
Chemometric Models of Differential Amino Acids at the Navα and Navβ Interface of Mammalian Sodium Channel Isoforms


Abstract
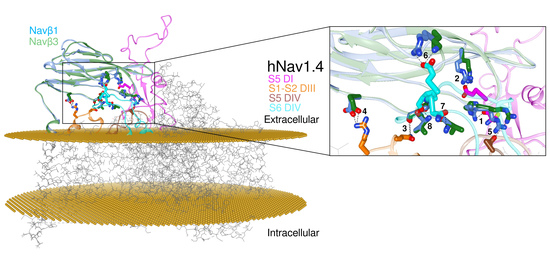
1. Introduction
1.1. The Sodium Channels
1.2. The Navα Subunit
1.3. The Navβ Subunits
1.4. The Present Contributions
2. Results
2.1. Determination of PPI in the Isoforms of the Navs
2.1.1. PPI Analysis in the Structural Complex eeNav1.4α/eeNavβ1
2.1.2. PPI Analysis of the hNav1.4α/hNavβ1 and hNav1.4α/hNavβ3 Models
2.1.3. Identification of the Interacting Residues
2.1.4. Structure Alignment of all Navβs Subunits
2.2. Pore Modulation by Navβ Concerning the Acceleration for Fast Gating Inactivation
2.3. Determination of Relevant ECL Properties
2.4. PPI Patterns on Navs Isoforms
3. Discussion
3.1. Hypothetical Acceleration of Fast Inactivation of Gating of the Navs
3.2. Properties of the ECL Residues for Navα Subunits
3.3. Volume and Surface Properties of the ECL on Navα Subunits
3.4. Interface Properties Between Navα and Navβ1 or Navβ3
4. Materials and Methods
4.1. In Silico Homology Modeling
4.2. Identification of Interacting Residues at the Interface Between Navα and Navβ
4.3. The Input Structures as Templates for the PPI Models
4.4. Determination of the Extracellular Regions of all Navα Subunits
4.5. Calculation of the Properties for all ECLs
4.6. Calculation of the Chemical Surface Properties for All IF-ECLs
4.7. Electrostatic Interactions of the IF-ECLs Surfaces (MEPS)
4.8. Calculation of the ECL Surface Properties at the Interface with Navβ1 and Navβ3 Subunits
4.9. External Model Validation of the PPI Models
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Catterall, W.A.; Goldin, A.L.; Waxman, S.G. International Union of Pharmacology. XLVII. Nomenclature and Structure-Function Relationships of Voltage-Gated Sodium Channels. Pharmacol. Rev. 2005, 57, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Fux, J.E.; Mehta, A.; Moffat, J.; Spafford, J.D. Eukaryotic Voltage-Gated Sodium Channels: On Their Origins, Asymmetries, Losses, Diversification and Adaptations. Front. Physiol. 2018, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Waxman, S.G. Sodium channels, the electrogenisome and the electrogenistat: Lessons and questions from the clinic. J. Physiol. 2012, 590, 2601–2612. [Google Scholar] [CrossRef] [PubMed]
- Goldin, A.L.; Barchi, R.L.; Caldwell, J.H.; Hofmann, F.; Howe, J.R.; Hunter, J.C.; Kallen, R.G.; Mandel, G.; Meisler, M.H.; Netter, Y.B.; et al. Nomenclature of voltage-gated sodium channels. Neuron 2000, 28, 365–368. [Google Scholar] [CrossRef]
- O’Malley, H.A.; Isom, L.L. Sodium channel β subunits: Emerging targets in channelopathies. Annu. Rev. Physiol. 2015, 77, 481–504. [Google Scholar] [CrossRef]
- Catterall, W.A. From Ionic Currents to Molecular Mechanisms. Neuron 2000, 26, 13–25. [Google Scholar] [CrossRef]
- Ogata, N.; Ohishi, Y. Molecular diversity of structure and function of the voltage-gated Na+ channels. Jpn. J. Pharmacol. 2002, 88, 365–377. [Google Scholar] [CrossRef]
- Woolf, C.J.; Mannion, R.J. Neuropathic pain: Aetiology, symptoms, mechanisms, and management. Lancet 1999, 353, 1959–1964. [Google Scholar] [CrossRef]
- Catterall, W.A. Sodium Channels, Inherited Epilepsy, and Antiepileptic Drugs. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 317–338. [Google Scholar] [CrossRef]
- Claes, L.R.; Deprez, L.; Suls, A.; Baets, J.; Smets, K.; Van Dyck, T.; Deconinck, T.; Jordanova, A.; De Jonghe, P. The SCN1A variant database: A novel research and diagnostic tool. Hum. Mutat. 2009, 30, E904–E920. [Google Scholar] [CrossRef]
