
Generation and Characterization of a DNA-GCN4 Oligonucleotide-Peptide Conjugate: The Impact DNA/Protein Interactions on the Sensitization of DNA


Abstract
:1. Introduction
2. Results and Discussion
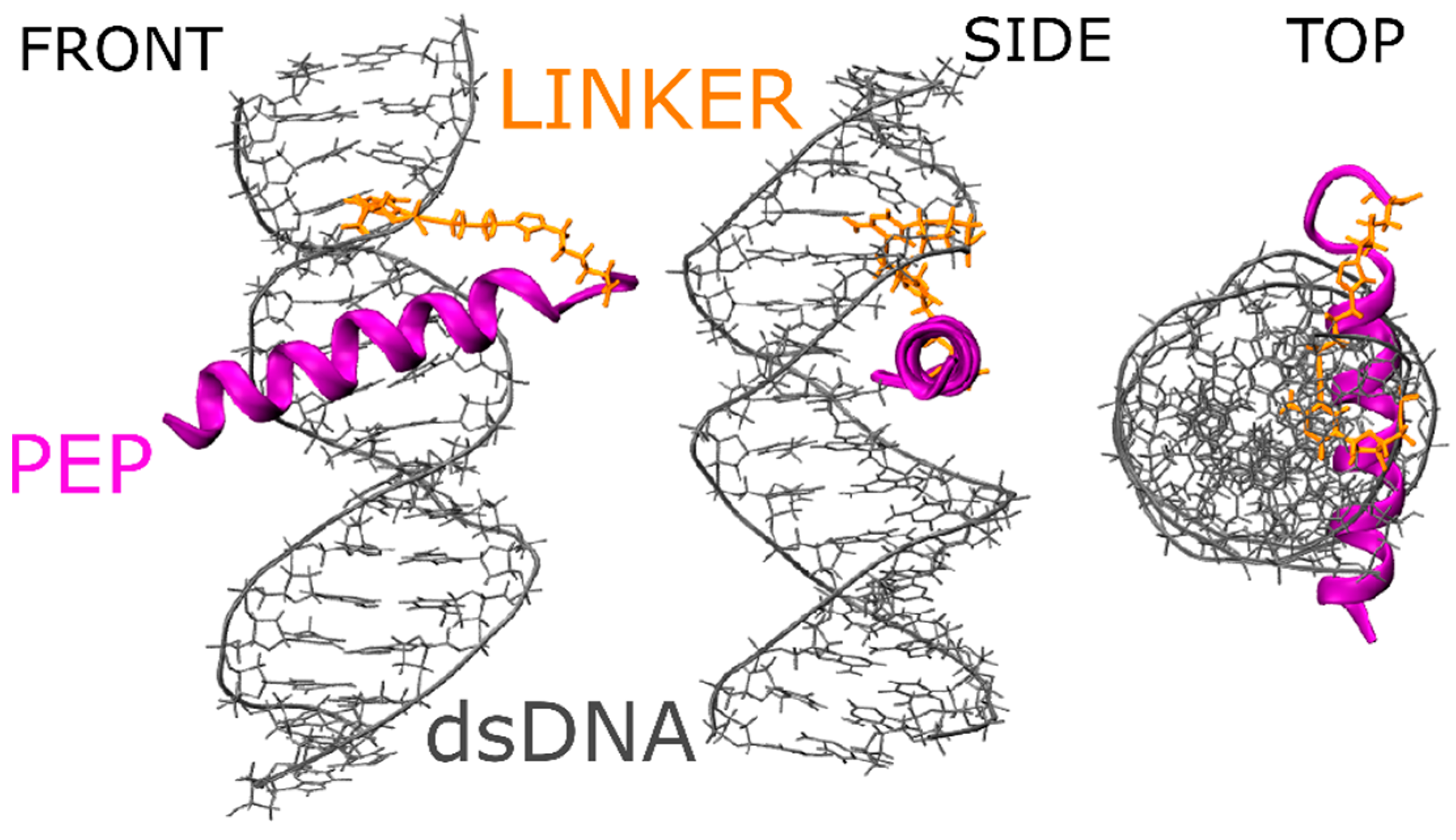
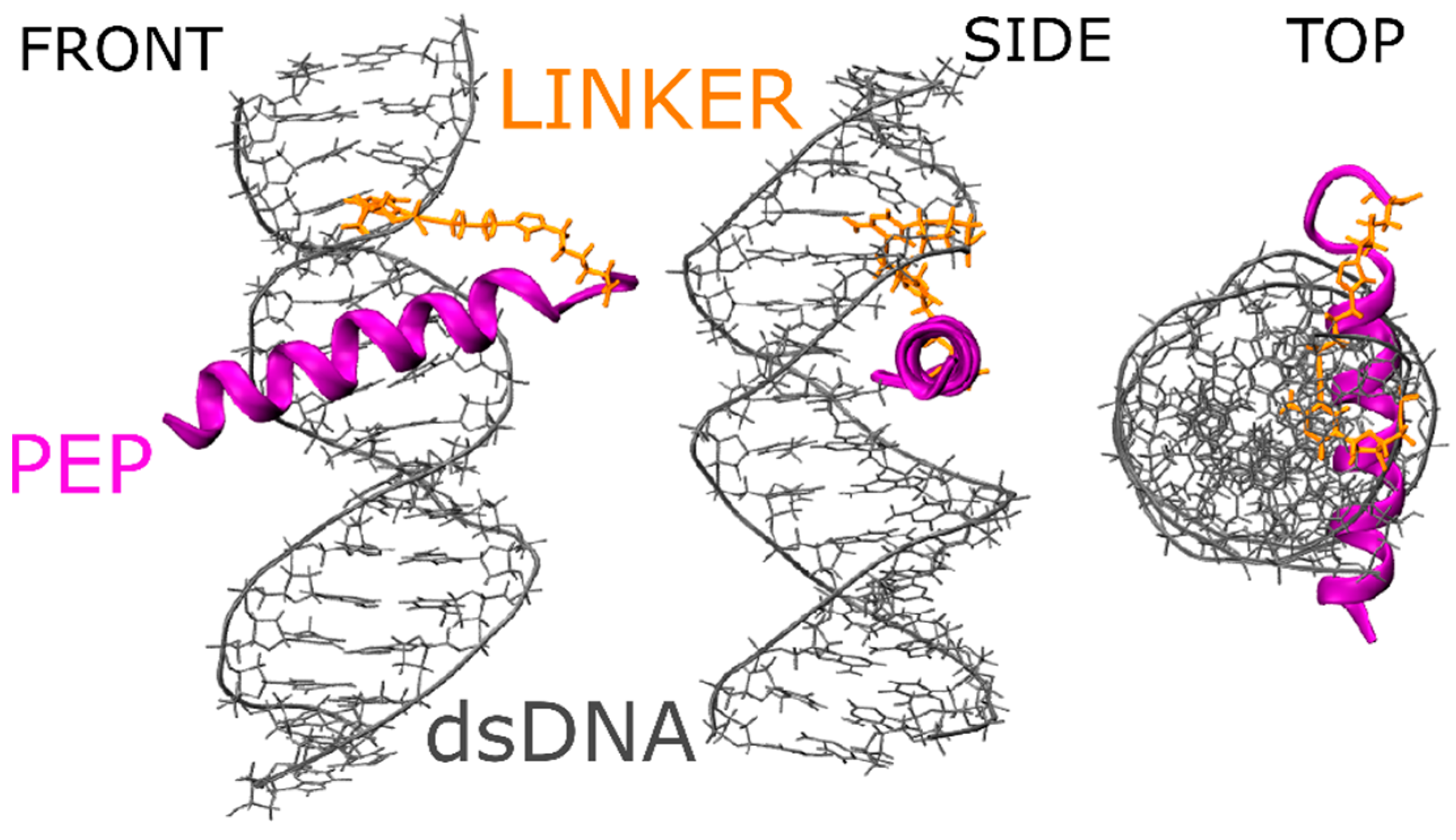
2.1. Molecular Properties of the GCN4-DNA Molecular System
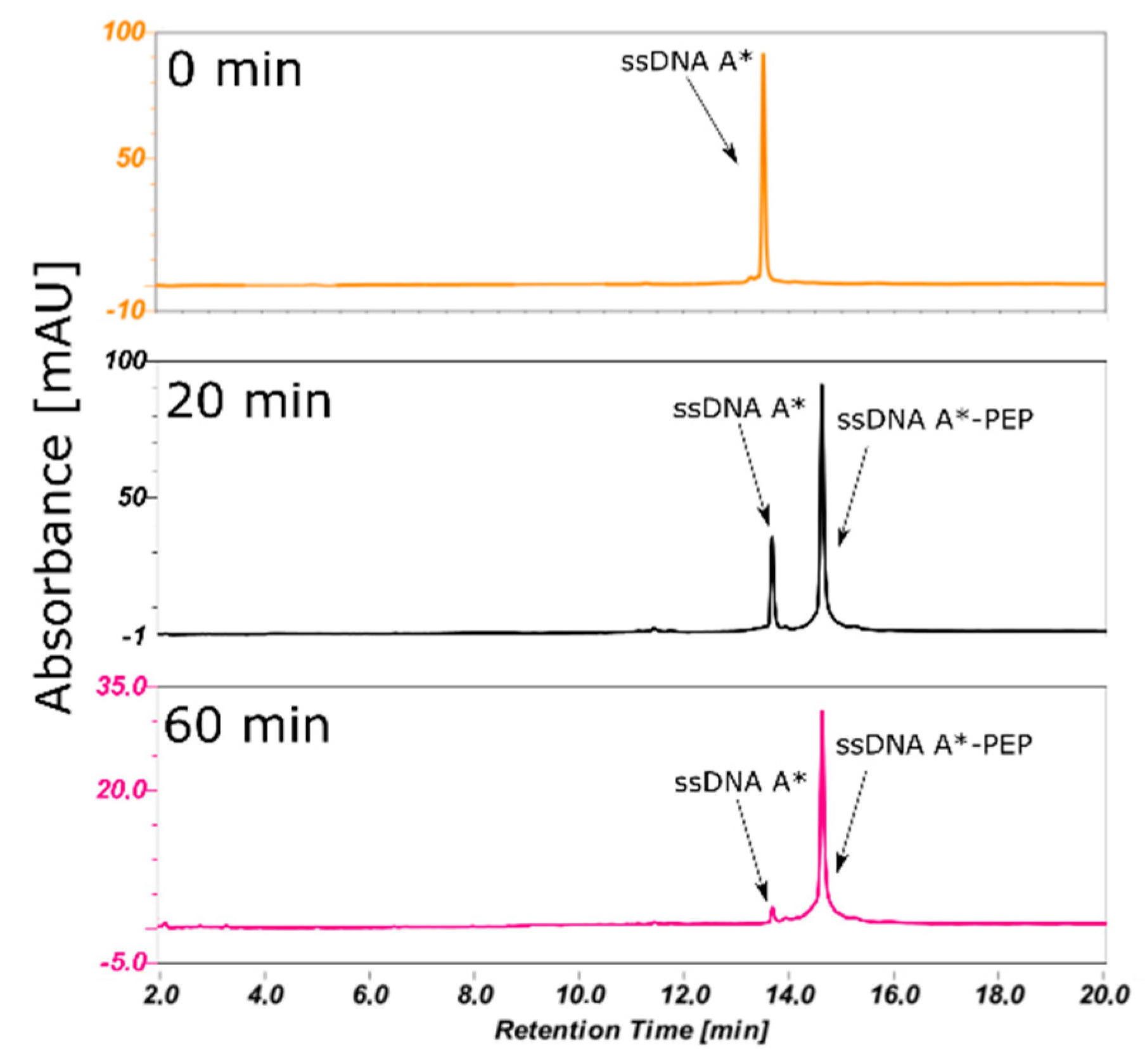
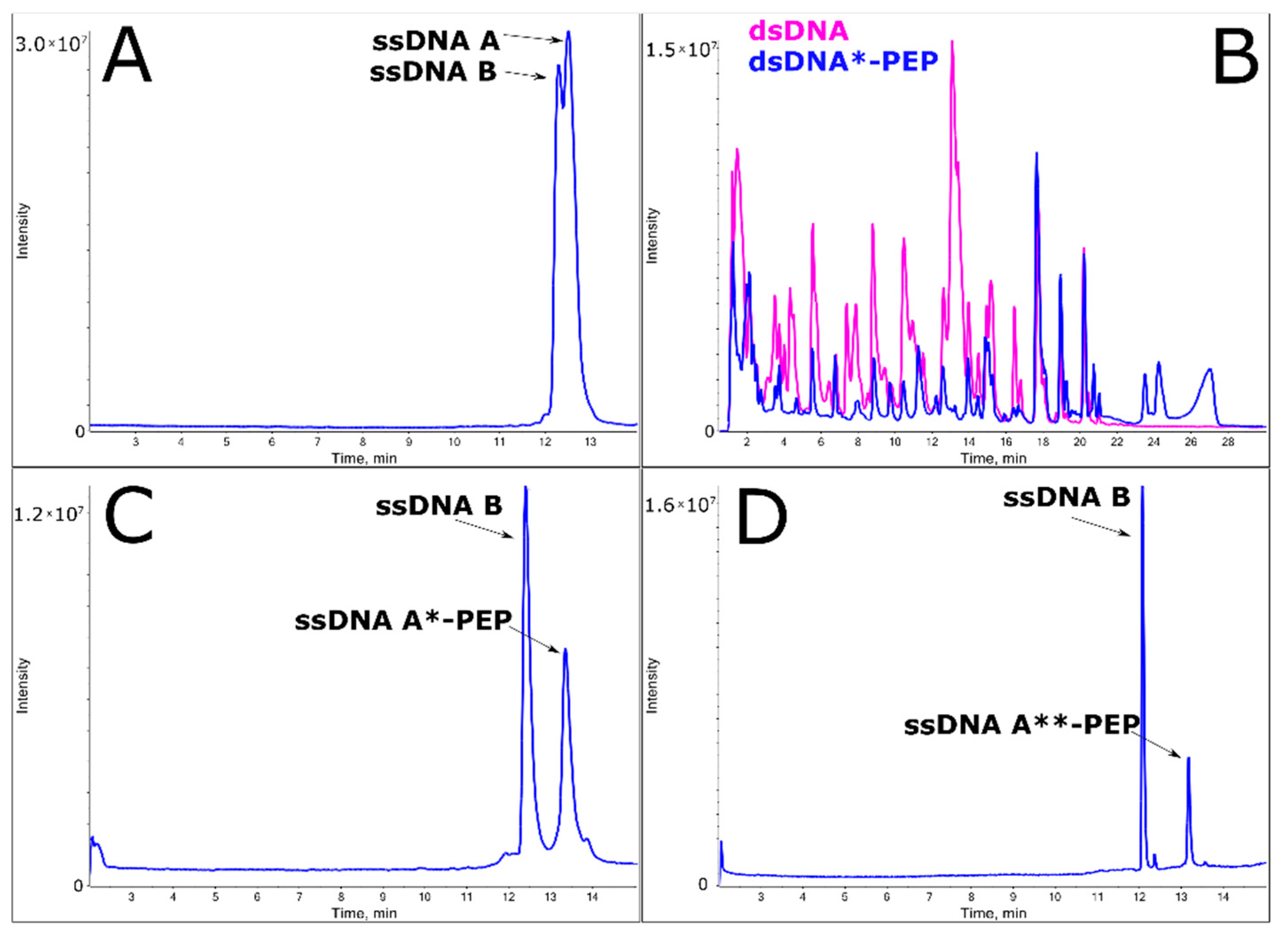
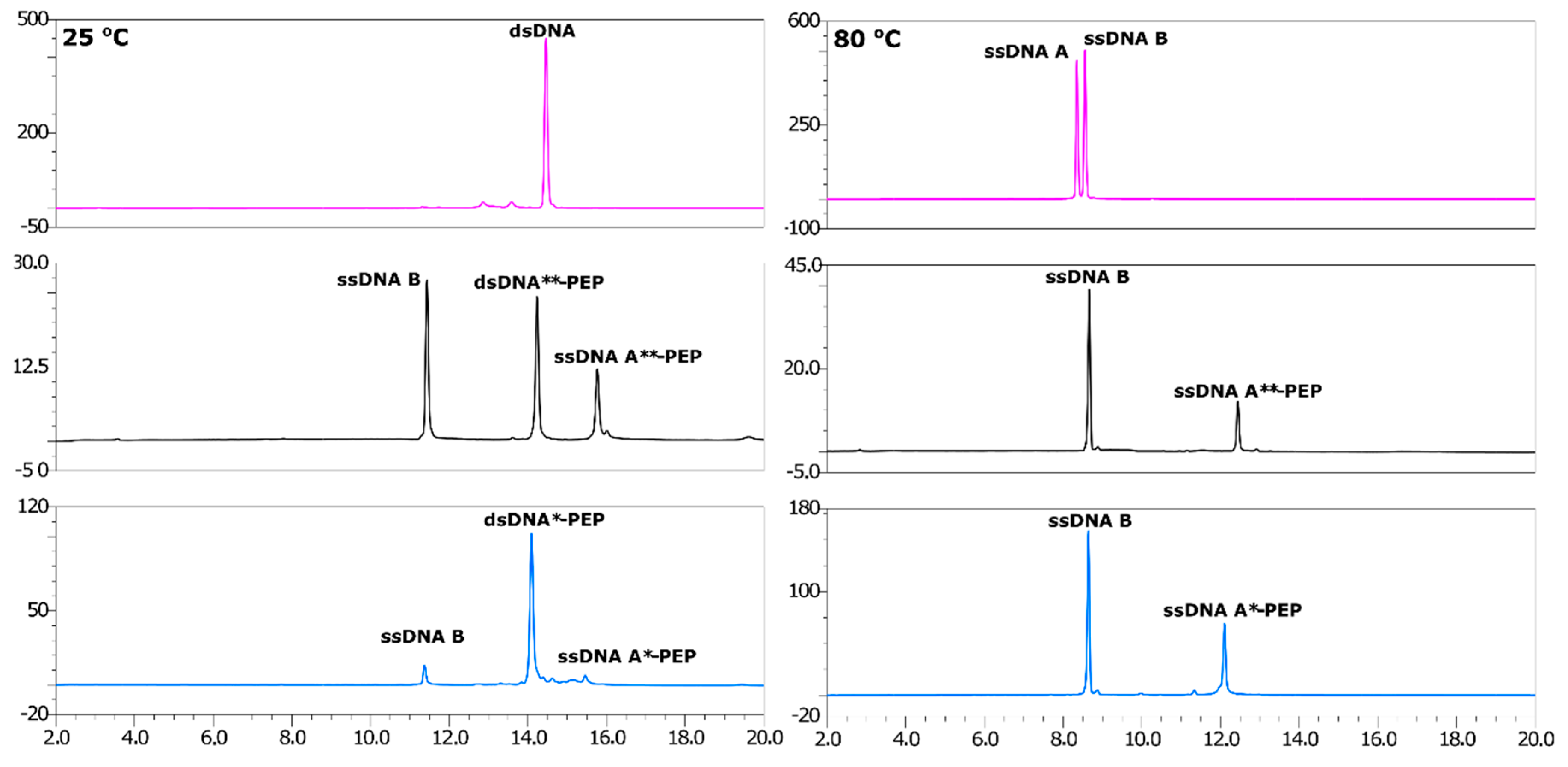
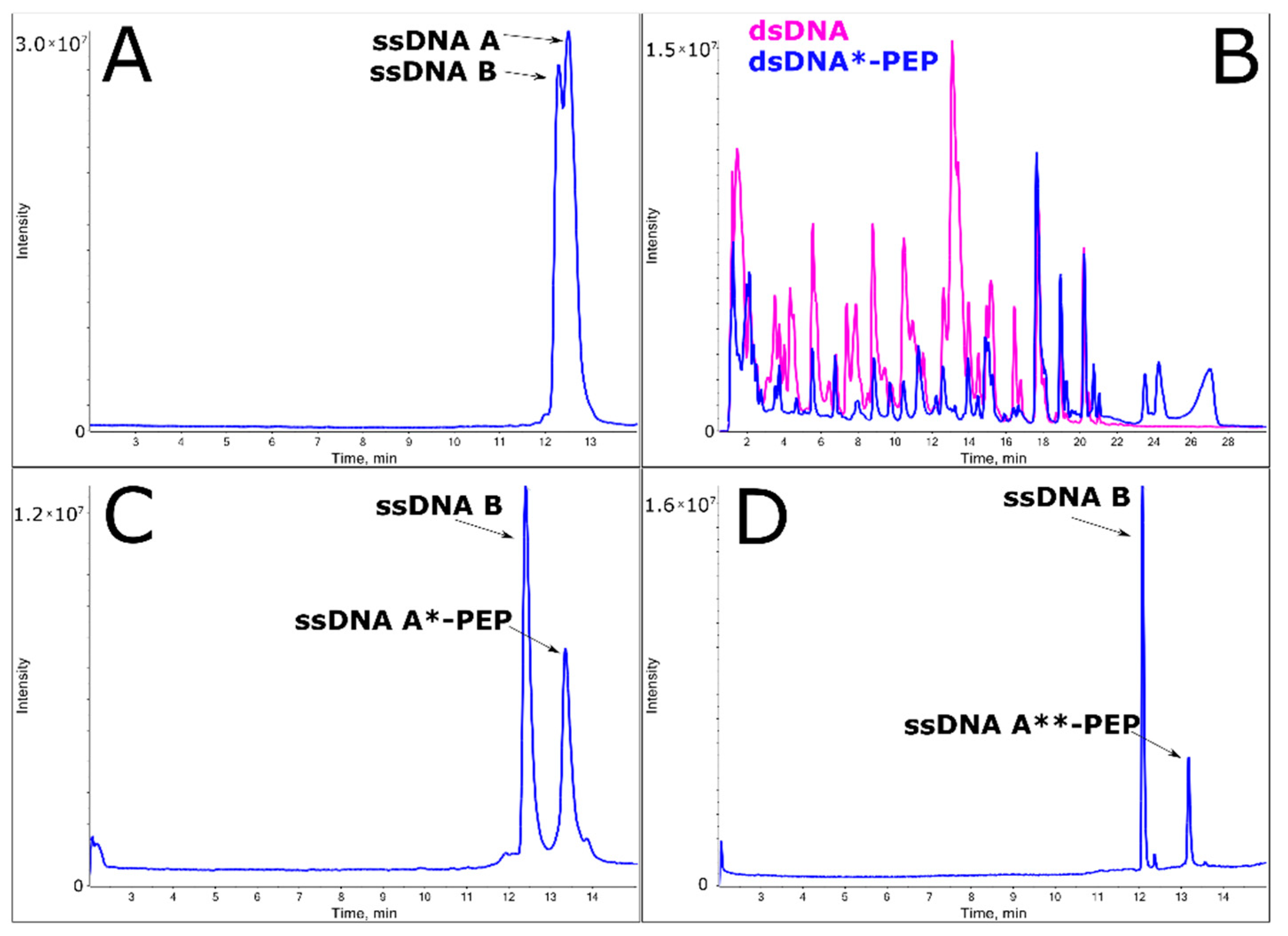
2.2. Efficient Synthesis of a ssDNA/Protein Complex
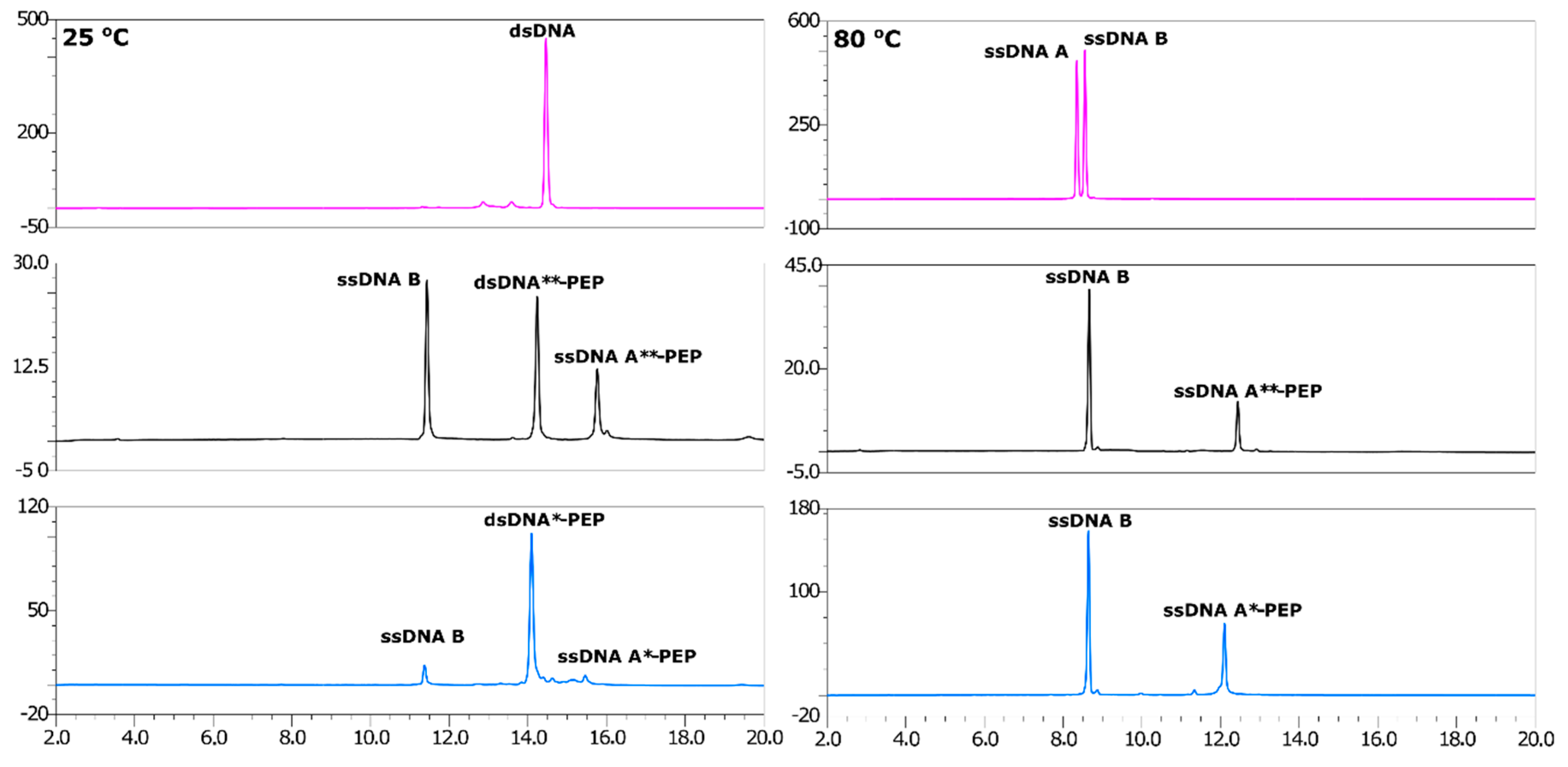
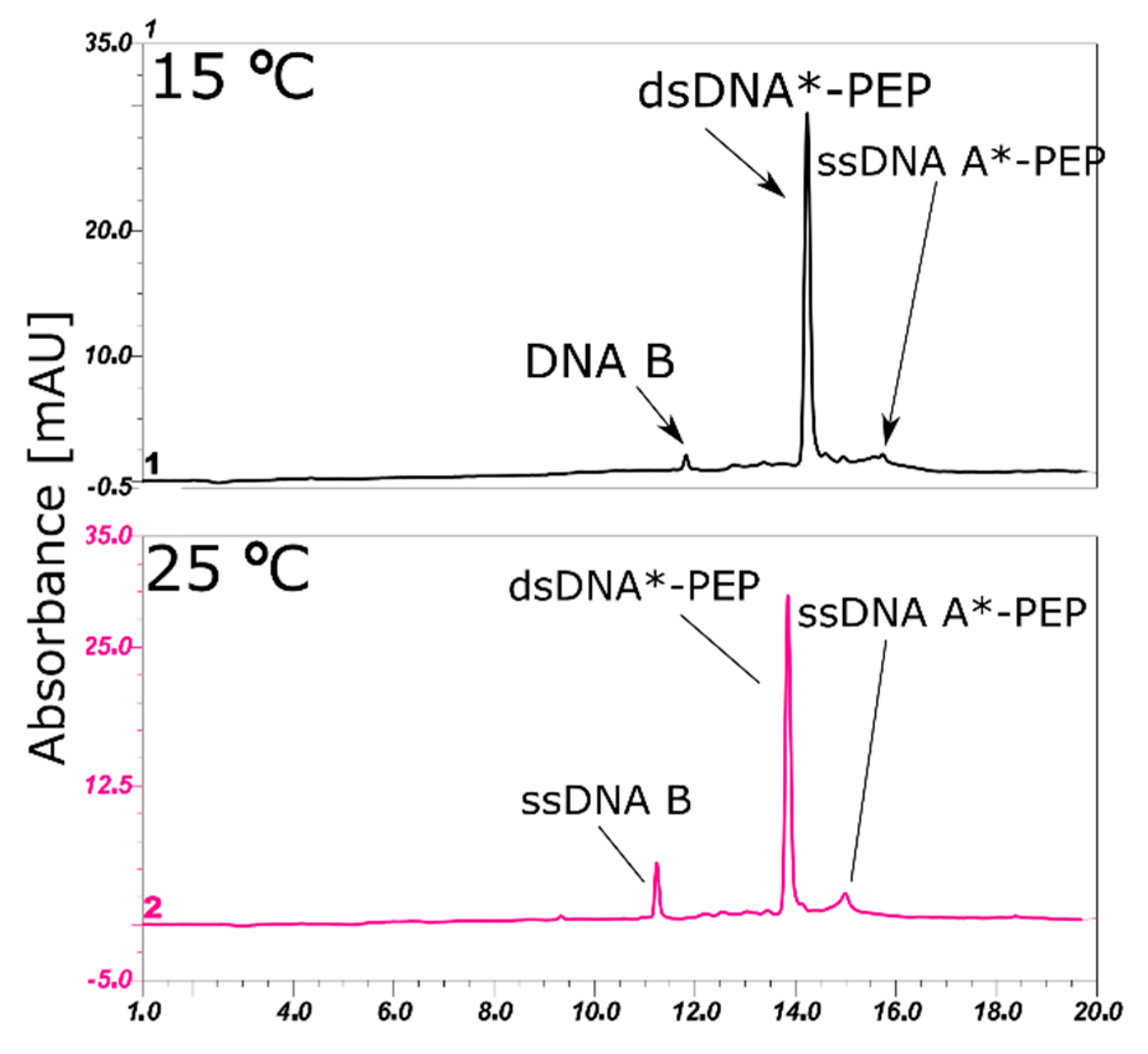
2.3. Annealing of ssDNA into dsDNA
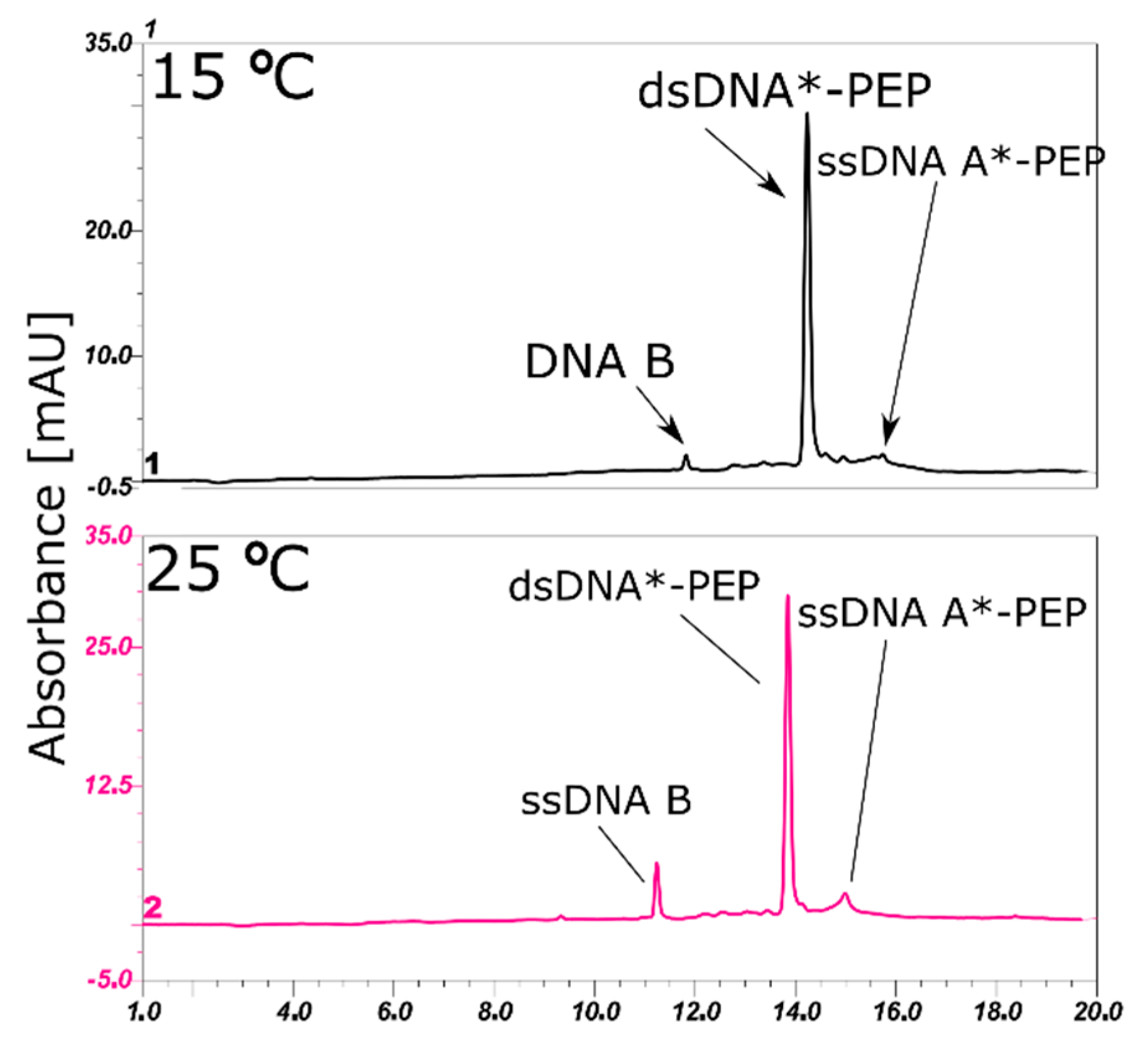
2.4. LC-MS Analysis of dsDNA-Peptide Conjugate
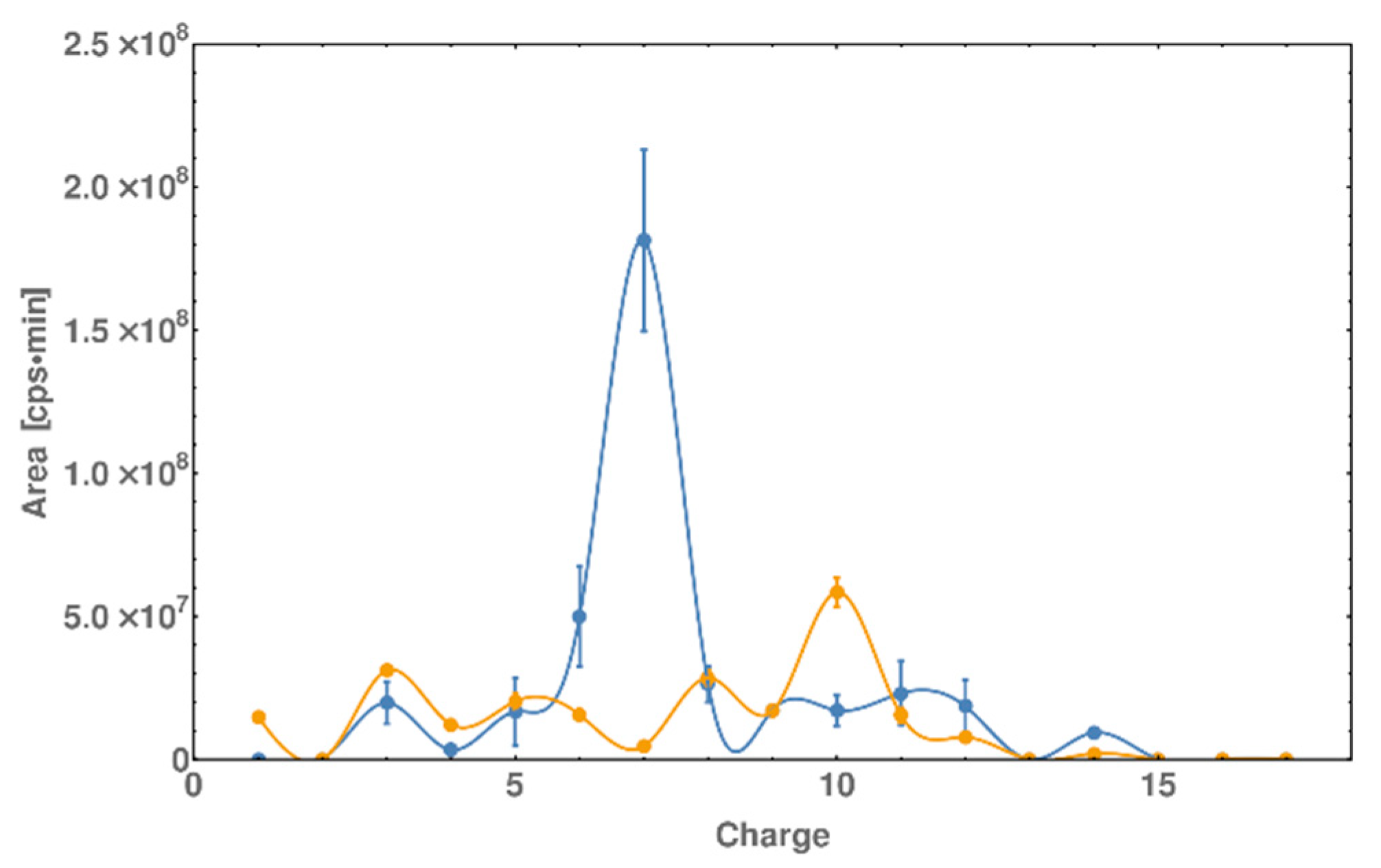
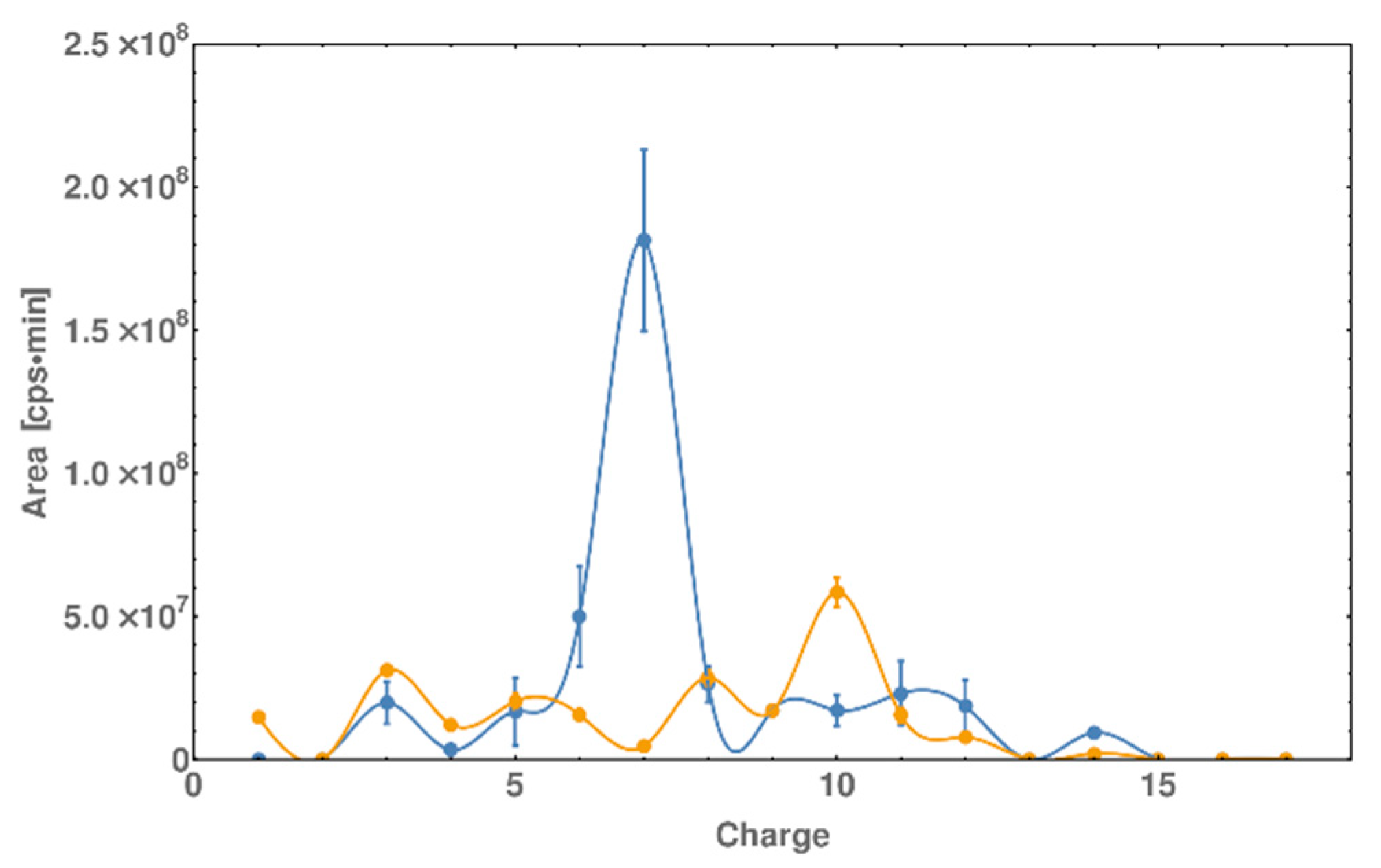
2.5. DNase I Footprinting Reveals the Binding Pattern of DNA and Peptide
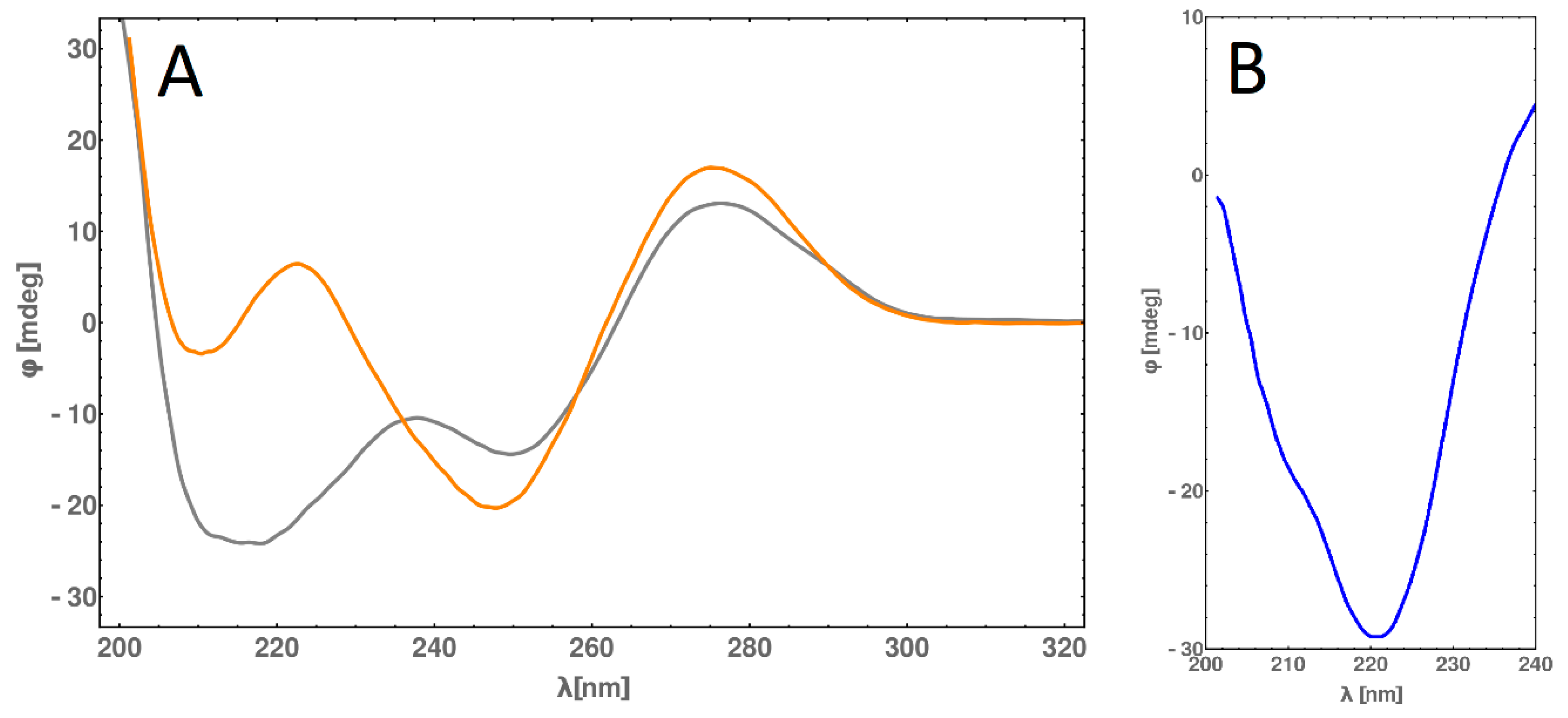
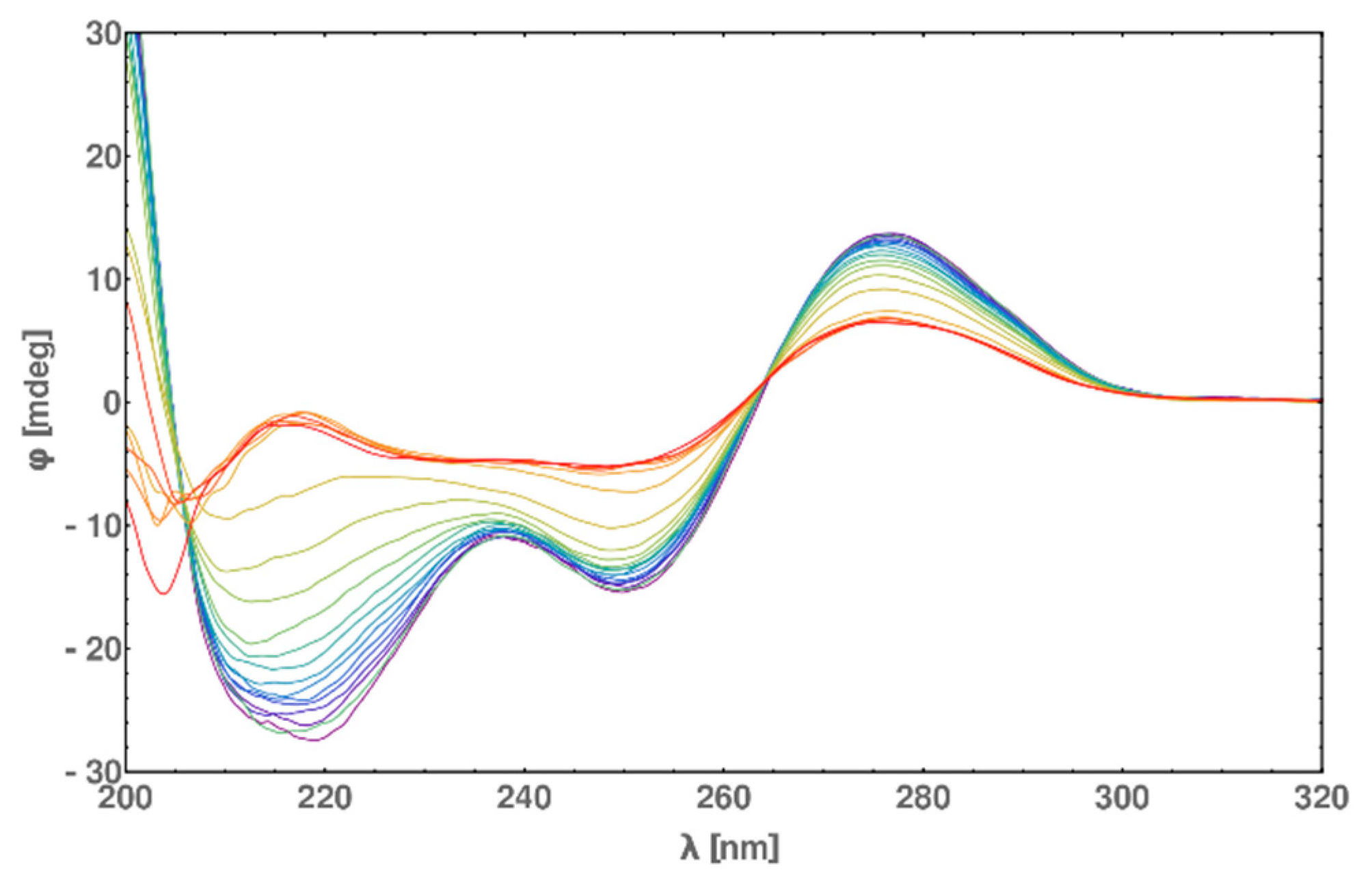
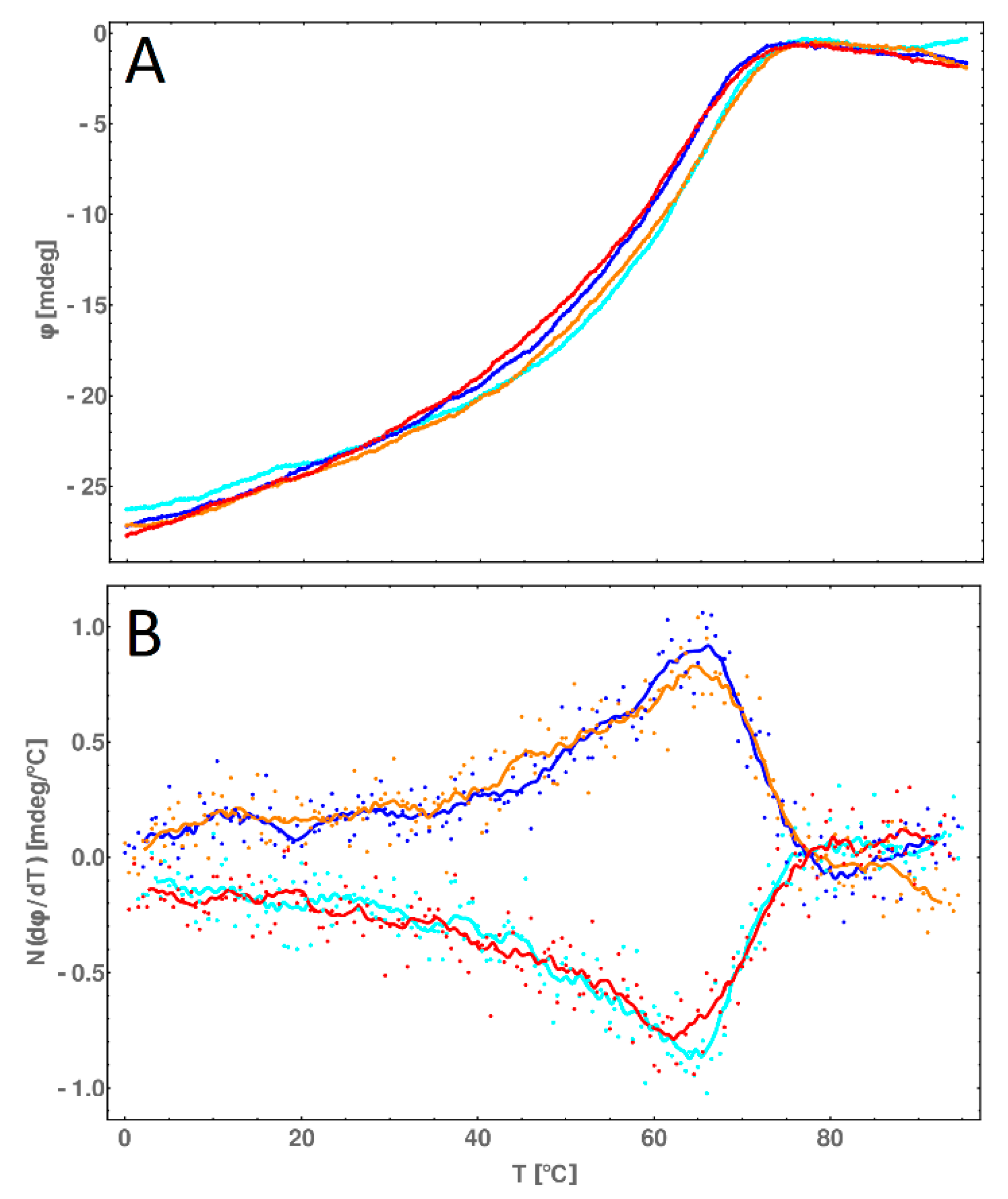
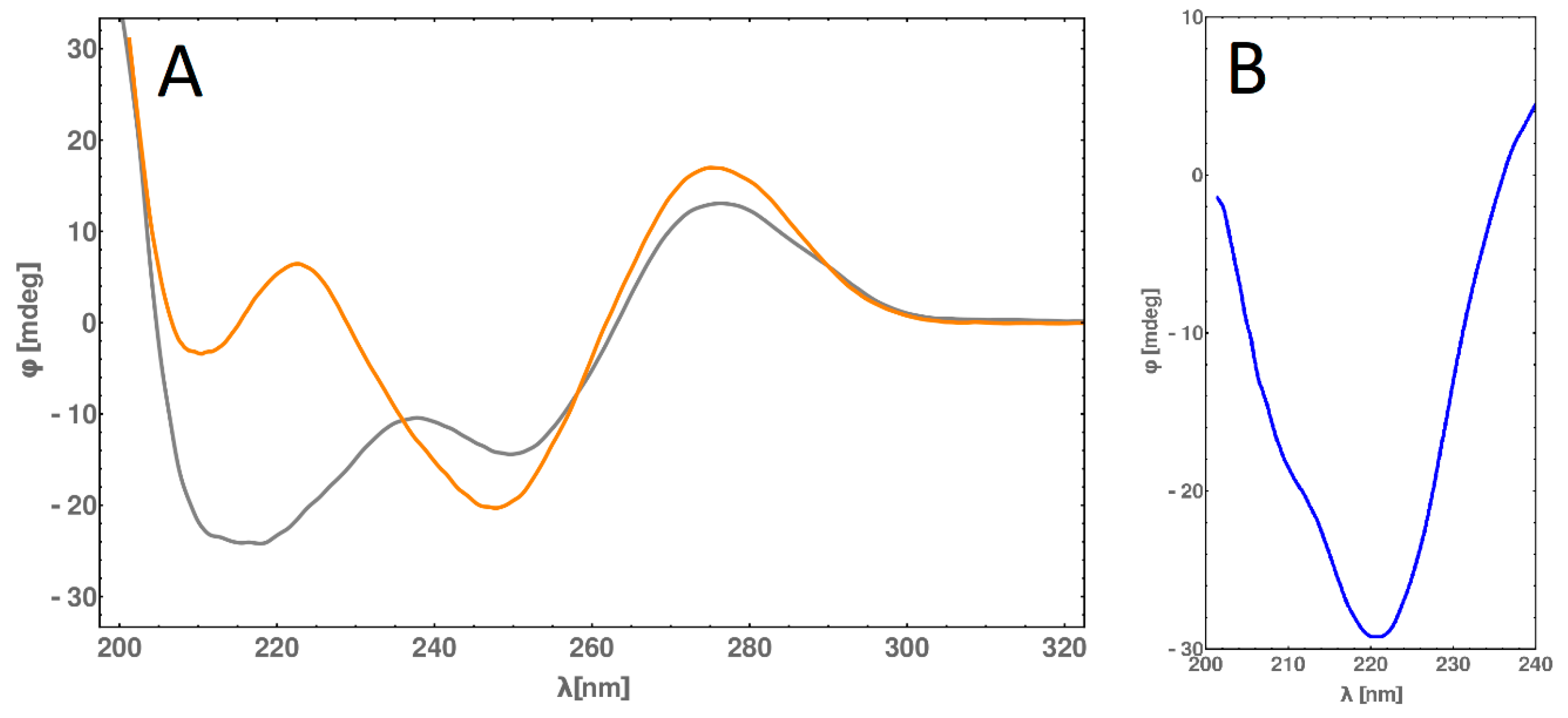
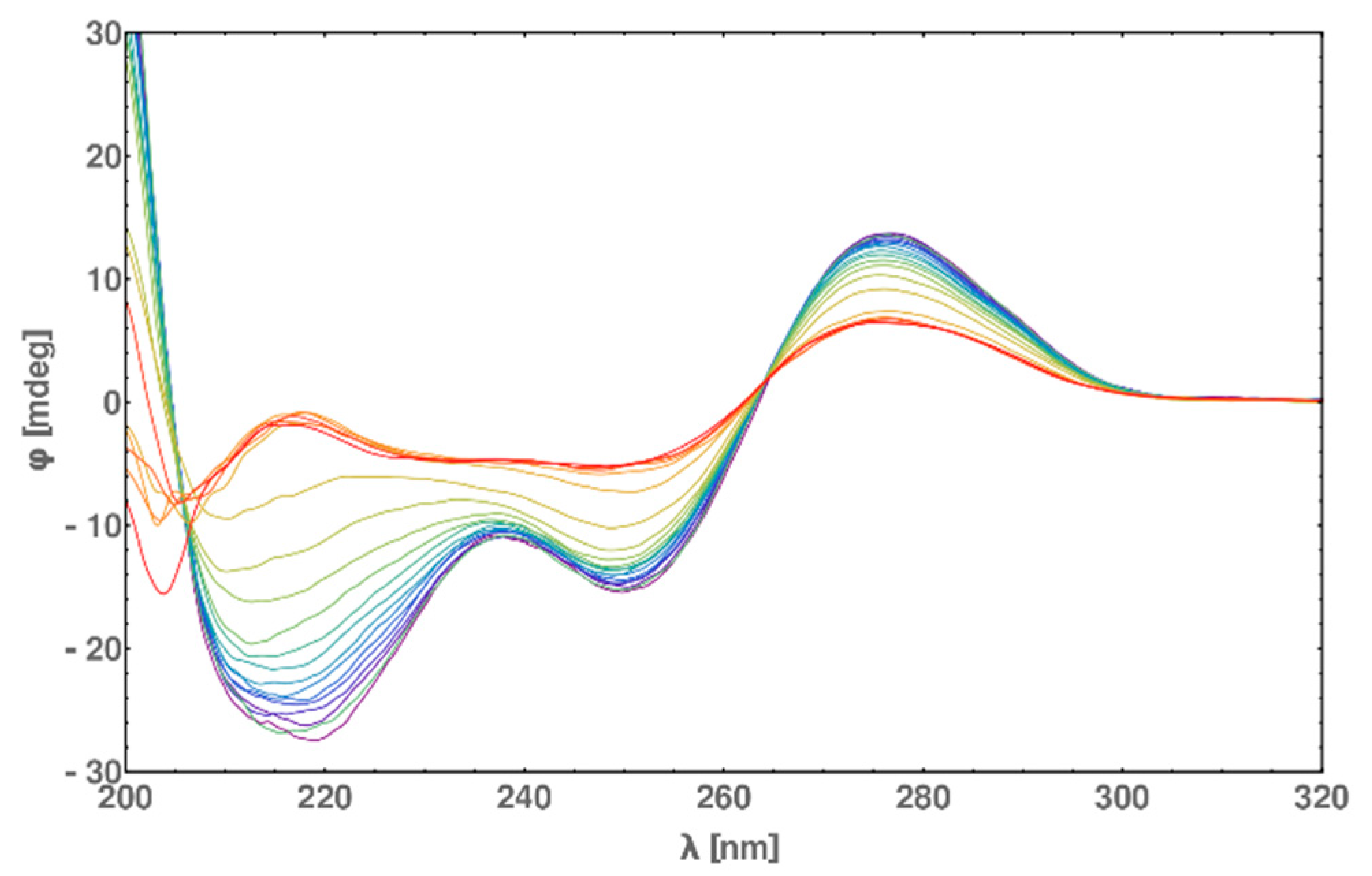
2.6. Circular Dichroism (CD) Spectra Measurement
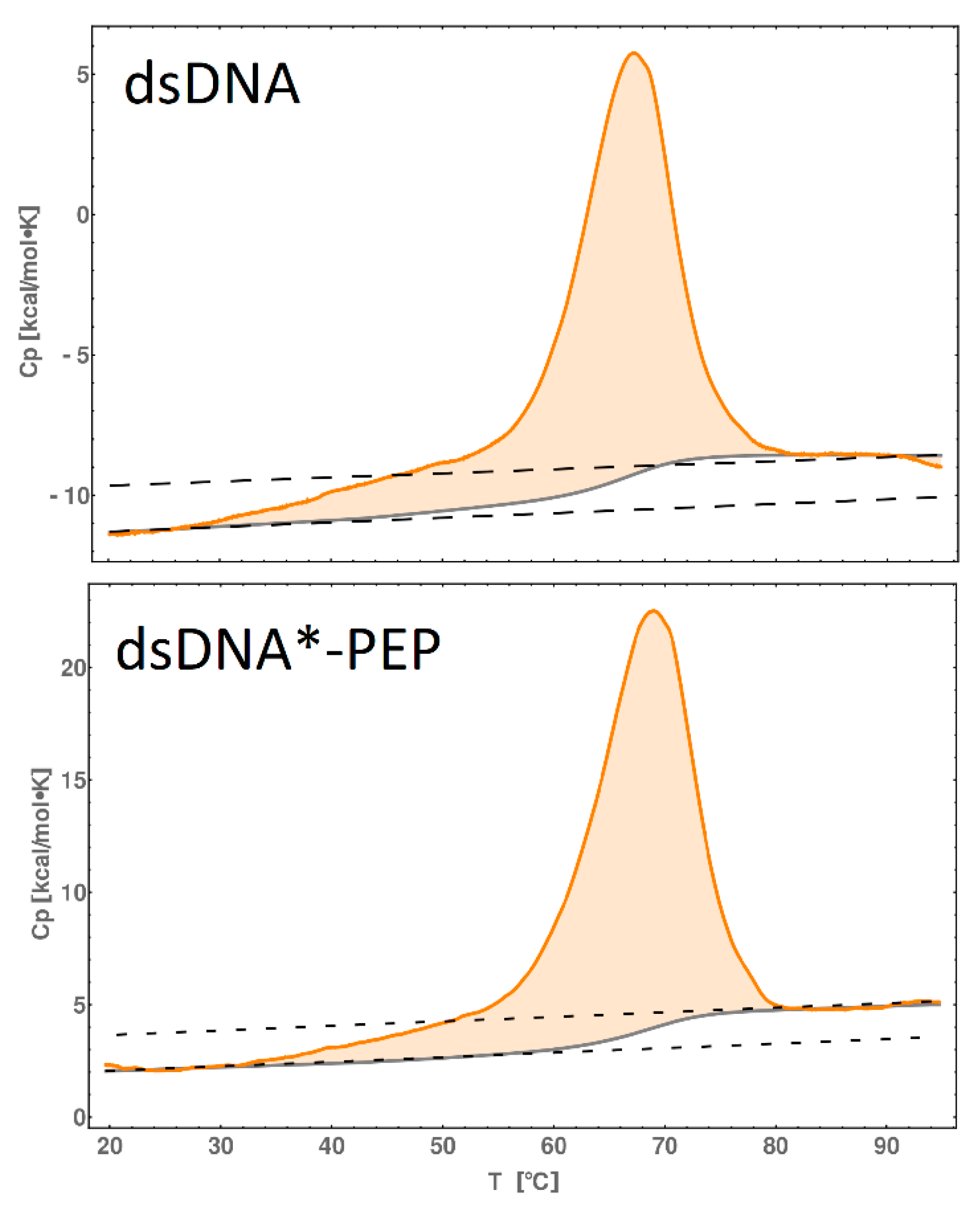
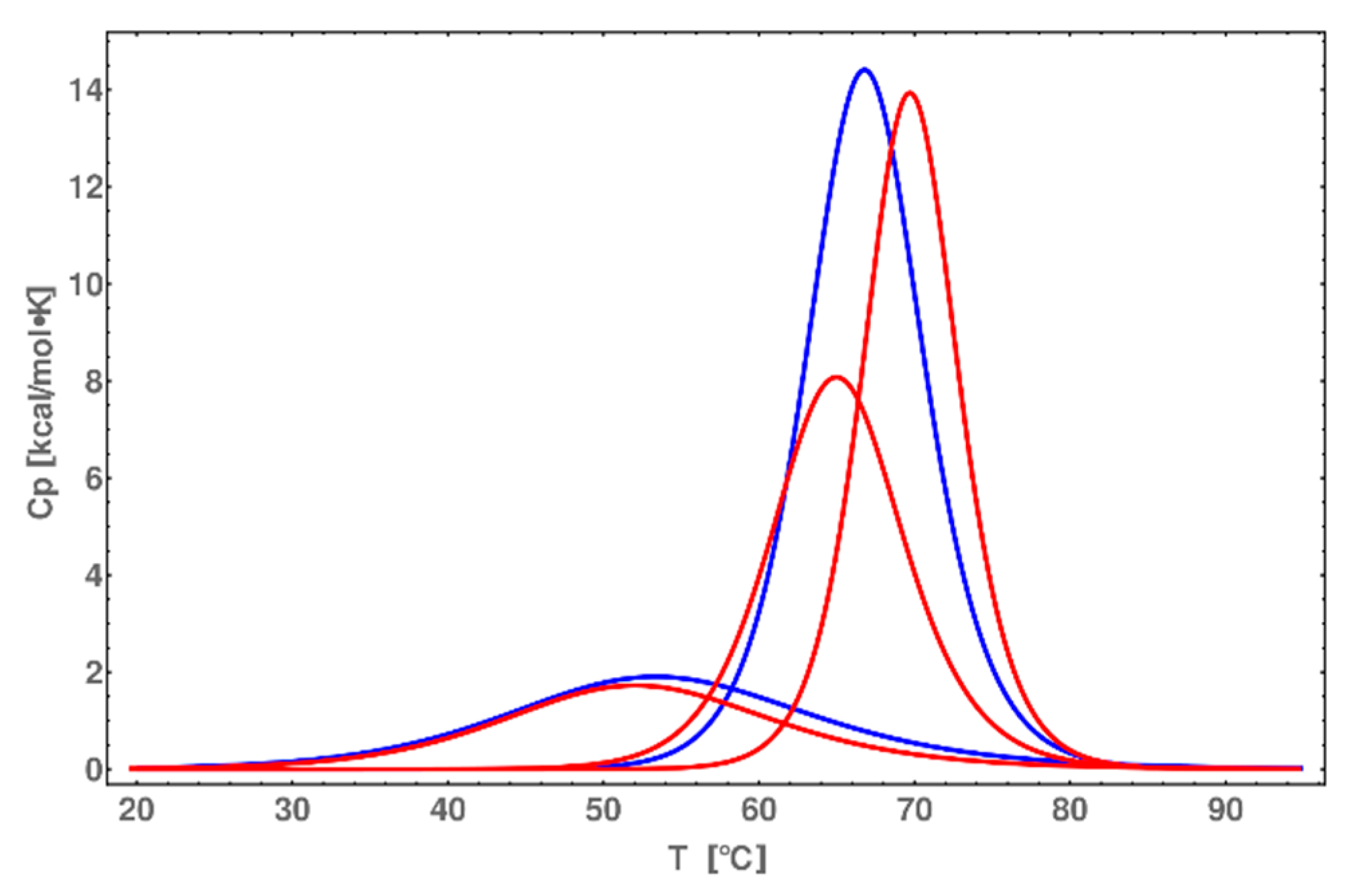
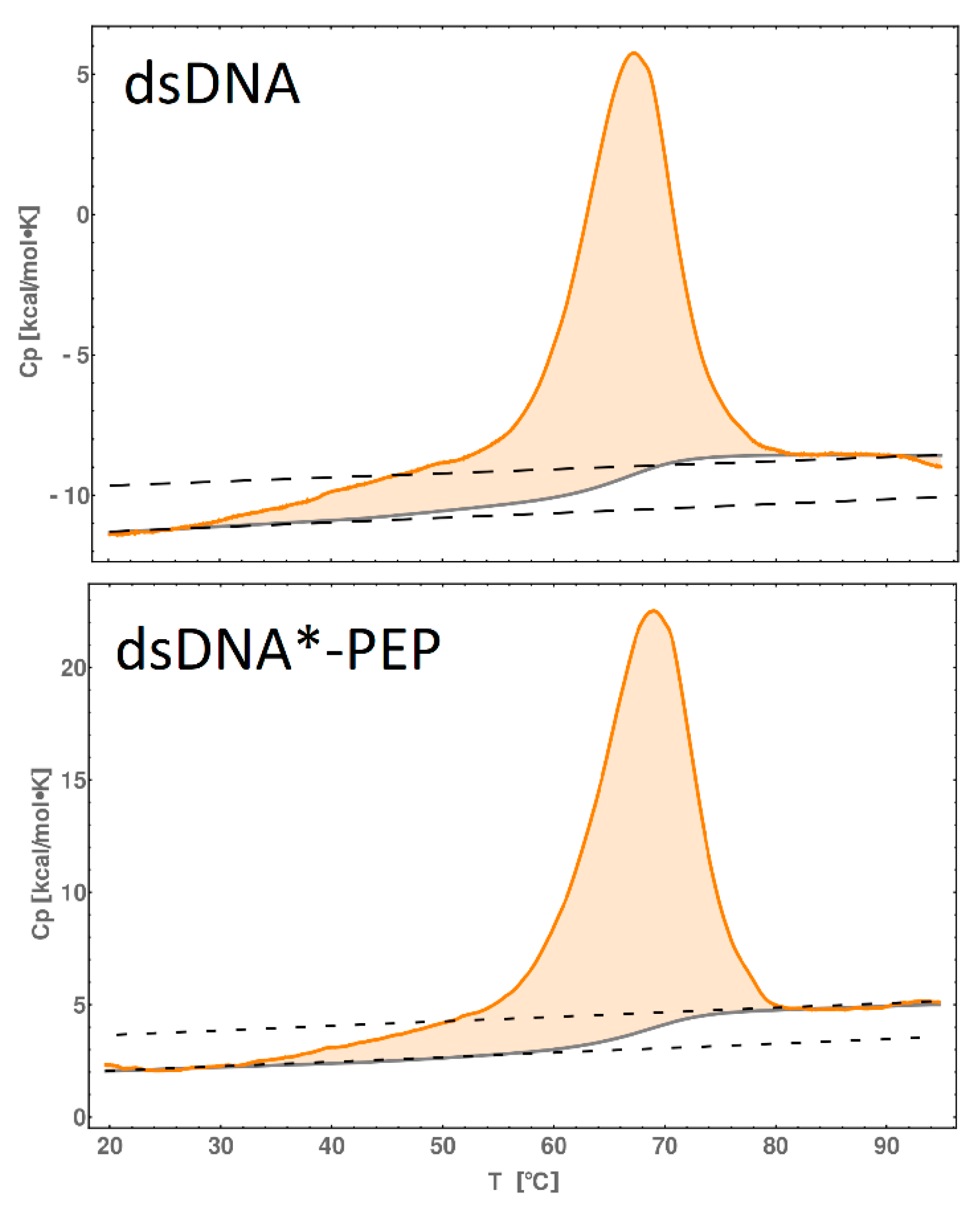
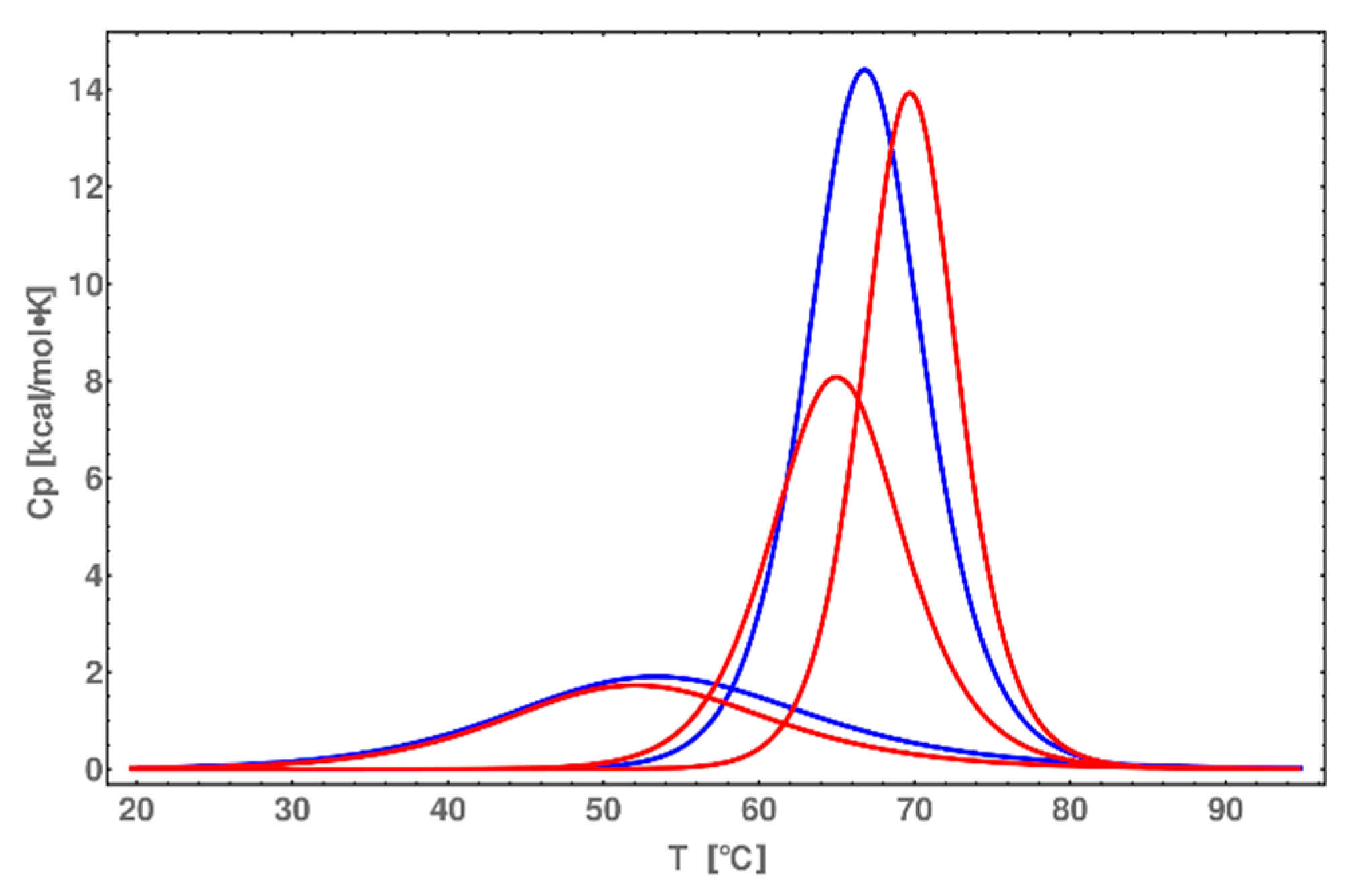
2.7. DSC Melting Profiles
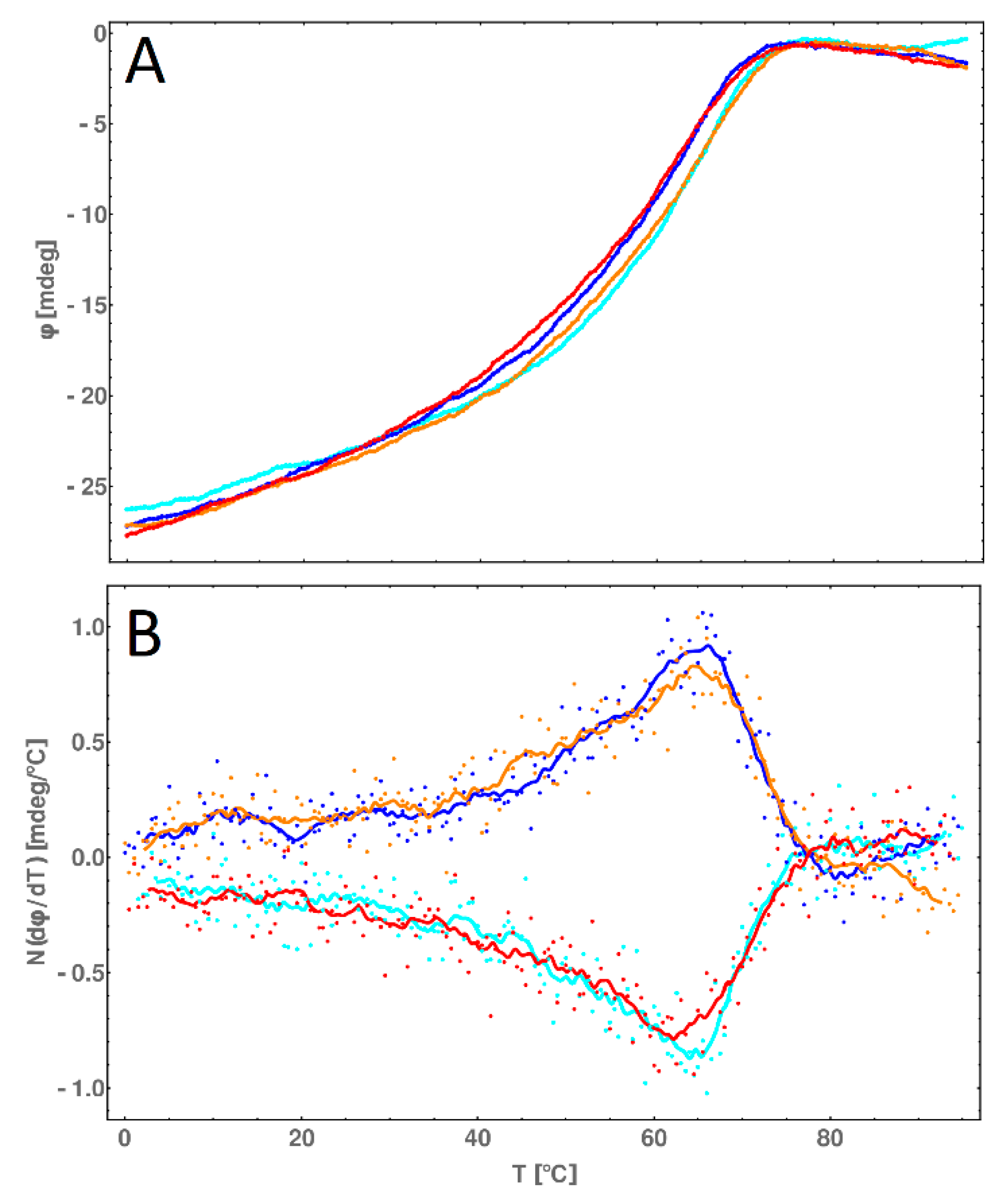
