
The Spectrum of Design Solutions for Improving the Activity-Selectivity Product of Peptide Antibiotics against Multidrug-Resistant Bacteria and Prostate Cancer PC-3 Cells

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals (Reagents)
2.2. Peptide Design Methods
- (a)
- physicochemical properties of helical peptides based on their sequence:HeliQuest tool [30]:
- (b)
- the probability for a peptide to be an antimicrobial peptide:CAMPR3 artificial intelligence algorithms for predicting AMPs [31]:
- (c)
- the probability for a peptide to be a cell-penetrating peptide:Cell-penetrating peptide (CPP) prediction, according to CPPrex-FL [32] and MLCPP [33] algorithms with respective links http://server.malab.cn/SkipCPP-Pred/Index.html and http://thegleelab.org/MLCPP/MLCPP.html.
- (d)
- the probability for a peptide to be an anticancer peptide:Anticancer probability servers used were that of:
2.3. Bacterial Strains and Antimicrobial Activity Assay
2.4. Cytotoxicity on Cancer Cells and Fibroblasts
2.5. Hemolysis of Human Erythrocytes
3. Results
3.1. Peptide Design

3.2. The Performance Parameters for Ranking Peptides When Antibacterial Activity and Toxicity to Human Erythrocytes are Both Taken into Account
3.3. Activity and Selectivity against Prostate Cancer Cells
4. Discussion
4.1. Effect of Charge and Helical Content on Activity
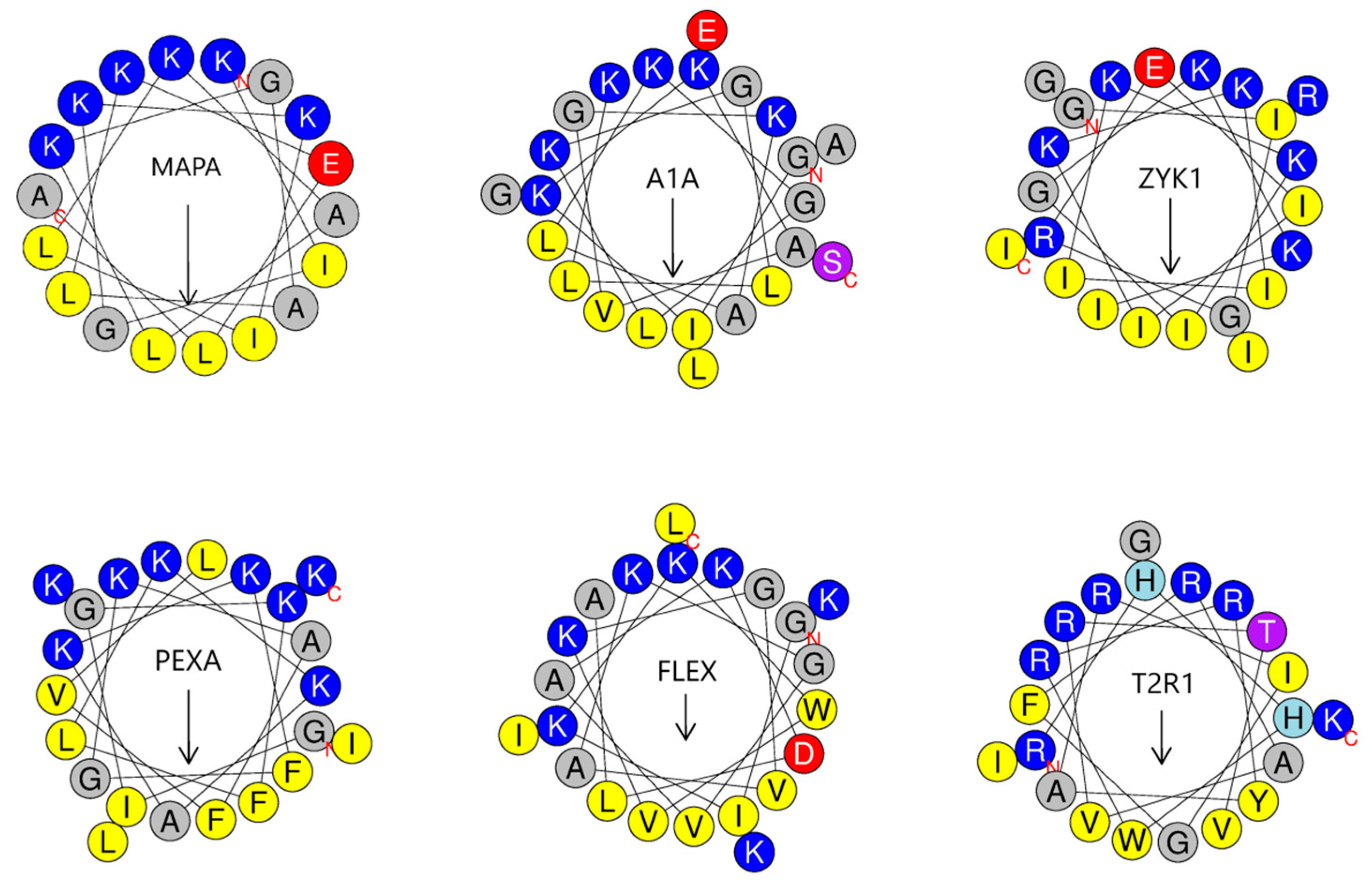
4.2. Effect of Amphipathic Motifs on Activity
4.3. Specific Advantages of Novel Folds
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schweizer, F. Cationic amphiphilic peptides with cancer-selective toxicity. Eur. J. Pharmacol. 2009, 625, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Hoskin, D.W.; Ramamoorthy, A. Studies on anticancer activities of antimicrobial peptides. Biochim. Biophys. Acta 2008, 1778, 357–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, H.L.; Yip, B.S.; Chen, K.H.; Yu, H.Y.; Chih, Y.H.; Cheng, H.T.; Chou, J.T.; Cheng, J.W. Novel antimicrobial peptides with high anticancer activity and selectivity. PLoS ONE 2015, 10, e0126390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felício, M.R.; Silva, O.N.; Gonçalves, S.; Santos, N.C.; Franco, O.L. Peptides with Dual Antimicrobial and Anticancer Activities. Front. Chem. 2017, 5, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Gaspar, D.; Veiga, A.S.; Castanho, M.A. From antimicrobial to anticancer peptides. A review. Front. Microbiol. 2013, 4, 294. [Google Scholar] [CrossRef] [Green Version]
- Boohaker, R.J.; Lee, M.W.; Vishnubhotla, P.; Perez, J.M.; Khaled, A.R. The use of therapeutic peptides to target and to kill cancer cells. Curr. Med. Chem. 2012, 19, 3794–3804. [Google Scholar] [CrossRef]
- Deslouches, B.; Di, Y.P. Antimicrobial peptides with selective antitumor mechanisms: Prospect for anticancer applications. Oncotarget 2017, 8, 46635–46651. [Google Scholar] [CrossRef] [Green Version]