- Huang, W.; Liu, M.; Yan, S.; Yan, N. Structure-based assessment of disease-related mutations in human voltage-gated sodium channels. Protein Cell 2017, 8, 401–438. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, M.; Patowary, A.; Stanaway, I.B.; Mccord, E.; Nesbitt, R.R.; Archer, M.; Scheuer, T.; Nickerson, D.; Raskind, W.H.; Wijsman, E.M.; et al. Association of rare missense variants in the second intracellular loop of NaV1.7 sodium channels with familial autism. Mol. Psychiatry 2016, 23, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A.; Cestele, S.; Yarov-Yarovoy, V.; Yu, F.H.; Konoki, K.; Scheuer, T. Voltage-gated ion channels and gating modifier toxins. Toxicon 2007, 49, 124–141. [Google Scholar] [CrossRef] [PubMed]
- Bagal, S.K.; E Marron, B.; Owen, R.M.; Storer, R.I.; A Swain, N. Voltage gated sodium channels as drug discovery targets. Channels 2015, 9, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, M.D.L.; Kraus, R.L. Voltage-Gated Sodium Channels: Structure, Function, Pharmacology, and Clinical Indications. J. Med. Chem. 2015, 58, 7093–7118. [Google Scholar] [CrossRef] [PubMed]
- Stevens, M.; Peigneur, S.; Tytgat, J. Neurotoxins and Their Binding Areas on Voltage-Gated Sodium Channels. Front. Pharmacol. 2011, 2, 71. [Google Scholar] [CrossRef] [PubMed]
- Mantegazza, M.; Catterall, W.A. Voltage-Gated Na+ Channels; Oxford University Press (OUP): Oxford, UK, 2012; Volume 80, pp. 41–54. [Google Scholar]
- Ahern, C.A.; Payandeh, J.; Bosmans, F.; Chanda, B. The hitchhiker’s guide to the voltage-gated sodium channel galaxy. J. Gen. Physiol. 2015, 147, 1–24. [Google Scholar] [CrossRef]
- Payandeh, J.; Scheuer, T.; Zheng, N.; Catterall, W.A. The crystal structure of a voltage-gated sodium channel. Nature 2011, 475, 353–358. [Google Scholar] [CrossRef]
- Long, S.B.; Tao, X.; Campbell, E.B.; MacKinnon, R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 2007, 450, 376–382. [Google Scholar] [CrossRef]
- Long, S.B. Crystal Structure of a Mammalian Voltage-Dependent Shaker Family K+ Channel. Science 2005, 309, 897–903. [Google Scholar] [CrossRef]
- Yu, F.H.; Yarov-Yarovoy, V.; Gutman, G.A.; Catterall, W.A. Overview of Molecular Relationships in the Voltage-Gated Ion Channel Superfamily. Pharmacol. Rev. 2005, 57, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Chanda, B.; Bezanilla, F. Tracking Voltage-dependent Conformational Changes in Skeletal Muscle Sodium Channel during Activation. J. Gen. Physiol. 2002, 120, 629–645. [Google Scholar] [CrossRef] [PubMed]
- Bosmans, F.; Martin-Eauclaire, M.-F.; Swartz, K.J. Deconstructing voltage sensor function and pharmacology in sodium channels. Nature 2008, 456, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Capes, D.L.; Goldschen-Ohm, M.P.; Arcisio-Miranda, M.; Bezanilla, F.; Chanda, B. Domain IV voltage-sensor movement is both sufficient and rate limiting for fast inactivation in sodium channels. J. Gen. Physiol. 2013, 142, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A. Voltage-gated sodium channels at 60: Structure, function and pathophysiology. J. Physiol. 2012, 590, 2577–2589. [Google Scholar] [CrossRef]
- Vargas, E.; Yarov-Yarovoy, V.; Khalili-Araghi, F.; Catterall, W.A.; Klein, M.L.; Tarek, M.; Lindahl, E.; Schulten, K.; Perozo, E.; Bezanilla, F.; et al. An emerging consensus on voltage-dependent gating from computational modeling and molecular dynamics simulations. J. Gen. Physiol. 2012, 140, 587–594. [Google Scholar] [CrossRef]
- Brackenbury, W.J.; Isom, L.L. Na+ Channel? Subunits: Overachievers of the Ion Channel Family. Front. Pharmacol. 2011, 2, 53. [Google Scholar] [CrossRef]
- Cusdin, F.S.; Clare, J.J.; Jackson, A.P. Trafficking and Cellular Distribution of Voltage-Gated Sodium Channels. Traffic 2008, 9, 17–26. [Google Scholar] [CrossRef]
- Patino, G.A.; Isom, L.L. Electrophysiology and beyond: Multiple roles of Na+ channel β subunits in development and disease. Neurosci. Lett. 2010, 486, 53–59. [Google Scholar] [CrossRef]