2.8. CD Melting Temperature Comparative Analysis of dsDNA and dsDNA-PEP
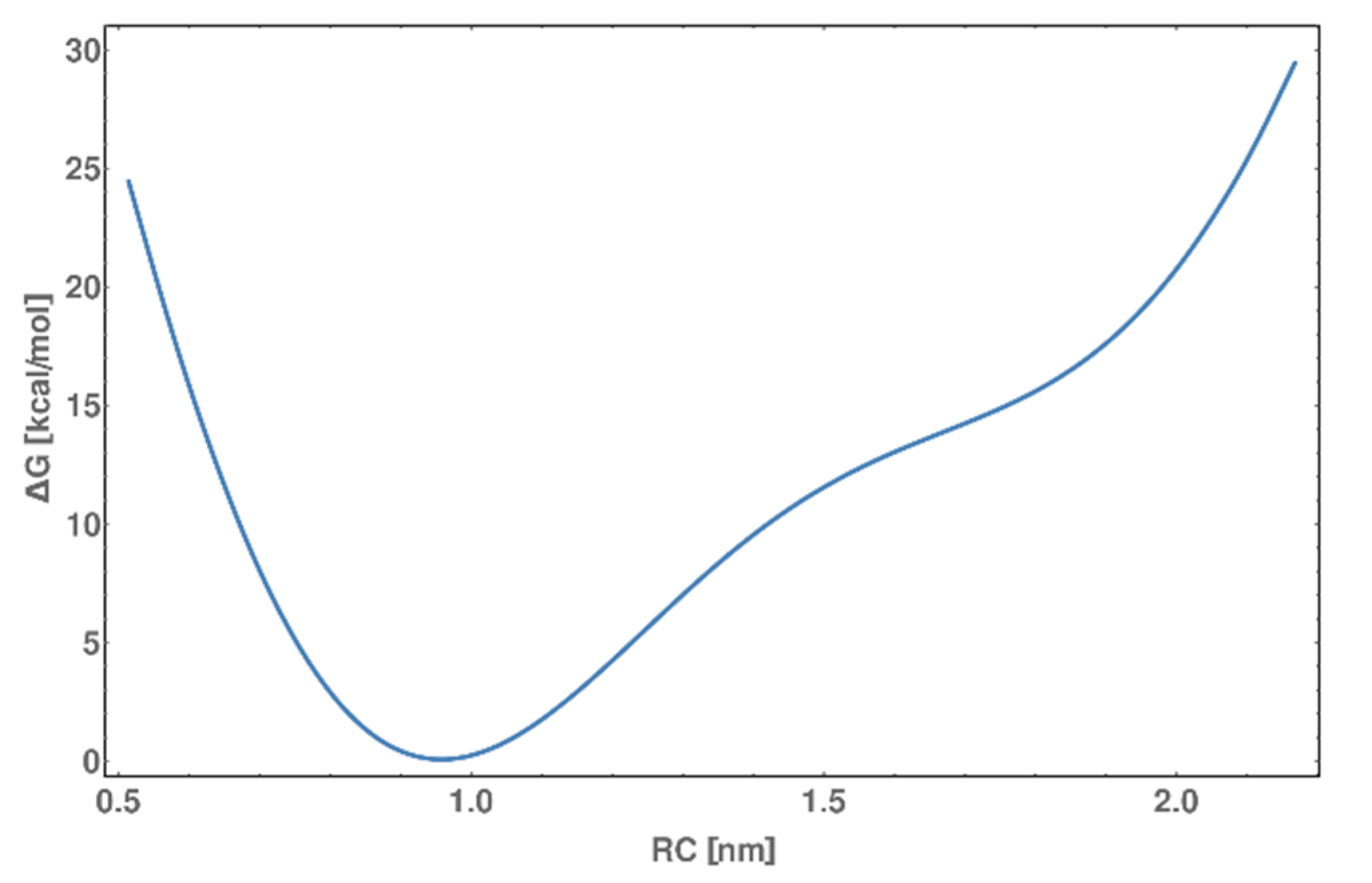
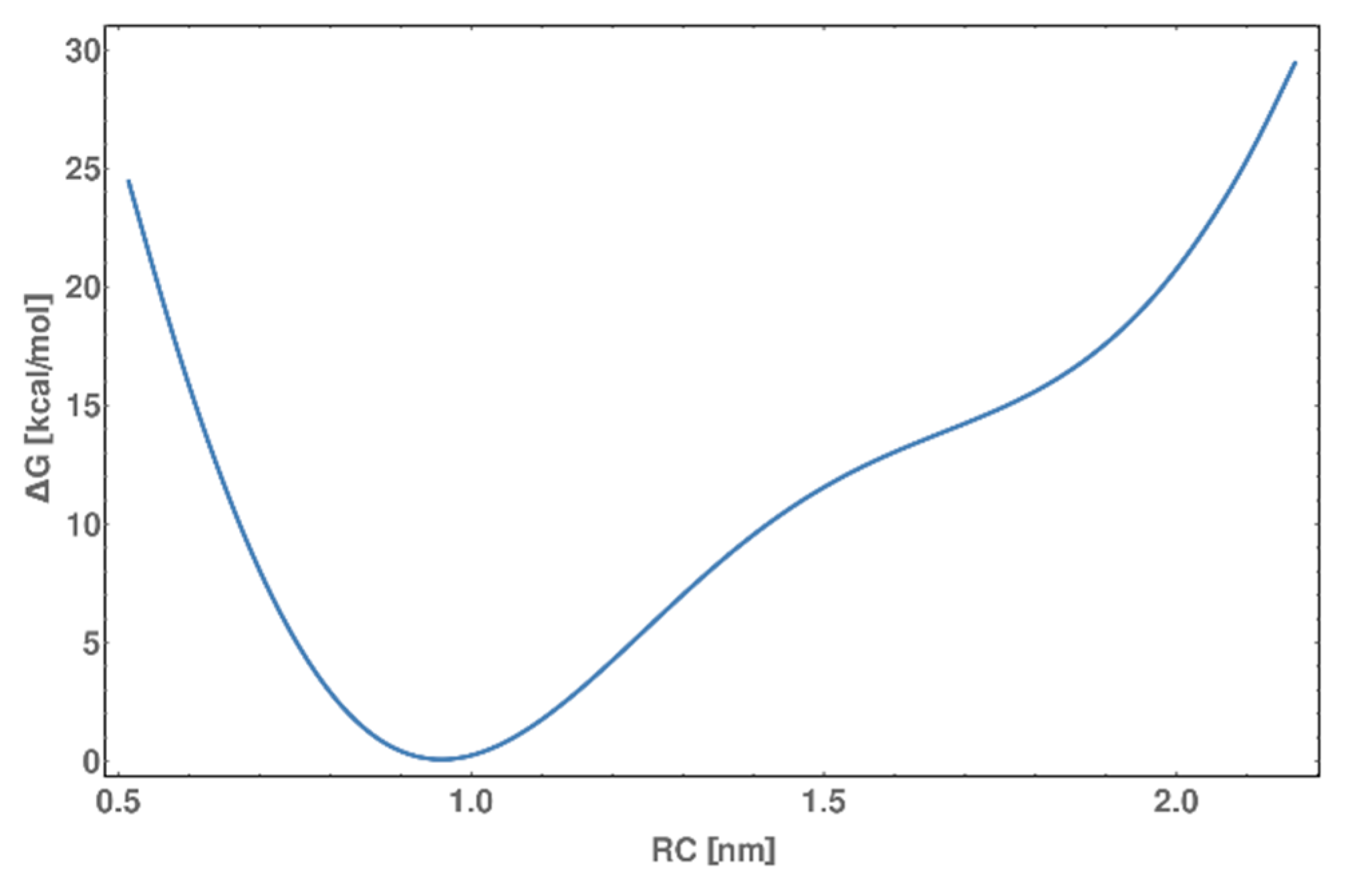
2.9. Metadynamics of dsDNA-PEP Complex
3. Materials and Methods
3.1. Materials
3.2. HPLC
3.3. LC-MS
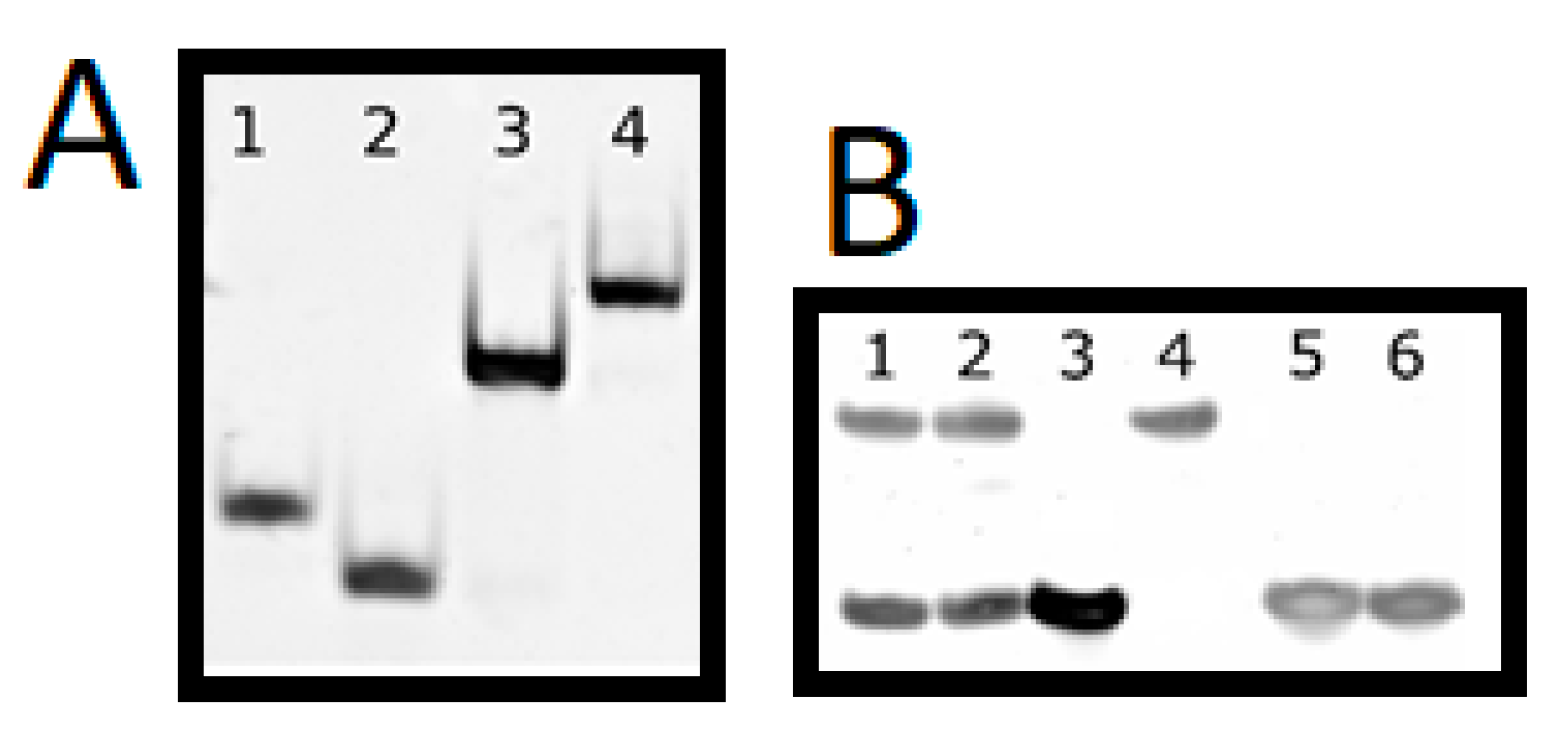
3.4. Native and Denaturing PAGE Electrophoresis
3.5. DNase Footprinting
3.6. Annealing and DNA-Peptide Ligation
3.7. Circular Dichroism (CD) Measurements
3.8. Differential Scanning Calorimetry (DSC) Measurements
3.9. Molecular Dynamics Structure Preparation
3.10. Simulation Procedure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rak, J.; Chomicz, L.; Wiczk, J.; Westphal, K.; Zdrowowicz, M.; Wityk, P.; Makurat, S.; Golon, Ł. Mechanisms of damage to DNA labeled with electrophilic nucleobases induced by ionizing or UV radiation. J. Phys. Chem. B 2015, 119, 8227–8238. [Google Scholar] [CrossRef]
- Hudson, W.H.; Ortlund, E.A. The structure, function and evolution of proteins that bind DNA and RNA. Nat. Rev. Mol. Cell Biol. 2014, 15, 749–760. [Google Scholar] [CrossRef] [Green Version]
- Bell, S.P.; Gann, A.; Levine, M.R.; Losick, J.D.; Watson, T.A. Baker. Chapter II. Structure and study of macromolecules. In Molecular Biology of the Gene, 7th ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2008. [Google Scholar]
