
Design and Synthesis of Novel Hybrid 8-Hydroxy Quinoline-Indole Derivatives as Inhibitors of Aβ Self-Aggregation and Metal Chelation-Induced Aβ Aggregation

 , ,
, ,  , and
, and 
Abstract
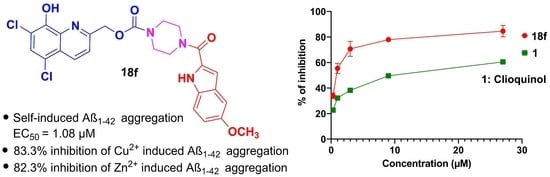
1. Introduction
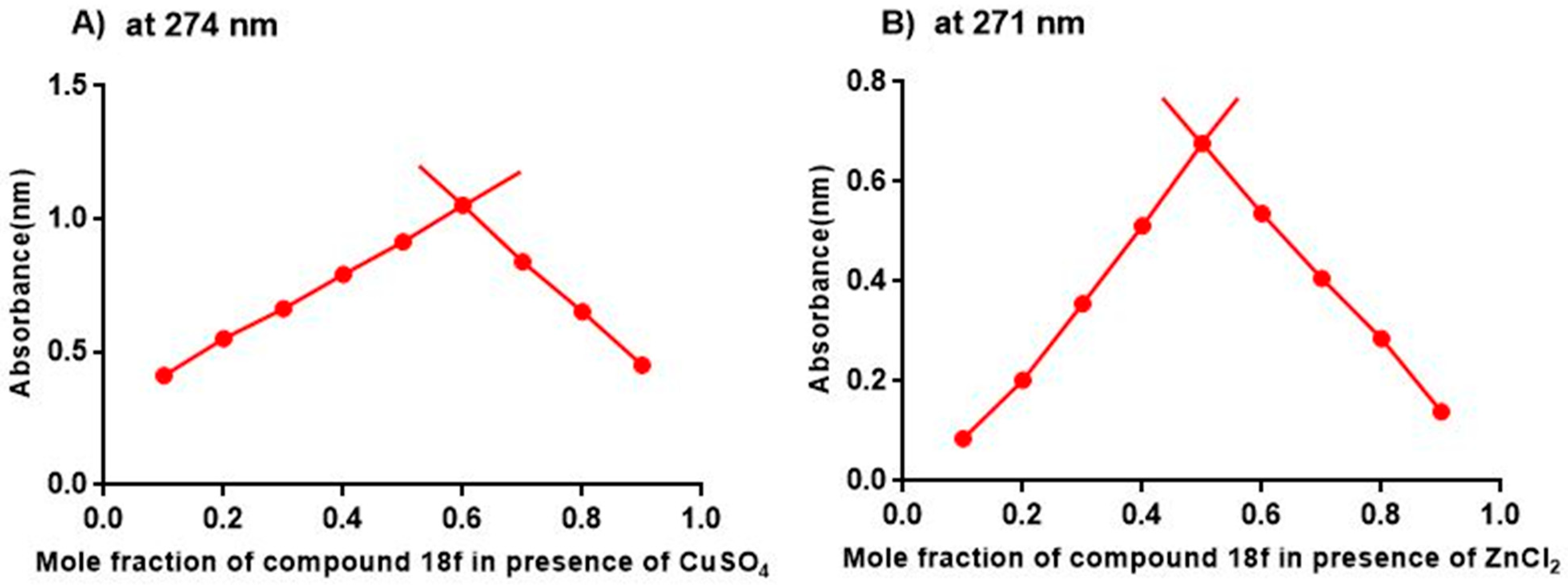
2. Results and Discussion
3. Experimental Section
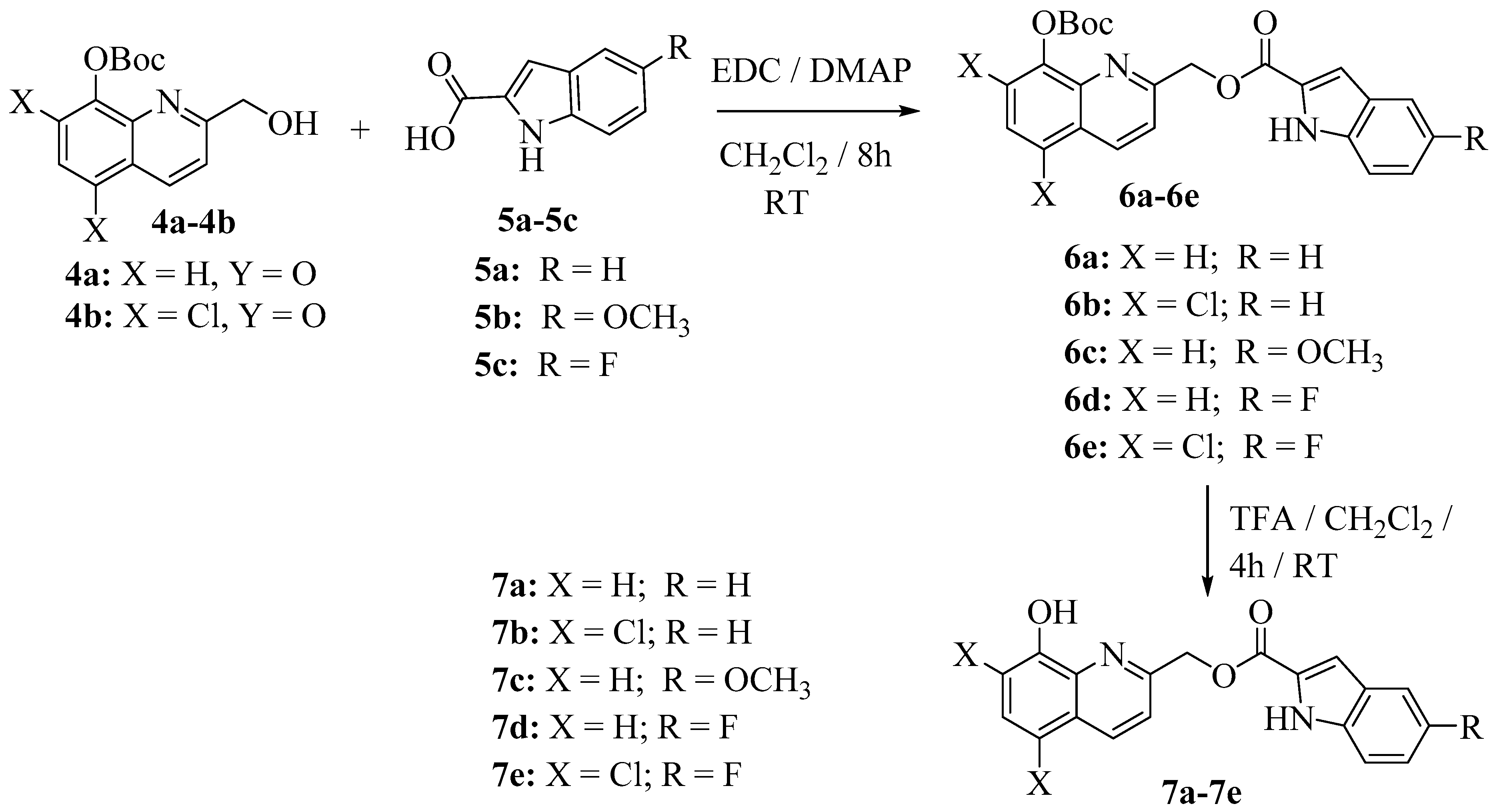
3.1. Chemistry
3.2. Experimental Procedure for the Synthesis of Hybrid 8-Hydroxyquinoline-Indole Analogs
3.3. Methodology for the In Vitro Self-Induced Aβ1–42 Aggregation Assay
3.4. Methodology for the In Vitro Metal-Induced Aβ1–42 Aggregation Assay
3.5. Methodology for the Metal-Chelation Study
3.6. Methodology for In Vitro HEK-Tau and SY5Y-APPSw Cell Aggregation Assays
3.7. Methodology for Molecular Docking
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Förstl, H.; Kurz, A. Clinical features of Alzheimer’s disease. Eur. Arch. Psy. Clin. Neurosci. 1999, 249, 288–290. [Google Scholar] [CrossRef] [PubMed]
- Torre, J.D.; Aliev, G.; Perry, G. Drug Therapy in Alzheimer’s Disease. N. Engl. J. Med. 2004, 351, 1911–1913. [Google Scholar]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef]
- Walsh, D.M.; Selkoe, D.J. Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron 2004, 44, 181–193. [Google Scholar] [CrossRef]
- Munoz-Torrero, D. Acetylcholinesterase inhibitors as disease-modifying therapies for Alzheimer’s disease. Curr. Med. Chem. 2008, 15, 2433–2455. [Google Scholar] [CrossRef]
- Young, A.B. Four decades of neurodegenerative disease research: How far we have come! J. Neurosci. 2009, 29, 12722–12728. [Google Scholar] [CrossRef]
- Spangenberg, E.E.; Green, K.N. Inflammation in Alzheimer’s disease: Lessons learned from microglia-depletion models. Brain Behav. Immun. 2017, 61, 1–11. [Google Scholar] [CrossRef]
- Deora, G.S.; Kantham, S.; Chan, S.; Dighe, S.N.; Veliyath, S.K.; McColl, G.; Parat, M.O.; McGeary, R.P.; Ross, B.P. Multifunctional Analogs of Kynurenic Acid for the Treatment of Alzheimer’s Disease: Synthesis, Pharmacology, and Molecular Modeling Studies. ACS Chem. Neurosci. 2017, 8, 2667–2675. [Google Scholar] [CrossRef] [PubMed]