- Cruciani, R.A.; Barker, J.L.; Zasloff, M.; Chen, H.C.; Colamonici, O. Antibiotic magainins exert cytolytic activity against transformed-cell lines through channel formation. Proc. Natl. Acad. Sci. USA 1991, 88, 3792–3796. [Google Scholar] [CrossRef] [Green Version]
- Ohsaki, Y.; Gazdar, A.F.; Chen, H.C.; Johnson, B.E. Antitumor-activity of magainin analogs against human lung-cancer cell-lines. Cancer Res. 1992, 52, 3534–3538. [Google Scholar]
- Jacob, L.; Zasloff, M. Potential therapeutic applications of magainins and other antimicrobial agents of animal origin. Antimicrob. Pept. 1994, 186, 197–216. [Google Scholar]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novković, M.; Simunić, J.; Bojović, V.; Tossi, A.; Juretić, D. DADP: The database of anuran defense peptides. Bioinformatics 2012, 28, 1406–1407. [Google Scholar] [CrossRef] [PubMed]
- Pirtskhalava, M.; Gabrielian, A.; Cruz, P.; Griggs, H.L.; Squires, R.B.; Hurt, D.E.; Grigolava, M.; Chubinidze, M.; Gogoladze, G.; Vishnepolsky, B.; et al. DBAASP v.2: An enhanced database of structure and antimicrobial/cytotoxic activity of natural and synthetic peptides. Nucleic Acids Res. 2016, 44, 6503. [Google Scholar] [CrossRef] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Shoombuatong, W.; Schaduangrat, N.; Nantasenamat, C. Unraveling the bioactivity of anticancer peptides as deduced from machine learning. EXCLI J. 2018, 17, 734–752. [Google Scholar] [CrossRef]
- Huerta-Cantillo, J.; Navarro-Garcia, F. Properties and design of antimicrobial peptides as potential tools against pathogens and malignant cells. Medigr. Investig. Discapac. 2016, 5, 96–115. [Google Scholar]
- Papo, N.; Shai, Y. Host defense peptides as new weapons in cancer treatment. Cell. Mol. Life. Sci. 2005, 62, 784–790. [Google Scholar] [CrossRef]
- Gottesman, M.M. Mechanisms of cancer drug resistance. Annu. Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef] [Green Version]
- Hancock, R.E. Cationic peptides: Effectors in innate immunity and novel antimicrobials. Lancet Infect. Dis. 2001, 1, 156–164. [Google Scholar] [CrossRef]
- Johnstone, S.A.; Gelmon, K.; Mayer, L.D.; Hancock, R.E.; Bally, M.B. In vitro characterization of the anticancer activity of membrane-active cationic peptides. I. Peptide-mediated cytotoxicity and peptide-enhanced cytotoxic activity of doxorubicin against wild-type and p-glycoprotein over-expressing tumor cell lines. Anticancer Drug Des. 2000, 15, 151–160. [Google Scholar] [PubMed]
- Juretić, D.; Sonavane, Y.; Ilić, N.; Gajski, G.; Goić-Barišić, I.; Tonkić, M.; Kozic, M.; Maravić, A.; Pellay, F.X.; Zoranić, L. Designed peptide with a flexible central motif from ranatuerins adapts its conformation to bacterial membranes. Biochim. Biophys. Acta 2018, 1860, 2655–2668. [Google Scholar] [CrossRef] [PubMed]
- Juretić, D.; Vukičević, D.; Petrov, D.; Novković, M.; Bojović, V.; Lučić, B.; Ilić, N.; Tossi, A. Knowledge-based computational methods for identifying or designing novel, non-homologous antimicrobial peptides. Eur. Biophys. J. 2011, 40, 371–385. [Google Scholar] [CrossRef] [PubMed]