- Mukherjee, S.; Majumdar, S.; Bhattacharyya, D. Role of hydrogen bonds in protein−DNA recognition: Effect of nonplanar amino groups. J. Phys. Chem. B 2005, 109, 10484–10492. [Google Scholar] [CrossRef] [PubMed]
- Mandel-Gutfreund, Y.; Schueler, O.; Margalit, H. Comprehensive analysis of hydrogen bonds in regulatory protein DNA-complexes: In search of common principles. J. Mol. Biol. 1995, 253, 370–382. [Google Scholar] [CrossRef] [Green Version]
- Pabo, C.O.; Sauer, R.T. Transcription factors: Structural families and principles of DNA recognition. Annu. Revi. Biochem. 1992, 61, 1053–1095. [Google Scholar] [CrossRef]
- Suzuki, M.; Yagi, N. DNA recognition code of transcription factors in the helix-turn-helix, probe helix, hormone receptor, and zinc finger families. Proc. Natl. Acad. Sci. USA 1994, 91, 12357–12361. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Xie, Y.; Schaefer, H.F. Electron attachment to DNA single strands: Gas phase and aqueous solution. Nucleic Acids Res. 2007, 35, 5165–5172. [Google Scholar] [CrossRef] [Green Version]
- Rak, J.; Mazurkiewicz, K.; Kobyłecka, M.; Storoniak, P.; Haranczyk, M.; Dąbkowska, I.; Bachorz, R.A.; Gutowski, M.; Radisic, D.; Stokes, S.T.; et al. Radiation Induced Molecular Phenomena in Nucleic Acids; Springer: Dordrecht, The Netherlands, 2008; pp. 619–667. [Google Scholar]
