
Structural Basis of Beneficial Design for Effective Nicotinamide Phosphoribosyltransferase Inhibitors


 , ,
, , 
Abstract
1. Introduction
2. Results and Discussion
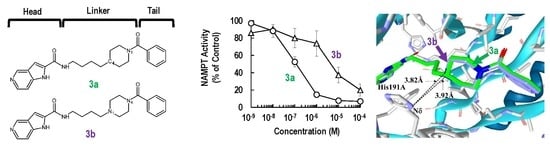
2.1. Synthesis and Biochemical Properties of 3a and 3b
2.2. Binding Modes of 3a and 3b to the Tunnel Cavity of NAMPT
2.3. Influence of Enthalpic Effects of 3a and 3b on the Interactions with Tunnel Cavity of NAMPT
3. Materials and Methods
3.1. Enzymatic NAMPT Assay
3.2. Cells and Cell Culture
3.3. Cell Activity Assay
3.4. In Silico Binding Mode and Binding Affinity Score Analyses
3.5. Synthetic Procedures of the Target Compounds
3.5.1. General Experimental Methods
3.5.2. General Procedures for Synthesizing the Target Compounds 3a and 3b
3.5.3. (4-(4-aminobutyl)piperidin-1-yl)(phenyl)methanone (2a)
3.5.4. N-(4-(1-benzoylpiperidin-4-yl)butyl)-1H-pyrrolo[3,2-c]pyridine-2-carboxamide (3a)
3.5.5. 4-(4-Aminobutyl)piperazin-1-yl)(phenyl)methanone (2b)
3.5.6. N-(4-(4-Benzoylpiperazin-1-yl)butyl)-1H-pyroro[3,2-c]pyridine-2-carboxamide (3b)
3.6. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Belenky, P.; Bogan, K.L.; Brenner, C. NAD+ metabolism in health and disease. Trends Biochem. Sci. 2007, 32, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Menzies, K.J.; Auwerx, J. NAD(+) Metabolism and the control of energy homeostasis: A balancing act between mitochondria and the nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef] [PubMed]
- Tanuma, S.; Sato, A.; Oyama, T.; Yoshimori, A.; Abe, H.; Uchiumi, F. New Insights into the roles of NAD+-poly(ADP-ribose) metabolism and poly(ADP-ribose) glycohydrolase. Curr. Protein Pept. Sci. 2016, 17, 668–682. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Su, X.; Quinn, W.J., 3rd; Hui, S.; Krukenberg, K.; Frederick, D.W.; Redpath, P.; Zhan, L.; Chellappa, K.; White, E.; et al. Quantitative analysis of NAD synthesis-breakdown fluxes. Cell Metab. 2018, 27, 1067–1080.e5. [Google Scholar] [CrossRef]
- Schreiber, V.; Dantzer, F.; Ame, J.C.; de Murcia, G. Poly(ADP-ribose): Novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006, 7, 517–528. [Google Scholar] [CrossRef]
- Tanuma, S.I.; Shibui, Y.; Oyama, T.; Uchiumi, F.; Abe, H. Targeting poly(ADP-ribose) glycohydrolase to draw apoptosis codes in cancer. Biochem. Pharmacol. 2019, 167, 163–172. [Google Scholar] [CrossRef]
- Revollo, J.R.; Grimm, A.A.; Imai, S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J. Biol. Chem. 2004, 279, 50754–50763. [Google Scholar] [CrossRef]
- Mei, S.C.; Brenner, C. NAD as a genotype-specific drug target. Chem. Biol. 2013, 20, 1307–1308. [Google Scholar] [CrossRef][Green Version]
- Preiss, J.; Handler, P. Enzymatic synthesis of nicotinamide mononucleotide. J. Biol. Chem. 1957, 225, 759–770. [Google Scholar]
- Bogan, K.L.; Brenner, C. Nicotinic acid, nicotinamide, and nicotinamide riboside: A molecular evaluation of NAD+ precursor vitamins in human nutrition. Ann. Rev. Nutr. 2008, 28, 115–130. [Google Scholar] [CrossRef]