- Calhoun, J.D.; Isom, L.L. The Role of Non-pore-Forming β Subunits in Physiology and Pathophysiology of Voltage-Gated Sodium Channels. Handb. Exp. Pharmacol. 2014, 221, 51–89. [Google Scholar] [CrossRef]
- Catterall, W.A. Structure and function of voltage-gated ion channels. Annu. Rev. Biochem. 1995, 64, 493–531. [Google Scholar] [CrossRef] [PubMed]
- Isom, L.L.; De Jongh, K.S.; Patton, D.E.; Reber, B.F.; Offord, J.; Charbonneau, H.; Walsh, K.; Goldin, A.L.; Catterall, W.A. Primary structure and functional expression of the β1 subunit of the rat brain sodium channel. Science 1992, 256, 839–842. [Google Scholar] [CrossRef] [PubMed]
- Isom, L.L.; Scheuer, T.; Brownstein, A.B.; Ragsdale, D.S.; Murphy, B.J.; Catterall, W.A. Functional Co-expression of the 1 and Type IIA Subunits of Sodium Channels in a Mammalian Cell Line. J. Boil. Chem. 1995, 270, 3306–3312. [Google Scholar] [CrossRef] [PubMed]
- McCormick, K.A.; Isom, L.L.; Ragsdale, D.; Smith, D.; Scheuer, T.; Catterall, W.A. Molecular Determinants of Na+Channel Function in the Extracellular Domain of the β1 Subunit. J. Boil. Chem. 1998, 273, 3954–3962. [Google Scholar] [CrossRef] [PubMed]
- Makita, N.; Bennett, P.B.; George, A.L., Jr. Molecular determinants of β1 subunit-induced gating modulation in voltage-dependent Na+ channels. J Neurosci. 1996, 22, 7117–7127. [Google Scholar] [CrossRef]
- E Patton, D.; Isom, L.L.; A Catterall, W.; Goldin, A.L. The adult rat brain beta 1 subunit modifies activation and inactivation gating of multiple sodium channel alpha subunits. J. Boil. Chem. 1994, 269, 17649–17655. [Google Scholar]
- Yu, E.J.; Ko, S.-H.; Lenkowski, P.W.; Pance, A.; Patel, M.K.; Jackson, A.P. Distinct domains of the sodium channel β3-subunit modulate channel-gating kinetics and subcellular location. Biochem. J. 2005, 392, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Cannon, S.C. Modulation of Na+channel inactivation by the ?1 subunit: A deletion analysis. Pflüg. Arch. Eur. J. Physiol. 1995, 431, 186–195. [Google Scholar] [CrossRef]
- Islas, A.A.; Sánchez-Solano, A.; Scior, T.; Millan-Perezpeña, L.; Salinas-Stefanon, E.M. Identification of Navβ1 Residues Involved in the Modulation of the Sodium Channel Nav1.4. PLoS ONE 2013, 8, e81995. [Google Scholar] [CrossRef]
- Scior, T.; Paiz-Candia, B.; Islas, Á.A.; Sánchez-Solano, A.; Peña,, L.M.-P.; Mancilla-Simbro, C.; Salinas-Stefanon, E.M. Predicting a double mutant in the twilight zone of low homology modeling for the skeletal muscle voltage-gated sodium channel subunit beta-1 (Na v 1.4 β1). Comput. Struct. Biotechnol. J. 2015, 13, 229–240. [Google Scholar] [CrossRef]
- Sánchez-Solano, A.; Islas, A.A.; Scior, T.; Paiz-Candia, B.; Millan-Perezpeña, L.; Salinas-Stefanon, E.M. Characterization of specific allosteric effects of the Na+ channel β1 subunit on the Nav1.4 isoform. Eur. Biophys. J. 2016, 46, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Chopra, S.S.; Watanabe, H.; Zhong, T.P.; Roden, D.M. Molecular cloning and analysis of zebrafish voltage-gated sodium channel beta subunit genes: Implications for the evolution of electrical signaling in vertebrates. BMC Evol. Boil. 2007, 7, 113. [Google Scholar] [CrossRef] [PubMed]
- Isom, L.L. Sodium channel beta subunits: Anything but auxiliary. Neuroscientist 2001, 7, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Buffington, S.A.; Rasband, M.N. Na+ channel-dependent recruitment of Navβ4 to axon initial segments and nodes of Ranvier. J. Neurosci. 2013, 33, 6191–6202. [Google Scholar] [CrossRef]
- Chen, C.; Calhoun, J.D.; Zhang, Y.; Lopez-Santiago, L.; Zhou, N.; Davis, T.H.; Salzer, J.L.; Isom, L.L. Identification of the Cysteine Residue Responsible for Disulfide Linkage of Na+Channel α and β2 Subunits. J. Boil. Chem. 2012, 287, 39061–39069. [Google Scholar] [CrossRef]