- Szyperska, A.; Gajewicz, A.; Mazurkiewicz, K.; Leszczynski, J.; Rak, J. Theoretical studies on interactions between low energy electrons and protein–DNA fragments: Valence anions of AT-amino acids side chain complexes. Phys. Chem. Chem. Phys. 2011, 13, 19499–19507. [Google Scholar] [CrossRef]
- Solomun, T.; Skalický, T. The interaction of a protein–DNA surface complex with low-energy electrons. Chem. Phys. Lett. 2008, 453, 101–104. [Google Scholar] [CrossRef]
- Ptasińska, S.; Li, Z.; Mason, N.J.; Sanche, L. Damage to amino acid–nucleotide pairs induced by 1 eV electrons. Phys. Chem. Chem. Phys. 2010, 12, 9367–9372. [Google Scholar]
- von Sonntag, C. Free-Radical-Induced DNA Damage and Its Repair; Springer: Heidelberg, Germany, 2006; pp. 282–284. [Google Scholar]
- Hall, D.B.; Holmlin, R.E.; Barton, J.K. Oxidative DNA damage through long-range electron transfer. Nature 1996, 382, 731. [Google Scholar] [CrossRef] [PubMed]
- Cupellini, L.; Wityk, P.; Mennucci, B.; Rak, J. Photoinduced electron transfer in 5-bromouracil labeled DNA. A contrathermodynamic mechanism revisited by electron transfer theories. J. Phys. Chem. Chem. Phys. 2019, 21, 4387–4393. [Google Scholar] [CrossRef] [PubMed]
- Ellenberger, T.E.; Brandl, C.J.; Struhl, K.; Harrison, S.C. The GCN4 basic region leucine zipper binds DNA as a dimer of uninterrupted α helices: Crystal structure of the protein-DNA complex. Cell 1992, 71, 1223–1237. [Google Scholar] [CrossRef]