- Hiremathad, A.; Keri, R.S.; Esteves, A.R.; Cardoso, S.M.; Chaves, S.; Santos, M.A. Novel Tacrine-Hydroxyphenylbenzimidazole hybrids as potential multitarget drug candidates for Alzheimer’s disease. Eur. J. Med. Chem. 2018, 148, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Dwivedi, A.R.; Sarkar, B.; Gupta, S.K.; Krishnamurthy, S.; Mantha, A.K.; Parkash, J.; Kumar, V. 4,6-Diphenylpyrimidine Derivatives as Dual Inhibitors of Monoamine Oxidase and Acetylcholinesterase for the Treatment of Alzheimer’s Disease. ACS Chem. Neurosci. 2019, 10, 252–265. [Google Scholar] [CrossRef]
- Li, X.; Wang, H.; Xu, Y.; Liu, W.; Gong, Q.; Wang, W.; Qiu, X.; Zhu, J.; Mao, F.; Zhang, H.; et al. Novel Vilazodone-Tacrine Hybrids as Potential Multitarget-Directed Ligands for the Treatment of Alzheimer’s Disease Accompanied with Depression: Design, Synthesis, and Biological Evaluation. ACS Chem. Neurosci. 2017, 8, 2708–2721. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, J.; Chiao, P.; Bussiere, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, T.; Shakeri, A.; Rao, P.P. Amyloid cascade in Alzheimer’s disease: Recent advances in medicinal chemistry. Eur. J. Med. Chem. 2016, 113, 258–272. [Google Scholar] [CrossRef] [PubMed]
- Viola, K.L.; Klein, W.L. Amyloid-β oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis. Acta Neuropathol. 2015, 129, 183–206. [Google Scholar] [CrossRef]
- Chang, L.; Cui, W.; Yang, Y.; Xu, S.; Zhou, W.; Fu, H.; Hu, S.; Mak, S.; Hu, J.; Wang, Q.; et al. Protection against β-amyloid-induced synaptic and memory impairments via altering β-amyloid assembly by bis(heptyl)-cognitin. Sci. Rep. 2015, 5, 10256. [Google Scholar] [CrossRef]
- Zhao, L.N.; Long, H.; Mu, Y.; Chew, L.Y. The toxicity of amyloid β oligomers. Int. J. Mol. Sci. 2012, 13, 7303–7327. [Google Scholar] [CrossRef]
- Barnham, K.J.; Bush, A.I. Metals in Alzheimer’s and Parkinson’s diseases. Curr. Opin. Chem. Biol. 2008, 12, 222–228. [Google Scholar] [CrossRef]
