Google Scholar] [CrossRef]
- Kraus, J.; Packman, S.; Fowler, B.; Rosenberg, L.E. Purification and properties of cystathionine beta-synthase from human liver. Evidence for identical subunits. J. Biol. Chem. 1978, 253, 6523–6528. [Google Scholar]
- Oyenarte, I.; Majtan, T.; Ereno, J.; Corral-Rodriguez, M.A.; Kraus, J.P.; Martinez-Cruz, L.A. Purification, crystallization and preliminary crystallographic analysis of human cystathionine beta-synthase. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2012, 68, 1318–1322. [Google Scholar] [CrossRef]
- Wingfield, P.T. Overview of the purification of recombinant proteins. Curr. Protoc. Protein Sci. 2015, 80, 6.1.1–6.1.35. [Google Scholar] [CrossRef]
- Moest, R.R. Hydrogen sulfide determination by the methylene blue method. Anal. Chem. 1975, 47, 1204–1205. [Google Scholar] [CrossRef]
- Reese, B.K.; Finneran, D.W.; Mills, H.J.; Zhu, M.-X.; Morse, J.W. Examination and refinement of the determination of aqueous hydrogen sulfide by the methylene blue method. Aquat. Geochem. 2011, 17, 567–582. [Google Scholar] [CrossRef]
- Hartle, M.D.; Pluth, M.D. A practical guide to working with H2S at the interface of chemistry and biology. Chem. Soc. Rev. 2016, 45, 6108–6117. [Google Scholar] [CrossRef] [PubMed]
- Michailidou, M.; Giannouli, V.; Kotsikoris, V.; Papadodima, O.; Kontogianni, G.; Kostakis, I.K.; Lougiakis, N.; Chatziioannou, A.; Kolisis, F.N.; Marakos, P.; et al. Novel pyrazolopyridine derivatives as potential angiogenesis inhibitors: Synthesis, biological evaluation and transcriptome-based mechanistic analysis. Eur. J. Med. Chem. 2016, 121, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Augsburger, F.; Randi, E.B.; Jendly, M.; Ascencao, K.; Dilek, N.; Szabo, C. Role of 3-mercaptopyruvate sulfurtransferase in the regulation of proliferation, migration, and bioenergetics in murine colon cancer cells. Biomolecules 2020, 10, 447. [Google Scholar] [CrossRef] [PubMed]
- Niesen, F.H.; Berglund, H.; Vedadi, M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protocols 2007, 2, 2212–2221. [Google Scholar] [CrossRef]
- Farid, R.; Day, T.; Friesner, R.A.; Pearlstein, R.A. New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorganic Med. Chem. 2006, 14, 3160–3173. [Google Scholar] [CrossRef]
- Sherman, W.; Beard, H.S.; Farid, R. Use of an induced fit receptor structure in virtual screening. Chem. Biol. Drug Des. 2006, 67, 83–84. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- Botou, M.; Yalelis, V.; Lazou, P.; Zantza, I.; Papakostas, K.; Charalambous, V.; Mikros, E.; Flemetakis, E.; Frillingos, S. Specificity profile of NAT/NCS2 purine transporters in Sinorhizobium (Ensifer) meliloti. Mol. Microbiol. 2020. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1, 3, 4, 5 and 7 are available (≤1 mg) from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | R1 | R2 | R3 | R4 | Inhibition (%) |
|---|---|---|---|---|---|
| 1 |  |  | H |  | 70 |
| 2 |  | | H | | 30 |
| 3 | | |  | | 20 |
| 4 | |  | H | | 20 |
| 5 | | N≡C | | | No inhibition |
| 6 | |  |  | H | No inhibition |
| 7 | | N≡C | H | H | No inhibition |
| 8 |  | Cl | H | | <20 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fantel, A.-M.; Myrianthopoulos, V.; Georgoulis, A.; Lougiakis, N.; Zantza, I.; Lamprinidis, G.; Augsburger, F.; Marakos, P.; Vorgias, C.E.; Szabo, C.; et al. Screening of Heteroaromatic Scaffolds against Cystathionine Beta-Synthase Enables Identification of Substituted Pyrazolo[3,4-c]Pyridines as Potent and Selective Orthosteric Inhibitors. Molecules 2020, 25, 3739. https://doi.org/10.3390/molecules25163739
Fantel A-M, Myrianthopoulos V, Georgoulis A, Lougiakis N, Zantza I, Lamprinidis G, Augsburger F, Marakos P, Vorgias CE, Szabo C, et al. Screening of Heteroaromatic Scaffolds against Cystathionine Beta-Synthase Enables Identification of Substituted Pyrazolo[3,4-c]Pyridines as Potent and Selective Orthosteric Inhibitors. Molecules. 2020; 25(16):3739. https://doi.org/10.3390/molecules25163739
Chicago/Turabian StyleFantel, Anna-Maria, Vassilios Myrianthopoulos, Anastasios Georgoulis, Nikolaos Lougiakis, Iliana Zantza, George Lamprinidis, Fiona Augsburger, Panagiotis Marakos, Constantinos E. Vorgias, Csaba Szabo, and et al. 2020. "Screening of Heteroaromatic Scaffolds against Cystathionine Beta-Synthase Enables Identification of Substituted Pyrazolo[3,4-c]Pyridines as Potent and Selective Orthosteric Inhibitors" Molecules 25, no. 16: 3739. https://doi.org/10.3390/molecules25163739
APA StyleFantel, A.-M., Myrianthopoulos, V., Georgoulis, A., Lougiakis, N., Zantza, I., Lamprinidis, G., Augsburger, F., Marakos, P., Vorgias, C. E., Szabo, C., Pouli, N., Papapetropoulos, A., & Mikros, E. (2020). Screening of Heteroaromatic Scaffolds against Cystathionine Beta-Synthase Enables Identification of Substituted Pyrazolo[3,4-c]Pyridines as Potent and Selective Orthosteric Inhibitors. Molecules, 25(16), 3739. https://doi.org/10.3390/molecules25163739