- Fountain, K.J.; Gilar, M.; Gebler, J.C. Analysis of native and chemically modified oligonucleotides by tandem ion-pair reversed-phase high-performance liquid chromatography/electrospray ionization mass spectrometry. Rap. Comm. Mass Spec. 2003, 17, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Huber, C.G.; Oberacher, H. Analysis of nucleic acids by on-line liquid chromatography–Mass spectrometry. Mass Spec. Rev. 2001, 20, 310–343. [Google Scholar] [CrossRef] [PubMed]
- Carroll, A.S.; Gilbert, D.E.; Liu, X.; Cheung, J.W.; Michnowicz, J.E.; Wagner, G.; Ellenberger, T.E.; Blackwell, T.K. SKN-1 domain folding and basic region monomer stabilization upon DNA binding. Genes Dev. 1997, 11, 2227–2238. [Google Scholar] [CrossRef] [Green Version]
- Stanojevic, D.; Verdine, G.L. Deconstruction of GCN4/GCRE into a monomeric peptide-DNA complex. Nat. Struc. Biol. 1995, 2, 450–457. [Google Scholar] [CrossRef]
- Oakley, M.G.; Dervan, P.B. Structural motif of the GCN4 DNA binding domain characterized by affinity cleaving. Science 1990, 248, 847–851. [Google Scholar] [CrossRef]
- Ahmad, F.A.A.; Azmi, F.; Skwarczynski, M.; Toth, I. Peptide Conjugation via CuAAC ‘Click’ Chemistry. Molecules 2013, 18, 13148–13174. [Google Scholar] [CrossRef] [Green Version]
- Amblard, F.; Cho, J.H.; Schinazi, R.F. Cu(I)-catalyzed Huisgen azide-alkyne 1,3-dipolar cycloaddition reaction in nucleoside, nucleotide, and oligonucleotide chemistry. Chem. Rev. 2009, 109, 4207–4220. [Google Scholar] [CrossRef] [Green Version]