- Himes, R.A.; Park, G.Y.; Siluvai, G.S.; Blackburn, N.J.; Karlin, K.D. Structural studies of copper(I) complexes of amyloid-β peptide fragments: Formation of two-coordinate bis(histidine) complexes. Angew. Chem. Int. Ed. Engl. 2008, 47, 9084–9087. [Google Scholar] [CrossRef]
- Hou, L.; Zagorski, M.G. NMR reveals anomalous copper(II) binding to the amyloid Abeta peptide of Alzheimer’s disease. J. Am. Chem. Soc. 2006, 128, 9260–9261. [Google Scholar] [CrossRef]
- Duce, J.A.; Bush, A.I. Biological metals and Alzheimer’s disease: Implications for therapeutics and diagnostics. Prog. Neurobiol. 2010, 92, 1–18. [Google Scholar] [CrossRef]
- Kepp, K.P. Bioinorganic Chemistry of Alzheimer’s Disease. Chem. Rev. 2012, 112, 5193–5239. [Google Scholar] [CrossRef] [PubMed]
- Savelieff, M.G.; DeToma, A.S.; Derrick, J.S.; Lim, M.H. The ongoing search for small molecules to study metal-associated amyloid-β species in Alzheimer’s disease. Acc. Chem. Res. 2014, 47, 2475–2482. [Google Scholar] [CrossRef]
- Kim, A.C.; Lim, S.; Kim, Y.K. Metal Ion Effects on Aβ and Tau Aggregation. Int. J. Mol. Sci. 2018, 19, 128. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, C.W.; Bush, A.I.; Mackinnon, A.; Macfarlane, S.; Mastwyk, M.; MacGregor, L.; Kiers, L.; Cherny, R.; Li, Q.X.; Tammer, A.; et al. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: A pilot phase 2 clinical trial. Arch. Neurol. 2003, 60, 1685–1691. [Google Scholar] [CrossRef] [PubMed]
- Cherny, R.A.; Atwood, C.S.; Xilinas, M.E.; Gray, D.N.; Jones, W.D.; McLean, C.A.; Barnham, K.J.; Volitakis, I.; Fraser, F.W.; Kim, Y.; et al. Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron 2001, 30, 665–676. [Google Scholar] [CrossRef]
- Adlard, P.A.; Cherny, R.A.; Finkelstein, D.I.; Gautier, E.; Robb, E.; Cortes, M.; Volitakis, I.; Liu, X.; Smith, J.P.; Perez, K.; et al. Rapid restoration of cognition in Alzheimer’s transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Abeta. Neuron 2008, 59, 43–55. [Google Scholar] [CrossRef]