- Chiarugi, A.; Dölle, C.; Felici, R.; Ziegler, M. The NAD metabolome—A key determinant of cancer cell biology. Nat. Rev. Cancer 2012, 12, 741–752. [Google Scholar] [CrossRef]
- Garten, A.; Petzold, S.; Körner, A.; Imai, S.; Kiess, W. Nampt: Linking NAD biology, metabolism and cancer. Trends Endocrinol. Metab. TEM 2009, 20, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Chowdhry, S.; Zanca, C.; Rajkumar, U.; Koga, T.; Diao, Y.; Raviram, R.; Liu, F.; Turner, K.; Yang, H.; Brunk, E.; et al. NAD metabolic dependency in cancer is shaped by gene amplification and enhancer remodelling. Nature 2019, 569, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.K.; Kim, S.R.; Kim, J.G.; Kim, J.Y.; Koo, T.H.; Jang, H.O.; Yun, I.; Yoo, M.A.; Bae, M.K. Hypoxic induction of human visfatin gene is directly mediated by hypoxia-inducible factor-1. FEBS Lett. 2006, 580, 4105–4113. [Google Scholar] [CrossRef] [PubMed]
- Bauer, L.; Venz, S.; Junker, H.; Brandt, R.; Radons, J. Nicotinamide phosphoribosyltransferase and prostaglandin H2 synthase 2 are up-regulated in human pancreatic adenocarcinoma cells after stimulation with interleukin-1. Int. J. Oncol. 2009, 35, 97–107. [Google Scholar] [PubMed]
- Shackelford, R.E.; Bui, M.M.; Coppola, D.; Hakam, A. Over-expression of nicotinamide phosphoribosyltransferase in ovarian cancers. Int. J. Clin. Exp. Pathol. 2010, 3, 522–527. [Google Scholar] [PubMed]
- Wang, B.; Hasan, M.K.; Alvarado, E.; Yuan, H.; Wu, H.; Chen, W.Y. NAMPT overexpression in prostate cancer and its contribution to tumor cell survival and stress response. Oncogene 2011, 30, 907–921. [Google Scholar] [CrossRef] [PubMed]
- Hasmann, M.; Schemainda, I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 2003, 63, 7436–7442. [Google Scholar]
- Hjarnaa, P.J.; Jonsson, E.; Latini, S.; Dhar, S.; Larsson, R.; Bramm, E.; Skov, T.; Binderup, L. CHS 828, a novel pyridyl cyanoguanidine with potent antitumor activity in vitro and in vivo. Cancer Res. 1999, 59, 5751–5757. [Google Scholar]
- Micheli, V.; Simmonds, H.A.; Sestini, S.; Ricci, C. Importance of nicotinamide as an NAD precursor in the human erythrocyte. Arch. Biochem. Biophys. 1990, 283, 40–45. [Google Scholar] [CrossRef]
- Khan, J.A.; Tao, X.; Tong, L. Molecular basis for the inhibition of human NMPRTase, a novel target for anticancer agents. Nat. Struct. Mol. Biol. 2006, 13, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Olesen, U.H.; Christensen, M.K.; Björkling, F.; Jäättelä, M.; Jensen, P.B.; Sehested, M.; Nielsen, S.J. Anticancer agent CHS-828 inhibits cellular synthesis of NAD. Biochem. Biophys. Res. Commun. 2008, 367, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Von Heideman, A.; Berglund, A.; Larsson, R.; Nygren, P. Safety and efficacy of NAD depleting cancer drugs: Results of a phase I clinical trial of CHS 828 and overview of published data. Cancer Chemother. Pharmacol. 2010, 65, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Bi, T.Q.; Che, X.M. Nampt/PBEF/visfatin and cancer. Cancer Biol. Ther. 2010, 10, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Galli, U.; Travelli, C.; Massarotti, A.; Fakhfouri, G.; Rahimian, R.; Tron, G.C.; Genazzani, A.A. Medicinal chemistry of nicotinamide phosphoribosyltransferase (NAMPT) inhibitors. J. Med. Chem. 2013, 56, 6279–6296. [Google Scholar]