- Luscombe, N.M.; Austin, S.E.; Berman, H.M.; Thornton, J.M. An overview of the structures of protein-DNA complexes. Gen. Biol. 2000, 1, Reviews001-1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Presolski, S.I.; Hong, V.P.; Finn, M.G. Copper-Catalyzed Azide-Alkyne Click Chemistry for Bioconjugation. Curr. Protoc. Chem. Biol. 2011, 3, 153–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piechowski, M.V.; Thelen, M.A.; Hoigné, J.; Bühler, R.E. tert-Butanol as an OH-Scavenger in the Pulse Radiolysis of Oxygenated Aqueous Systems. Ber. Bunsenges. Physik. Chem. 1992, 96, 1448–1454. [Google Scholar] [CrossRef]
- Đurović, A.D.; Stojanović, Z.S.; Kravić, S.Ž.; Suturović, Z.J.; Brezo, T.Ž.; Grahovac, N.L.; Milanović, S.D. A comparison of different methods to remove dissolved oxygen: Application to the electrochemical determination of imidacloprid. Acta Period. Technol. 2015, 46, 149–155. [Google Scholar] [CrossRef]
- Seldeen, K.L.; McDonald, C.B.; Deegan, B.J.; Farooq, A. Evidence that the bZIP domains of the Jun transcription factor bind to DNA as monomers prior to folding and homodimerization. Arch. Biochem. Biophys. 2008, 473, 48–60. [Google Scholar] [CrossRef]