- Rončević, T.; Gajski, G.; Ilić, N.; Goić-Barišić, I.; Tonkić, M.; Zoranić, L.; Simunić, J.; Benincasa, M.; Mijatović, M.; Tossi, A.; et al. PGLa-H tandem-repeat peptides active against multidrug resistant clinical bacterial isolates. Biochim. Biophys. Acta 2017, 1859, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Kamech, N.; Vukičević, D.; Ladram, A.; Piesse, C.; Vasseur, J.; Bojović, V.; Simunić, J.; Juretić, D. Improving the selectivity of antimicrobial peptides from anuran skin. J. Chem. Inf. Model. 2012, 52, 3341–3351. [Google Scholar] [CrossRef] [PubMed]
- Juretić, D.; Vukicević, D.; Ilić, N.; Antcheva, N.; Tossi, A. Computational design of highly selective antimicrobial peptides. J. Chem. Inf. Model. 2009, 49, 2873–2882. [Google Scholar] [CrossRef] [PubMed]
- Kozić, M.; Vukičević, D.; Simunić, J.; Rončević, T.; Antcheva, N.; Tossi, A.; Juretić, D. Predicting the Minimal Inhibitory Concentration for Antimicrobial Peptides with Rana-Box Domain. J. Chem. Inf. Model. 2015, 55, 2275–2287. [Google Scholar] [CrossRef]
- Juretić, D.; Zucić, D.; Lucić, B.; Trinajstić, N. Preference functions for prediction of membrane-buried helices in integral membrane proteins. Comput. Chem. 1998, 22, 279–294. [Google Scholar] [CrossRef]
- Juretić, D.; Zoranić, L.; Zucić, D. Basic charge clusters and predictions of membrane protein topology. J. Chem. Inf. Comput. Sci. 2002, 42, 620–632. [Google Scholar] [CrossRef]
- Gautier, R.; Douguet, D.; Antonny, B.; Drin, G. HELIQUEST: A web server to screen sequences with specific α-helical properties. Bioinformatics 2008, 24, 2101–2102. [Google Scholar] [CrossRef]
- Waghu, F.H.; Barai, R.S.; Gurung, P.; Idicula-Thomas, S. CAMPR3: A database on sequences, structures and signatures of antimicrobial peptides. Nucleic Acids Res. 2016, 44, D1094–D1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiang, X.; Zhou, C.; Ye, X.; Du, P.F.; Su, R.; Wei, L. CPPred-FL: A sequence-based predictor for large-scale identification of cell-penetrating peptides by feature representation learning. Brief. Bioinform. 2020, 21, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Subramaniyam, S.; Shin, T.H.; Kim, M.O.; Lee, G. Machine-Learning-Based Prediction of Cell-Penetrating Peptides and Their Uptake Efficiency with Improved Accuracy. J. Proteome Res. 2018, 17, 2715–2726. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, A.; Kapoor, P.; Kumar, R.; Chaudhary, K.; Gautam, A.; Raghava, G.P.S. In Silico Models for Designing and Discovering Novel Anticancer Peptides. Sci. Rep. 2013, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Schaduangrat, N.; Nantasenamat, C.; Prachayasittikul, V.; Shoombuatong, W. ACPred: A Computational Tool for the Prediction and Analysis of Anticancer Peptides. Molecules 2019, 24, 1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boopathi, V.; Subramaniyam, S.; Malik, A.; Lee, G.; Manavalan, B.; Yang, D.C. mACPpred: A Support Vector Machine-Based Meta-Predictor for Identification of Anticancer Peptides. Int. J. Mol. Sci. 2019, 20, 1964. [Google Scholar] [CrossRef] [Green Version]