- Shen, H.; Zhou, Q.; Pan, X.; Li, Z.; Wu, J.; Yan, N. Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. Science 2017, 355, eaal4326. [Google Scholar] [CrossRef]
- Yan, Z.; Zhou, Q.; Wang, L.; Wu, J.; Zhao, Y.; Huang, G.; Peng, W.; Shen, H.; Lei, J.; Yan, N. Structure of the Na v 1.4-β1 Complex from Electric Eel. Cell 2017, 170, 470–482.e11. [Google Scholar] [CrossRef]
- Pan, X.; Li, Z.; Zhou, Q.; Shen, H.; Wu, K.; Huang, X.; Chen, J.; Zhang, J.; Zhu, X.; Lei, J.; et al. Structure of the human voltage-gated sodium channel Nav1.4 in complex with β1. Science 2018, 362, eaau2486. [Google Scholar] [CrossRef]
- Pan, X.; Li, Z.; Huang, X.; Huang, G.; Gao, S.; Shen, H.; Liu, L.; Lei, J.; Yan, N. Molecular basis for pore blockade of human Na+ channel Nav1.2 by the μ-conotoxin KIIIA. Science 2019, 363, 1309–1313. [Google Scholar] [CrossRef]
- Shen, H.; Liu, D.; Wu, K.; Lei, J.; Yan, N. Structures of human Nav1.7 channel in complex with auxiliary subunits and animal toxins. Science 2019, 363, 1303–1308. [Google Scholar] [CrossRef]
- Jiang, D.; Shi, H.; Tonggu, L.; El-Din, T.M.G.; Lenaeus, M.J.; Zhao, Y.; Yoshioka, C.; Zheng, N.; Catterall, W.A. Structure of the Cardiac Sodium Channel. Cell 2020, 180, 122–134.e10. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.H.; Westenbroek, R.E.; Silos-Santiago, I.; McCormick, K.A.; Lawson, D.; Ge, P.; Ferriera, H.; Lilly, J.; Distefano, P.S.; Catterall, W.A.; et al. Sodium Channel β4, a New Disulfide-Linked Auxiliary Subunit with Similarity to β2. J. Neurosci. 2003, 23, 7577–7585. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, J.M.; Das, S.; Van Petegem, F.; Bosmans, F. Crystallographic insights into sodium-channel modulation by the 4 subunit. Proc. Natl. Acad. Sci. USA 2013, 110, E5016–E5024. [Google Scholar] [CrossRef] [PubMed]
- Namadurai, S.; Balasuriya, D.; Rajappa, R.; Wiemhöfer, M.; Stott, K.; Klingauf, J.; Edwardson, J.M.; Chirgadze, D.Y.; Jackson, A.P. Crystal structure and molecular imaging of the Nav channel β3 subunit indicates a trimeric assembly. J. Boil. Chem. 2014, 289, 10797–10811. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Gilchrist, J.; Bosmans, F.; Van Petegem, F. Binary architecture of the Nav1.2- β2 signaling complex. Elife 2016, 5, e10960. [Google Scholar] [CrossRef]
- McCormick, K.A.; Srinivasan, J.; White, K.; Scheuer, T.; Catterall, W.A. The Extracellular Domain of the β1 Subunit Is Both Necessary and Sufficient for β1-like Modulation of Sodium Channel Gating. J. Boil. Chem. 1999, 274, 32638–32646. [Google Scholar] [CrossRef]
- Yereddi, N.R.; Cusdin, F.S.; Namadurai, S.; Packman, L.C.; Monie, T.P.; Slavny, P.; Clare, J.J.; Powell, A.J.; Jackson, A.P. The immunoglobulin domain of the sodium channel β3 subunit contains a surface-localized disulfide bond that is required for homophilic binding. FASEB J. 2012, 27, 568–580. [Google Scholar] [CrossRef]
- Shapovalov, M.V.; Dunbrack, R.L. A Smoothed Backbone-Dependent Rotamer Library for Proteins Derived from Adaptive Kernel Density Estimates and Regressions. Struct. 2011, 19, 844–858. [Google Scholar] [CrossRef]
- Scouras, A.D.; Daggett, V. The dynameomics rotamer library: Amino acid side chain conformations and dynamics from comprehensive molecular dynamics simulations in water. Protein Sci. 2011, 20, 341–352. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Available online. Met. Powder Rep. 2008, 63, 11. [CrossRef]
- Dolinsky, T.J.; Czodrowski, P.; Li, H.; Nielsen, J.E.; Jensen, J.H.; Klebe, G.; Baker, N. PDB2PQR: Expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007, 35, W522–W525. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2017, 27, 14–25. [Google Scholar] [CrossRef]
- Microsoft Corporation. Microsoft Excel. Available online: https://office.microsoft.com/excel (accessed on 20 May 2016).