- Faux, N.G.; Ritchie, C.W.; Gunn, A.; Rembach, A.; Tsatsanis, A.; Bedo, J.; Harrison, J.; Lannfelt, L.; Blennow, K.; Zetterberg, H.; et al. PBT2 rapidly improves cognition in Alzheimer’s Disease: Additional phase II analyses. J. Alzheimers Dis. 2010, 20, 509–516. [Google Scholar] [CrossRef]
- Fernandez-Bachiller, M.I.; Perez, C.; Gonzalez-Munoz, G.C.; Conde, S.; Lopez, M.G.; Villarroya, M.; Garcia, A.G.; Rodriguez-Franco, M.I. Novel tacrine-8-hydroxyquinoline hybrids as multifunctional agents for the treatment of Alzheimer’s disease, with neuroprotective, cholinergic, antioxidant, and copper-complexing properties. J. Med. Chem. 2010, 53, 4927–4937. [Google Scholar] [CrossRef]
- Zheng, H.; Gal, S.; Weiner, L.M.; Bar-Am, O.; Warshawsky, A.; Fridkin, M.; Youdim, M.B. Novel multifunctional neuroprotective iron chelator-monoamine oxidase inhibitor drugs for neurodegenerative diseases: In vitro studies on antioxidant activity, prevention of lipid peroxide formation and monoamine oxidase inhibition. J. Neurochem. 2005, 95, 68–78. [Google Scholar] [CrossRef]
- Zheng, H.; Youdim, M.B.; Fridkin, M. Site-activated multifunctional chelator with acetylcholinesterase and neuroprotective-neurorestorative moieties for Alzheimer’s therapy. J. Med. Chem. 2009, 52, 4095–4098. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, Y.; Wang, B.; Li, W.; Huang, L.; Li, X. Design, Synthesis, and Evaluation of Orally Available Clioquinol-Moracin M Hybrids as Multitarget-Directed Ligands for Cognitive Improvement in a Rat Model of Neurodegeneration in Alzheimer’s Disease. J. Med. Chem. 2015, 58, 8616–8637. [Google Scholar] [CrossRef] [PubMed]
- López-Iglesias, B.; Pérez, C.; Morales-García, J.A.; Alonso-Gil, S.; Pérez-Castillo, A.; Romero, A.; López, M.G.; Villarroya, M.; Conde, S.; Rodríguez-Franco, M.I. New Melatonin–N,N-Dibenzyl(N-methyl)amine Hybrids: Potent Neurogenic Agents with Antioxidant, Cholinergic, and Neuroprotective Properties as Innovative Drugs for Alzheimer’s Disease. J. Med. Chem. 2014, 57, 3773–3785. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Rodriguez, G.; Klempin, F.; Babu, H.; Benitez-King, G.; Kempermann, G. Melatonin modulates cell survival of new neurons in the hippocampus of adult mice. Neuropsychopharmacology 2009, 34, 2180–2191. [Google Scholar] [CrossRef]