- Liang, L.; Astruc, D. The copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) “click” reaction and its applications. An overview. Coord. Chem. Rev. 2011, 255, 2933–2945. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds dsDNA**-PEP are available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviation | Composition | Experimental Mass [Da] | Theoretical Mass [Da] |
|---|---|---|---|
| PEP | H-ASP-PRO-ALA-ALA-LEU-LYS-ARG-ALA-ARG-ASN-THR-GLU-ALA-ALA-ARG-ARG-SER-ARG-ALA-ARG-LYS-GLY-GLY-LYS(N3)-NH2 | 2661.5001 | 2661.4320 |
| ssDNA A | 5′-GCA CGT CAT CCG TATAG-3′ | 5170.9401 | 5169.4020 |
| ssDNA A* | 5′-GCA XGT CAT CCG TATAG-3′ (X = C8-Alkyne dC) | 5273.8241 | 5273.4591 |
| ssDNA A** | 5′-GCA XYT CAT CCG TATAG-3′ (X = C8-Alkyne dC, Y = 8-bromo-2′-deoxyguanosine) | 5352.4231 | 5352.1046 |
| ssDNA A*-PEP | 5′-GCA XGT CAT CCG TATAG-3′ (X = PEP conjugated by CLICLK chemistry | 7936.9349 | 7936.4653 |
| ssDNA A**-PEP | 5′-GCA XYT CAT CCG TATAG-3′ (X = PEP conjugated by CLICLK chemistry, Y = 8-bromo-2′-deoxyguanosine) | 8013.9383 | 8013.4521 |
| ssDNA B | 5′-CTA TAC GGA TGA CGT GC-3′ | 5209.9135 | 5210.4530 |
| dsDNA | ssDNA A: 5′-GCA CGT CAT CCG TATAG-3′ and ssDNA B: 5′-CTA TAC GGA TGA CGT GC-3′ | - | - |
| dsDNA* | ssDNA A*: 5′-GCA XGT CAT CCG TATAG-3′ (X = C8-Alkyne dC) and ssDNA B: 5′-CTA TAC GGA TGA CGT GC-3′ | - | - |
| dsDNA*-PEP | ssDNA A*-PEP: 5′-GCA XGT CAT CCG TATAG-3′ (X = PEP conjugated by CLICLK chemistry) and ssDNA B: 5′-CTA TAC GGA TGA CGT GC-3′ | - | - |