- Hermann, J.C.; Marti-Arbona, R.; Fedorov, A.A.; Fedorov, E.; Almo, S.C.; Shoichet, B.K.; Raushel, F.M. Structure-based activity prediction for an enzyme of unknown function. Nature 2007, 448, 775–779. [Google Scholar] [CrossRef] [PubMed]
- Zahiri, J.; Bozorgmehr, J.H.; Masoudi-Nejad, A. Computational Prediction of Protein–Protein Interaction Networks: Algo-rithms and Resources. Curr. Genom. 2013, 14, 397–414. [Google Scholar] [CrossRef]
- Ezkurdia, I.; Bartoli, L.; Fariselli, P.; Casadio, R.; Valencia, A.; Tress, M.L. Progress and challenges in predicting protein-protein interaction sites. Briefings Bioinform. 2008, 10, 233–246. [Google Scholar] [CrossRef]
- Whisstock, J.; Lesk, A. Prediction of protein function from protein sequence and structure. Q. Rev. Biophys. 2003, 36, 307–340. [Google Scholar] [CrossRef]
- Izidoro, S.; De Melo-Minardi, R.C.; Pappa, G.L. GASS: Identifying enzyme active sites with genetic algorithms. Bioinformatics 2014, 31, 864–870. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Kalyanaraman, C.; Zhao, S.; Tian, B. Leveraging structure for enzyme function prediction: Methods, opportunities, and challenges. Trends Biochem. Sci. 2014, 39, 363–371. [Google Scholar] [CrossRef]
- Ma, B.; Elkayam, T.; Wolfson, H.; Nussinov, R. Protein-protein interactions: Structurally conserved residues distinguish between binding sites and exposed protein surfaces. Proc. Natl. Acad. Sci. USA 2003, 100, 5772–5777. [Google Scholar] [CrossRef] [PubMed]
- Keskin, O.; Ma, B.; Nussinov, R. Hot Regions in Protein–Protein Interactions: The Organization and Contribution of Structurally Conserved Hot Spot Residues. J. Mol. Boil. 2005, 345, 1281–1294. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Keskin, O.; Ma, B.; Nussinov, R.; Liang, J. Protein–Protein Interactions: Hot Spots and Structurally Conserved Residues often Locate in Complemented Pockets that Pre-organized in the Unbound States: Implications for Docking. J. Mol. Boil. 2004, 344, 781–795. [Google Scholar] [CrossRef] [PubMed]
- Kruger, L.C.; Isom, L.L. Voltage-Gated Na+Channels: Not Just for Conduction. Cold Spring Harb. Perspect. Boil. 2016, 8, a029264. [Google Scholar] [CrossRef] [PubMed]
- Salvage, C.S.; Huang, C.L.H.; Jackson, A.P. Cell-Adhesion Properties of β-Subunits in the Regulation of Cardiomyocyte Sodium Channels. Biomolecules 2020, 10, 989. [Google Scholar] [CrossRef]
- Isom, L.L.; Ragsdale, D.S.; De Jongh, K.S.; E Westenbroek, R.; Reber, B.F.; Scheuer, T.; A Catterall, W. Structure and function of the beta 2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell 1995, 83, 90121–90123. [Google Scholar] [CrossRef]
- Molinarolo, S.; Granata, D.; Carnevale, V.; Ahern, C.A. Mining Protein Evolution for Insights into Mechanisms of Voltage-Dependent Sodium Channel Auxiliary Subunits. Handb. Exp. Pharmacol. 2018, 246, 33–49. [Google Scholar] [CrossRef]
- Zhu, W.; Voelker, T.L.; Varga, Z.; Schubert, A.R.; Nerbonne, J.M.; Silva, J. Mechanisms of noncovalent β subunit regulation of NaV channel gating. J. Gen. Physiol. 2017, 149, 813–831. [Google Scholar] [CrossRef]
- Messner, D.J.; A Catterall, W. The sodium channel from rat brain. Separation and characterization of subunits. J. Boil. Chem. 1985, 260, 10597–10604. [Google Scholar]
- Morgan, K.; Stevens, E.B.; Shah, B.; Cox, P.J.; Dixon, A.K.; Lee, K.; Pinnock, R.D.; Hughes, J.; Richardson, P.J.; Mizuguchi, K.; et al. beta 3: An additional auxiliary subunit of the voltage-sensitive sodium channel that modulates channel gating with distinct kinetics. Proc. Natl. Acad. Sci. USA 2000, 97, 2308–2313. [Google Scholar] [CrossRef]
- Glass, W.G.; Duncan, A.L.; Biggin, P.C. Computational Investigation of Voltage-Gated Sodium Channel β3 Subunit Dynamics. Front. Mol. Biosci. 2020, 7, 40. [Google Scholar] [CrossRef] [PubMed]