- Yang, X.; Cai, P.; Liu, Q.; Wu, J.; Yin, Y.; Wang, X.; Kong, L. Novel 8-hydroxyquinoline derivatives targeting β-amyloid aggregation, metal chelation and oxidative stress against Alzheimer’s disease. Bioorg. Med. Chem. 2018, 26, 3191–3201. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.H.; Southon, A.G.; Fraser, B.H.; Krause-Heuer, A.M.; Zhang, B.; Shoup, T.M.; Lewis, R.; Volitakis, I.; Han, Y.; Greguric, I.; et al. Novel Fluorinated 8-Hydroxyquinoline Based Metal Ionophores for Exploring the Metal Hypothesis of Alzheimer’s Disease. ACS Med. Chem. Lett. 2015, 6, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Gil, V.M.S.; Oliveira, N.C. On the use of the method of continuous variations. J. Chem. Educ. 1990, 67, 473. [Google Scholar] [CrossRef]
- Sensi, S.L.; Paoletti, P.; Koh, J.Y.; Aizenman, E.; Bush, A.I.; Hershfinkel, M. The neurophysiology and pathology of brain zinc. J. Neurosci. 2011, 31, 16076–16085. [Google Scholar] [CrossRef]
- Crouch, P.J.; Savva, M.S.; Hung, L.W.; Donnelly, P.S.; Mot, A.I.; Parker, S.J.; Greenough, M.A.; Volitakis, I.; Adlard, P.A.; Cherny, R.A.; et al. The Alzheimer’s therapeutic PBT2 promotes amyloid-β degradation and GSK3 phosphorylation via a metal chaperone activity. J. Neurochem. 2011, 119, 220–230. [Google Scholar] [CrossRef]
- Biancalana, M.; Koide, S. Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochim. Biophys. Acta. 2010, 1804, 1405–1412. [Google Scholar] [CrossRef]
- Crescenzi, O.; Tomaselli, S.; Guerrini, R.; Salvadori, S.; D’Ursi, A.M.; Temussi, P.A.; Picone, D. Solution structure of the Alzheimer amyloid β-peptide (1–42) in an apolar microenvironment. Similarity with a virus fusion domain. Eur. J. Biochem. 2002, 269, 5642–5648. [Google Scholar] [CrossRef]