- Montecucco, F.; Cea, M.; Bauer, I.; Soncini, D.; Caffa, I.; Lasigliè, D.; Nahimana, A.; Uccelli, A.; Bruzzone, S.; Nencioni, A. Nicotinamide phosphoribosyltransferase (NAMPT) inhibitors as therapeutics: Rationales, controversies, clinical experience. Curr. Drug Targets 2013, 14, 637–643. [Google Scholar] [CrossRef]
- Oh, A.; Ho, Y.C.; Zak, M.; Liu, Y.; Chen, X.; Yuen, P.W.; Zheng, X.; Liu, Y.; Dragovich, P.S.; Wang, W. Structural and biochemical analyses of the catalysis and potency impact of inhibitor phosphoribosylation by human nicotinamide phosphoribosyltransferase. ChemBioChem Eur. J. Chem. Biol. 2014, 15, 1121–1130. [Google Scholar] [CrossRef]
- Adams, D.J.; Ito, D.; Rees, M.G.; Seashore-Ludlow, B.; Puyang, X.; Ramos, A.H.; Cheah, J.H.; Clemons, P.A.; Warmuth, M.; Zhu, P.; et al. NAMPT is the cellular target of STF-31-like small-molecule probes. ACS Chem. Biol. 2014, 9, 2247–2254. [Google Scholar] [CrossRef]
- Sampath, D.; Zabka, T.S.; Misner, D.L.; O’Brien, T.; Dragovich, P.S. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) as a therapeutic strategy in cancer. Pharmacol. Ther. 2015, 151, 16–31. [Google Scholar] [CrossRef]
- Preyat, N.; Leo, O. Complex role of nicotinamide adenine dinucleotide in the regulation of programmed cell death pathways. Biochem. Pharmacol. 2016, 101, 13–26. [Google Scholar] [CrossRef]
- Asawa, Y.; Katsuragi, K.; Sato, A.; Yoshimori, A.; Tanuma, S.I.; Nakamura, H. Structure-based drug design of novel carborane-containing nicotinamide phosphoribosyltransferase inhibitors. Bioorg. Med. Chem. 2019, 27, 2832–2844. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zhang, X.; Bheda, P.; Revollo, J.R.; Imai, S.; Wolberger, C. Structure of Nampt/PBEF/visfatin, a mammalian NAD+ biosynthetic enzyme. Nat. Struct. Mol. Biol. 2006, 13, 661–662. [Google Scholar] [CrossRef]
- Rongvaux, A.; Galli, M.; Denanglaire, S.; Van Gool, F.; Drèze, P.L.; Szpirer, C.; Bureau, F.; Andris, F.; Leo, O. Nicotinamide phosphoribosyl transferase/pre-B cell colony-enhancing factor/visfatin is required for lymphocyte development and cellular resistance to genotoxic stress. J. Immunol. (Baltimore) 2008, 181, 4685–4695. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.Q.; Heruth, D.P.; Ye, S.Q. Nicotinamide phosphoribosyltransferase in human diseases. J. Bioanal. Biomed. 2011, 3, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Holen, K.; Saltz, L.B.; Hollywood, E.; Burk, K.; Hanauske, A.R. The pharmacokinetics, toxicities, and biologic effects of FK866, a nicotinamide adenine dinucleotide biosynthesis inhibitor. Investig. N. Drugs 2008, 26, 45–51. [Google Scholar] [CrossRef]
- Ogino, Y.; Sato, A.; Uchiumi, F.; Tanuma, S.I. Cross resistance to diverse anticancer nicotinamide phosphoribosyltransferase inhibitors induced by FK866 treatment. Oncotarget 2018, 9, 16451–16461. [Google Scholar] [CrossRef]