- Namadurai, S.; Yereddi, N.R.; Cusdin, F.S.; Huang, C.L.-H.; Chirgadze, D.Y.; Jackson, A.P. A new look at sodium channel β subunits. Open Boil. 2015, 5, 140192. [Google Scholar] [CrossRef] [PubMed]
- Lenkowski, P.W.; Shah, B.S.; E Dinn, A.; Lee, K.; Patel, M.K. Lidocaine block of neonatal Nav1.3 is differentially modulated by co-expression of β1 and β3 subunits. Eur. J. Pharmacol. 2003, 467, 23–30. [Google Scholar] [CrossRef]
- Ragsdale, D.; McPhee, J.; Scheuer, T.; Catterall, W. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science 1994, 265, 1724–1728. [Google Scholar] [CrossRef] [PubMed]
- Ferrera, L.; Moran, O. β1-subunit modulates the Nav1.4 sodium channel by changing the surface charge. Exp. Brain Res. 2006, 172, 139–150. [Google Scholar] [CrossRef]
- Kinch, L.N.; Grishin, N.V. Evolution of protein structures and functions. Curr. Opin. Struct. Biol. 2002, 12, 400–408. [Google Scholar] [CrossRef]
- Starr, T.; Thornton, J.W. Epistasis in protein evolution. Protein Sci. 2016, 25, 1204–1218. [Google Scholar] [CrossRef]
- Kauvar, L.M.; O Villar, H. Deciphering cryptic similarities in protein binding sites. Curr. Opin. Biotechnol. 1998, 9, 390–394. [Google Scholar] [CrossRef]
- Russell, R.B.; Sasieni, P.D.; Sternberg, M.J. Supersites within superfolds. Binding site similarity in the absence of homology 1 1Edited by J. Thornton. J. Mol. Boil. 1998, 282, 903–918. [Google Scholar] [CrossRef]
- Spitzer, R.; Cleves, A.E.; Varela, R.; Jain, A.N. Protein function annotation by local binding site surface similarity. Proteins Struct. Funct. Bioinform. 2013, 82, 679–694. [Google Scholar] [CrossRef]
- Richards, F.M. Areas, volumes, packing and protein structure. Annu. Rev. Biophys. Bioeng. 1977, 6, 151–176. [Google Scholar] [CrossRef]
- Gainza, P.; Sverrisson, F.; Monti, F.; Rodolà, E.; Boscaini, D.; Bronstein, M.M.; Correia, B.E. Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning. Nat. Methods 2019, 17, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Illergård, K.; Ardell, D.H.; Elofsson, A. Structure is three to ten times more conserved than sequence-A study of structural response in protein cores. Proteins: Struct. Funct. Bioinform. 2009, 77, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Sousounis, K.; E Haney, C.; Cao, J.; Sunchu, B.; Tsonis, P.A. Conservation of the three-dimensional structure in non-homologous or unrelated proteins. Hum. Genom. 2012, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Gibrat, J.-F.; Madej, T.; Bryant, S.H. Surprising similarities in structure comparison. Curr. Opin. Struct. Boil. 1996, 6, 377–385. [Google Scholar] [CrossRef]
- Caffrey, D.R.; Somaroo, S.; Hughes, J.D.; Mintseris, J.; Huang, E.S. Are protein–protein interfaces more conserved in sequence than the rest of the protein surface? Protein Sci. 2004, 13, 190–202. [Google Scholar] [CrossRef]
- Grishin, N.V.; Phillips, M.A. The subunit interfaces of oligomeric enzymes are conserved to a similar extent to the overall protein sequences. Protein Sci. 1994, 3, 2455–2458. [Google Scholar] [CrossRef] [PubMed]
- Binkowski, T.A.; Joachimiak, A. Protein Functional Surfaces: Global Shape Matching and Local Spatial Alignments of Ligand Binding Sites. BMC Struct. Boil. 2008, 8, 45. [Google Scholar] [CrossRef]
- A Marshall, S.; Morgan, C.S.; Mayo, S.L. Electrostatics significantly affect the stability of designed homeodomain variants. J. Mol. Boil. 2002, 316, 189–199. [Google Scholar] [CrossRef]
- Chothia, C.; Janin, J. Principles of protein–protein recognition. Nature 1975, 256, 705–708. [Google Scholar] [CrossRef]
- Sheinerman, F.B.; Norel, R.; Honig, B. Electrostatic aspects of protein–protein interactions. Curr. Opin. Struct. Boil. 2000, 10, 153–159. [Google Scholar] [CrossRef]