- Kondejewski, L.H.; Jelokhani-Niaraki, M.; Farmer, S.W.; Lix, B.; Kay, C.M.; Sykes, B.D.; Hancock, R.E.W.; Hodges, R.S. Dissociation of antimicrobial and hemolytic activities in cyclic peptide diastereomers by systematic alterations in amphipathicity. J. Biol. Chem. 1999, 274, 13181–13192. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.X.; Mant, C.T.; Farmer, S.W.; Hancock, R.E.W.; Vasil, M.L.; Hodges, R.S. Rational design of alpha-helical antimicrobial peptides with enhanced activities and specificity/therapeutic index. J. Biol. Chem. 2005, 280, 12316–12329. [Google Scholar] [CrossRef] [Green Version]
- Panteleev, P.V.; Bolosov, I.A.; Balandin, S.V.; Ovchinnikova, T.V. Design of antimicrobial peptide arenicin analogs with improved therapeutic indices. J. Pept. Sci. 2015, 21, 105–113. [Google Scholar] [CrossRef]
- Munk, J.K.; Ritz, C.; Fliedner, F.P.; Frimodt-Moller, N.; Hansen, P.R. Novel Method to Identify the Optimal Antimicrobial Peptide in a Combination Matrix, Using Anoplin as an Example. Antimicrob. Agents Chemother. 2014, 58, 1063–1070. [Google Scholar] [CrossRef] [Green Version]
- Reinhardt, A. Antimicrobial Peptides as New Potential Antibiotics. Ph.D. Thesis, Universität zu Köln, Köln, Germany, 2017. [Google Scholar]
- Bobone, S.; Stella, L. Selectivity of Antimicrobial Peptides: A Complex Interplay of Multiple Equilibria. In Antimicrobial Peptides: Basics for Clinical Application. Advances in Experimental Medicine and Biology; Matsuzaki, K., Ed.; Springer-Verlag Singapore Pte Ltd.: Singapore, 2019; Volume 1117, pp. 175–214. [Google Scholar]
- Xu, W.; Zhu, X.; Tan, T.T.; Li, W.Z.; Shan, A.S. Design of Embedded-Hybrid Antimicrobial Peptides with Enhanced Cell Selectivity and Anti-Biofilm Activity. PLoS ONE 2014, 9, e98935. [Google Scholar] [CrossRef] [PubMed]
- Juretic, D.; Simunic, J. Design of alpha-helical antimicrobial peptides with a high selectivity index. Expert Opin. Drug Discov. 2019, 14, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Souza, B.M.; Mendes, M.A.; Santos, L.D.; Marques, M.R.; César, L.M.; Almeida, R.N.; Pagnocca, F.C.; Konno, K.; Palma, M.S. Structural and functional characterization of two novel peptide toxins isolated from the venom of the social wasp Polybia paulista. Peptides 2005, 26, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, M.; Begovic, E.; Chapman, J.; Putnam, N.H.; Hellsten, U.; Kawashima, T.; Kuo, A.; Mitros, T.; Salamov, A.; Carpenter, M.L.; et al. The Trichoplax genome and the nature of placozoans. Nature 2008, 454, 955–960. [Google Scholar] [CrossRef] [Green Version]
- Osigus, H.J.; Rolfes, S.; Herzog, R.; Kamm, K.; Schierwater, B. Polyplacotoma mediterranea is a new ramified placozoan species. Curr. Biol. 2019, 29, R148–R149. [Google Scholar] [CrossRef] [Green Version]