- Ogino, Y.; Sato, A.; Uchiumi, F.; Tanuma, S.I. Genomic and tumor biological aspects of the anticancer nicotinamide phosphoribosyltransferase inhibitor FK866 in resistant human colorectal cancer cells. Genomics 2019, 111, 1889–1895. [Google Scholar] [CrossRef]
- Burgos, E.S. NAMPT in regulated NAD biosynthesis and its pivotal role in human metabolism. Curr. Med. Chem. 2011, 18, 1947–1961. [Google Scholar] [CrossRef]
- Zheng, X.; Bauer, P.; Baumeister, T.; Buckmelter, A.J.; Caligiuri, M.; Clodfelter, K.H.; Han, B.; Ho, Y.C.; Kley, N.; Lin, J.; et al. Structure-based identification of ureas as novel nicotinamide phosphoribosyltransferase (Nampt) inhibitors. J. Med. Chem. 2013, 56, 4921–4937. [Google Scholar]
- Zheng, X.; Bauer, P.; Baumeister, T.; Buckmelter, A.J.; Caligiuri, M.; Clodfelter, K.H.; Han, B.; Ho, Y.C.; Kley, N.; Lin, J.; et al. Structure-based discovery of novel amide-containing nicotinamide phosphoribosyltransferase (nampt) inhibitors. J. Med. Chem. 2013, 56, 6413–6433. [Google Scholar]
- Gunzner-Toste, J.; Zhao, G.; Bauer, P.; Baumeister, T.; Buckmelter, A.J.; Caligiuri, M.; Clodfelter, K.H.; Fu, B.; Han, B.; Ho, Y.C.; et al. Discovery of potent and efficacious urea-containing nicotinamide phosphoribosyltransferase (NAMPT) inhibitors with reduced CYP2C9 inhibition properties. Bioorg. Med. Chem. Lett. 2013, 23, 3531–3538. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Bair, K.W.; Bauer, P.; Baumeister, T.; Bowman, K.K.; Buckmelter, A.J.; Caligiuri, M.; Clodfelter, K.H.; Feng, Y.; Han, B.; et al. Identification of amides derived from 1H-pyrazolo[3,4-b]pyridine-5-carboxylic acid as potent inhibitors of human nicotinamide phosphoribosyltransferase (NAMPT). Bioorg. Med. Chem. Lett. 2013, 23, 5488–5497. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Backus, K.M.; Correia, B.E.; Lum, K.M.; Forli, S.; Horning, B.D.; González-Páez, G.E.; Chatterjee, S.; Lanning, B.R.; Teijaro, J.R.; Olson, A.J.; et al. Proteome-wide covalent ligand discovery in native biological systems. Nature 2016, 534, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Bair, K.W.; Baumeister, T.; Buckmelter, A.J.; Clodfelter, K.H.; Han, B.; Kuntz, J.D.; Lin, J.; Reynolds, D.J.; Smith, C.C.; Wang, Z.; et al. Piperidine Derivatives and Compositions for the Inhibition of Nicotinamide Phosphoribosyltransferase (Nampt). U.S. Patent No 9,555,039, 2017. [Google Scholar]
- Vogel, P.; Duchosal, M.; Aimable, N.; Inmaculada, R.; Mollinedo, F.; Nencioni, A. Piperidine Derivatives for use In the Treatment of Pancreatic Cancer. U.S. Patent Application No. 16/323,473, 2019. [Google Scholar]
- Lockman, J.W.; Murphy, B.R.; Zigar, D.F.; Judd, W.R.; Slattum, P.M.; Gao, Z.H.; Ostanin, K.; Green, J.; McKinnon, R.; Terry-Lorenzo, R.T.; et al. Analogues of 4-[(7-Bromo-2-methyl-4-oxo-3H-quinazolin-6-yl)methylprop-2-ynylamino]-N-(3-pyridylmethyl)benzamide (CB-30865) as potent inhibitors of nicotinamide phosphoribosyltransferase (Nampt). J. Med. Chem. 2010, 53, 8734–8746. [Google Scholar] [PubMed]
- RDKit. Open-Source Cheminformatics Software. Available online: http://www.rdkit.org (accessed on 7 August 2020).