- Heifetz, A.; Katchalski-Katzir, E.; Eisenstein, M. Electrostatics in protein–protein docking. Protein Sci. 2002, 11, 571–587. [Google Scholar] [CrossRef] [PubMed]
- Vizcarra, C.L.; Mayo, S.L. Electrostatics in computational protein design. Curr. Opin. Chem. Boil. 2005, 9, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Protein Structure Modeling with MODELLER. Adv. Struct. Saf. Stud. 2014, 1137, 1–15. [Google Scholar] [CrossRef]
- Lomize, M.A.; Lomize, A.L.; Pogozheva, I.D.; Mosberg, H.I. OPM: Orientations of Proteins in Membranes database. Bioinformatics 2006, 22, 623–625. [Google Scholar] [CrossRef] [PubMed]
- Pedretti, A.; Villa, L.; Vistoli, G. VEGA--an open platform to develop chemo-bio-informatics applications, using plug-in architecture and script programming. J. Comput. Mol. Des. 2004, 18, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Sarvagalla, S.; Cheung, C.H.A.; Tsai, J.Y.; Hsiehd, H.P.; Coumar, M.S. Disruption of protein-protein interactions: Hot spot detection, structure-based virtual screening and in vitro testing for the anti-cancer drug target-surviving. RSC. Adv. 2016, 6, 31947–31959. [Google Scholar] [CrossRef]
- Cukuroglu, E.; Gursoy, A.; Keskin, O. HotRegion: A database of predicted hot spot clusters. Nucleic Acids Res. 2011, 40, D829–D833. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isoform | IF-ECLs 6 | S5 DI | S1-S2 DIII | S5 DIV | S6 DIV | PPI Pattern | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| PPI-Id 3 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | ||
| hNav1.1α 5 | P35498 2 | Y | Y | Y | Y | Y | N | Y | Y | I |
| mNav1.1α 5 | A2APX8 2 | Y | Y | Y | Y | Y | N | Y | Y | I |
| rNav1.1α 5 | P04774 2 | Y | Y | Y | Y | Y | N | Y | Y | I |
| hNav1.2α 4 | 6J8E1 | Y | Y | Y | Y | Y | Y | Y | Y | II |
| mNav1.2α 5 | B1AWN6 2 | Y | Y | Y | Y | Y | Y | Y | Y | II |
| rNav1.2α 5 | P04775 2 | Y | Y | Y | Y | Y | Y | Y | Y | II |
| hNav1.3α 5 | Q9NY46 2 | Y | Y | Y | Y | Y | N | Y | Y | I |
| mNav1.3α 5 | A2ASI5 2 | Y | Y | Y | Y | Y | N | Y | Y | I |
| rNav1.3α 5 | P08104 2 | Y | Y | Y | Y | Y | N | Y | Y | I |
| eeNav1.4α 4 | 5XSY1 | N | Y | Y | Y | Y | Y | Y | Y | IX |
| hNav1.4α 4 | 6AGF1 | Y | Y | Y | Y | Y | Y | Y | Y | II |
| mNav1.4α 5 | Q9ER60 2 | Y | Y | Y | Y | Y | Y | Y | Y | II |
| rNav1.4α 5 | P15390 2 | Y | Y | Y | Y | Y | Y | Y | Y | II |
| hNav1.5α 5 | Q14524 2 | Y | Y | N | Y | Y | N | Y | Y | III |
| mNav1.5α 5 | Q9JJV9 2 | Y | Y | N | Y | Y | N | Y | Y | III |
| rNav1.5α 4 | 6U701 | Y | Y | N | Y | Y | N | Y | Y | III |
| hNav1.6α 5 | Q9UQD0 2 | Y | Y | Y | Y | Y | Y | Y | Y | II |
| mNav1.6α 5 | Q9WTU3 2 | Y | Y | Y | Y | Y | Y | Y | Y | II |
| rNav1.6α 5 | O88420 2 | Y | Y | Y | Y | Y | Y | Y | Y | II |
| hNav1.7α 4 | 6J8G1 | Y | Y | Y | Y | Y | N | Y | Y | I |
| mNav1.7α 5 | Q62205 2 | Y | Y | Y | Y | Y | N | Y | Y | I |
| rNav1.7α 5 | O08562 2 | Y | Y | Y | Y | Y | N | Y | Y | I |
| hNav1.8α 5 | Q9Y5Y9 2 | Y | Y | Y | N | Y | N | Y | Y | IV |
| mNav1.8α 5 | Q6QIY3 2 | Y | Y | N | N | Y | N | Y | Y | V |
| rNav1.8α 5 | Q62968 2 | Y | N | N | N | Y | N | Y | Y | VI |
| hNav1.9α 5 | Q9UI33 2 | Y | N | Y | N | Y | N | N | N | VII |
| mNav1.9α 5 | Q9R053 2 | Y | Y | N | N | Y | N | N | N | VIII |
| rNav1.9α 5 | O88457 2 | Y | Y | N | N | Y | N | N | N | VIII |
| Isoform | UniProt Code 1 | ECL | |||
|---|---|---|---|---|---|
| S5 DI | S1-S2 DIII | S5 DIV | S6 DIV | ||
| 1, 2 | 3, 4 | 5 | 6, 7, 8 | ||
| hNav1.1α | P35498 | agqCpEgym | yidQrKtik | gidDmfn | pnkVNPgss |
| mNav1.1α | A2APX8 | agqCpEgym | yidQrKtik | gidDmfn | pnkVNPgss |
| rNav1.1α | P04774 | agqCpEgym | yidQrKtik | gidDmfn | pnkVNPgss |
| hNav1.2α | Q99250 | agqCpEgyi | yieQrKtik | gidDmfn | pdkDHPgss |
| mNav1.2α | B1AWN6 | agqCpEgyi | yieQrKtikd | gidDmfn | pekDHPgss |