| dsDNA*-PEP | ssDNA**-PEP: 5′-GCA XYT CAT CCG TATAG-3′ (X = PEP conjugated by CLICLK chemistry, Y = 8-bromo-2′-deoxyguanosine) and ssDNA B: 5′-CTA TAC GGA TGA CGT GC-3′ | - | - |
| [kcal/mol] | ΔG298 | ΔH298 | ΔS298 |
|---|---|---|---|
| dsDNA | 17 | 101 | 0.28 |
| dsDNA*-PEP | 27 | 186 | 0.53 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wityk, P.; Piątek, R.; Nowak, R.; Kostrzewa-Nowak, D. Generation and Characterization of a DNA-GCN4 Oligonucleotide-Peptide Conjugate: The Impact DNA/Protein Interactions on the Sensitization of DNA. Molecules 2020, 25, 3630. https://doi.org/10.3390/molecules25163630
Wityk P, Piątek R, Nowak R, Kostrzewa-Nowak D. Generation and Characterization of a DNA-GCN4 Oligonucleotide-Peptide Conjugate: The Impact DNA/Protein Interactions on the Sensitization of DNA. Molecules. 2020; 25(16):3630. https://doi.org/10.3390/molecules25163630
Chicago/Turabian StyleWityk, Paweł, Rafał Piątek, Robert Nowak, and Dorota Kostrzewa-Nowak. 2020. "Generation and Characterization of a DNA-GCN4 Oligonucleotide-Peptide Conjugate: The Impact DNA/Protein Interactions on the Sensitization of DNA" Molecules 25, no. 16: 3630. https://doi.org/10.3390/molecules25163630
APA StyleWityk, P., Piątek, R., Nowak, R., & Kostrzewa-Nowak, D. (2020). Generation and Characterization of a DNA-GCN4 Oligonucleotide-Peptide Conjugate: The Impact DNA/Protein Interactions on the Sensitization of DNA. Molecules, 25(16), 3630. https://doi.org/10.3390/molecules25163630