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef]
- Watson, M.; Roulston, A.; Bélec, L.; Billot, X.; Marcellus, R.; Bédard, D.; Bernier, C.; Branchaud, S.; Chan, H.; Dairi, K.; et al. The small molecule GMX1778 is a potent inhibitor of NAD+ biosynthesis: Strategy for enhanced therapy in nicotinic acid phosphoribosyltransferase 1-deficient tumors. Mol. Cell. Biol. 2009, 29, 5872–5888. [Google Scholar] [CrossRef]
- Olesen, U.H.; Petersen, J.G.; Garten, A.; Kiess, W.; Yoshino, J.; Imai, S.; Christensen, M.K.; Fristrup, P.; Thougaard, A.V.; Björkling, F.; et al. Target enzyme mutations are the molecular basis for resistance towards pharmacological inhibition of nicotinamide phosphoribosyltransferase. BMC Cancer 2010, 10, 677. [Google Scholar] [CrossRef]
- Wang, W.; Elkins, K.; Oh, A.; Ho, Y.C.; Wu, J.; Li, H.; Xiao, Y.; Kwong, M.; Coons, M.; Brillantes, B.; et al. Structural basis for resistance to diverse classes of NAMPT inhibitors. PLoS ONE 2014, 9, e109366. [Google Scholar] [CrossRef]
- Tosco, P.; Balle, T.; Shiri, F. Open3DALIGN: An open-source software aimed at unsupervised ligand alignment. J. Comput. Aided Mol. Des. 2011, 25, 777–783. [Google Scholar] [CrossRef]
- Nelson, D.J.; Brammer, C.N. Toward consistent terminology for cyclohexane conformers in introductory organic chemistry. J. Chem. Educ. 2011, 88, 292–294. [Google Scholar] [CrossRef]
- Gillig, A.; Majjigapu, S.R.; Sordat, B.; Vogel, P. Synthesis of a C-Iminoribofuranoside Analog of the Nicotinamide Phosphoribosyltransferase (NAMPT) Inhibitor FK866. Helv. Chim. Acta 2012, 95, 34–42. [Google Scholar] [CrossRef]
- Neumann, C.S.; Olivas, K.C.; Anderson, M.E.; Cochran, J.H.; Jin, S.; Li, F.; Loftus, L.V.; Meyer, D.W.; Neale, J.; Nix, J.C.; et al. Targeted Delivery of Cytotoxic NAMPT Inhibitors Using Antibody-Drug Conjugates. Mol. Cancer Ther. 2018, 17, 2633–2642. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 a (μM) | Binding Affinity Score b (kJ/mol) | |
|---|---|---|---|
| Non-Protonation | Protonation | ||
| 3a (piperidine) | 0.11 | −29.07 | - |
| 3b (piperazine) | 5.06 | −22.13 | −23.85 |
| 1 (piperidine) | 0.52 | −28.05 | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanuma, S.-i.; Katsuragi, K.; Oyama, T.; Yoshimori, A.; Shibasaki, Y.; Asawa, Y.; Yamazaki, H.; Makino, K.; Okazawa, M.; Ogino, Y.; et al. Structural Basis of Beneficial Design for Effective Nicotinamide Phosphoribosyltransferase Inhibitors. Molecules 2020, 25, 3633. https://doi.org/10.3390/molecules25163633
Tanuma S-i, Katsuragi K, Oyama T, Yoshimori A, Shibasaki Y, Asawa Y, Yamazaki H, Makino K, Okazawa M, Ogino Y, et al. Structural Basis of Beneficial Design for Effective Nicotinamide Phosphoribosyltransferase Inhibitors. Molecules. 2020; 25(16):3633. https://doi.org/10.3390/molecules25163633
Chicago/Turabian StyleTanuma, Sei-ichi, Kiyotaka Katsuragi, Takahiro Oyama, Atsushi Yoshimori, Yuri Shibasaki, Yasunobu Asawa, Hiroaki Yamazaki, Kosho Makino, Miwa Okazawa, Yoko Ogino, and et al. 2020. "Structural Basis of Beneficial Design for Effective Nicotinamide Phosphoribosyltransferase Inhibitors" Molecules 25, no. 16: 3633. https://doi.org/10.3390/molecules25163633
APA StyleTanuma, S.-i., Katsuragi, K., Oyama, T., Yoshimori, A., Shibasaki, Y., Asawa, Y., Yamazaki, H., Makino, K., Okazawa, M., Ogino, Y., Sakamoto, Y., Nomura, M., Sato, A., Abe, H., Nakamura, H., Takahashi, H., Tanuma, N., & Uchiumi, F. (2020). Structural Basis of Beneficial Design for Effective Nicotinamide Phosphoribosyltransferase Inhibitors. Molecules, 25(16), 3633. https://doi.org/10.3390/molecules25163633