| rNav1.2α | P04775 | agqCpEgyi | yieQrKtik | gidDmfn | pekDHPgss |
| hNav1.3α | Q9NY46 | agqCpEgyi | yieQrKtik | gidDmfn | pdtIHPgss |
| mNav1.3α | A2ASI5 | agqCpEgyi | yieQrKtik | gidDmfn | pdaIHPgss |
| rNav1.3α | P08104 | agqCpEgyi | yieQrKtik | gidDmfn | pdaIHPgss |
| * eeNav1.4α | P02719 | agkCpEgyt | yiwRrRvik | gvdDifn | pdvENPgtd |
| * hNav1.4α | P35499 | aghCpEgye | yieQrRvir | gidDmfn | pnlENPgts |
| mNav1.4α | Q9ER60 | aghCpEgye | yieQrRvir | gidDmfn | ptlENPgtn |
| rNav1.4α | P15390 | aghCpEgye | yieQrRvir | gidDmfn | ptlENPgtn |
| hNav1.5α | Q14524 | agtCpEgyr | yleErKtik | gidDmfn | ptlPNSngs |
| mNav1.5α | Q9JJV9 | agtCpEgyr | yleErKtik | gidDmfn | pnlPNSngs |
| rNav1.5α | P15389 | agtCpEgyr | yleErKtik | gidDmfn | pnlPNSngs |
| hNav1.6α | Q9UQD0 | agqCpEgyq | yieQrKtir | gidDmfn | ldkEHPgsg |
| mNav1.6α | Q9WTU3 | agqCpEgfq | yieQrKtir | gidDmfn | ldkEHPgsg |
| rNav1.6α | O88420 | agqCpEgfq | yieQrKtir | gidDmfn | ldkEHPgsg |
| hNav1.7α | Q15858 | sgqCpEgyt | yieRkKtik | ginDmfn | pkkVHPgss |
| mNav1.7α | Q62205 | sgqCpEgye | yieKkKtik | ginDmfn | pkkVHPgss |
| rNav1.7α | O08562 | sgqCpEgyi | yieKkKtik | ginDmfn | pkkVHPgss |
| hNav1.8α | Q9Y5Y9 | sghCpDgyi | yldQkPtvk | gidDmfn | pnlPNSngt |
| mNav1.8α | Q6QIY3 | aghCpNdyv | yleEkPrvk | gidDmfn | pnrPNSngs |
| rNav1.8α | Q62968 | aghCpGgyv | yleEkPrvk | gidDmfn | pnlPNSngs |
| hNav1.9α | Q9UI33 | nsaCsIqye | hleNqPkiq | gidDifn | rskESCnss |
| mNav1.9α | Q9R053 | rrsCpDgst | nlpSrPqve | gidDifn | eskASCnss |
| rNav1.9α | O88457 | srpCpNgst | nlpSrPqve | gidDifn | eakEHCnss |
| Subunit | UniProt code 1 | 7, 8, 3, 4 | 1,5 | 6 | 2 |
| * eeNavβ1 | A0A1L3MZ94 | sngAcVEvdsDtea | sckMRgev | mgsKntf | yfdRtlt |
| * hNavβ1 | Q07699 | acgGcVEvdsEtea | sckRRset | ngsRgtk | hvyRllf |
| mNavβ1 | P97952 | awgGcVEvdsDtea | sckRRset | ngsRgtk | hvyRllf |
| rNavβ1 | Q00954 | awgGcVEvdsEtea | sckRRset | ngsRgtk | hvyRllf |
| * hNavβ3 | Q9NY72 | cfpVcVEvpsEtea | scmKReev | ngsKdlq | nvsRefe |
| mNavβ3 | Q8BHK2 | cfpVcVEvpsEtea | scmKReev | ngsKdlq | nvsRefe |
| rNavβ3 | Q9JK00 | cfpVcVEvpsEtea | scmKReev | ngsKdlq | nvsRefe |
| hNavβ2 | O60939 | grsMeVTvpaTlnv | fnsCYtvn | sgnPsky | yimNppd |
| mNavβ2 | Q56A07 | grsMeVTaptTlsv | fnsCYtvn | sgnPsky | yitNppd |
| rNavβ2 | P54900 | grsMeVTvptTlsv | fnsCYtvn | sgnPsky | yitNppd |
| hNavβ4 | Q8IWT1 | sleVsVGkatDiya | fssCFgfe | vgsTkek | hvkNpke |
| mNavβ4 | Q7M729 | sleVsVGkatTiya | fssCYgfe | egsTkek | fvrNpke |
| rNavβ4 | Q7M730 | sleVsVGkatTiya | fssCYgfe | egsTkek | fvrNpke |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villa-Diaz, F.; Lopez-Nunez, S.; Ruiz-Castelan, J.E.; Salinas-Stefanon, E.M.; Scior, T. Chemometric Models of Differential Amino Acids at the Navα and Navβ Interface of Mammalian Sodium Channel Isoforms. Molecules 2020, 25, 3551. https://doi.org/10.3390/molecules25153551
Villa-Diaz F, Lopez-Nunez S, Ruiz-Castelan JE, Salinas-Stefanon EM, Scior T. Chemometric Models of Differential Amino Acids at the Navα and Navβ Interface of Mammalian Sodium Channel Isoforms. Molecules. 2020; 25(15):3551. https://doi.org/10.3390/molecules25153551
Chicago/Turabian StyleVilla-Diaz, Fernando, Susana Lopez-Nunez, Jordan E. Ruiz-Castelan, Eduardo Marcos Salinas-Stefanon, and Thomas Scior. 2020. "Chemometric Models of Differential Amino Acids at the Navα and Navβ Interface of Mammalian Sodium Channel Isoforms" Molecules 25, no. 15: 3551. https://doi.org/10.3390/molecules25153551
APA StyleVilla-Diaz, F., Lopez-Nunez, S., Ruiz-Castelan, J. E., Salinas-Stefanon, E. M., & Scior, T. (2020). Chemometric Models of Differential Amino Acids at the Navα and Navβ Interface of Mammalian Sodium Channel Isoforms. Molecules, 25(15), 3551. https://doi.org/10.3390/molecules25153551