- Du, H.; Liu, X.; Xie, J.; Ma, F. Novel Deoxyvasicinone–Donepezil Hybrids as Potential Multitarget Drug Candidates for Alzheimer’s Disease. ACS Chem. Neurosci. 2019, 10, 2397–2407. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Guo, Y.; Yan, J.; Luo, Z.; Luo, H.-B.; Yan, M.; Huang, L.; Li, X. Design, Synthesis, and Evaluation of Multitarget-Directed Resveratrol Derivatives for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2013, 56, 5843–5859. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | 1 | 7a | 7b | 7c | 7d | 7e | 12a | 12b |
|---|---|---|---|---|---|---|---|---|
| Aβ1–42 | 9.95 | 4.26 | 9.28 | 3.22 | 6.34 | 9.52 | >10 | >10 |
| Compd. | 18a | 18b | 18c | 18d | 18e | 18f | 18g | 18h |
| Aβ1–42 | >10 | >10 | 1.72 | 1.48 | 2.49 | 1.08 | >10 | 2.58 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bowroju, S.K.; Mainali, N.; Ayyadevara, S.; Penthala, N.R.; Krishnamachari, S.; Kakraba, S.; Reis, R.J.S.; Crooks, P.A. Design and Synthesis of Novel Hybrid 8-Hydroxy Quinoline-Indole Derivatives as Inhibitors of Aβ Self-Aggregation and Metal Chelation-Induced Aβ Aggregation. Molecules 2020, 25, 3610. https://doi.org/10.3390/molecules25163610
Bowroju SK, Mainali N, Ayyadevara S, Penthala NR, Krishnamachari S, Kakraba S, Reis RJS, Crooks PA. Design and Synthesis of Novel Hybrid 8-Hydroxy Quinoline-Indole Derivatives as Inhibitors of Aβ Self-Aggregation and Metal Chelation-Induced Aβ Aggregation. Molecules. 2020; 25(16):3610. https://doi.org/10.3390/molecules25163610
Chicago/Turabian StyleBowroju, Suresh K., Nirjal Mainali, Srinivas Ayyadevara, Narsimha R. Penthala, Sesha Krishnamachari, Samuel Kakraba, Robert J. Shmookler Reis, and Peter A. Crooks. 2020. "Design and Synthesis of Novel Hybrid 8-Hydroxy Quinoline-Indole Derivatives as Inhibitors of Aβ Self-Aggregation and Metal Chelation-Induced Aβ Aggregation" Molecules 25, no. 16: 3610. https://doi.org/10.3390/molecules25163610
APA StyleBowroju, S. K., Mainali, N., Ayyadevara, S., Penthala, N. R., Krishnamachari, S., Kakraba, S., Reis, R. J. S., & Crooks, P. A. (2020). Design and Synthesis of Novel Hybrid 8-Hydroxy Quinoline-Indole Derivatives as Inhibitors of Aβ Self-Aggregation and Metal Chelation-Induced Aβ Aggregation. Molecules, 25(16), 3610. https://doi.org/10.3390/molecules25163610