- Simunić, J.; Petrov, D.; Bouceba, T.; Kamech, N.; Benincasa, M.; Juretić, D. Trichoplaxin—A new membrane-active antimicrobial peptide from placozoan cDNA. Biochim. Biophys. Acta 2014, 1838, 1430–1438. [Google Scholar] [CrossRef]
- Zelezetsky, I.; Pontillo, A.; Puzzi, L.; Antcheva, N.; Segat, L.; Pacor, S.; Crovella, S.; Tossi, A. Evolution of the Primate Cathelicidin Correlation between Structural Variations and Antimicrobial Activity. J. Biol. Chem. 2006, 281, 19861–19871. [Google Scholar] [CrossRef] [Green Version]
- Masso-Silva, J.; Diamond, G. Antimicrobial peptides from fish. Pharmaceuticals 2014, 7, 265–310. [Google Scholar] [CrossRef] [Green Version]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Lee, H.T.; Lee, C.C.; Yang, J.R.; Lai, J.Z.; Chang, K.Y. A large-scale structural classification of antimicrobial peptides. BioMed Res. Int. 2015, 2015, 475062. [Google Scholar] [CrossRef]
- Ilić, N.; Novković, M.; Guida, F.; Xhindoli, D.; Benincasa, M.; Tossi, A.; Juretić, D. Selective antimicrobial activity and mode of action of adepantins, glycine-rich peptide antibiotics based on anuran antimicrobial peptide sequences. Biochim. Biophys. Acta 2013, 1828, 1004–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.C.; Brown, J.H.; Morell, J.L.; Huang, C.M. Synthetic magainin analogues with improved antimicrobial activity. FEBS Lett. 1988, 236, 462–466. [Google Scholar] [CrossRef] [Green Version]
- Maloy, W.L.; Kari, U.P. Structure-activity studies on magainins and other host-defense peptides. Biopolymers 1995, 37, 105–122. [Google Scholar] [CrossRef] [PubMed]
- Gottler, L.M.; Ramamoorthy, A. Structure, membrane orientation, mechanism, and function of pexiganan—A highly potent antimicrobial peptide designed from magainin. Biochim. Biophys. Acta 2009, 1788, 1680–1686. [Google Scholar] [CrossRef] [Green Version]
- Ge, Y.G.; MacDonald, D.L.; Holroyd, K.J.; Thornsberry, C.; Wexler, H.; Zasloff, M. In vitro antibacterial properties of pexiganan, an analog of magainin. Antimicrob. Agents Chemother. 1999, 43, 782–788. [Google Scholar] [CrossRef] [Green Version]
- Rotem, S.; Radzishevsky, I.; Mor, A. Physicochemical properties that enhance discriminative antibacterial activity of short dermaseptin derivatives. Antimicrob. Agents Chemother. 2006, 50, 2666–2672. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Hu, J.; Yang, C.; Zhang, Y.; Wang, F.; Mu, Q.; Pan, F.; Xu, H.; Lu, J.R. Amino acid side chains affect the bioactivity of designed short peptide amphiphiles. J. Mater. Chem. B 2016, 4, 2359–2368. [Google Scholar] [CrossRef]
- Tytler, E.M.; Anantharamaiah, G.M.; Walker, D.E.; Mishra, V.K.; Palgunachari, M.N.; Segrest, J.P. Molecular basis for prokaryotic specificity of magainin-induced lysis. Biochemistry 1995, 34, 4393–4401. [Google Scholar] [CrossRef]
- Nicolas, P.; Vanhoye, D.; Amiche, M. Molecular strategies in biological evolution of antimicrobial peptides. Peptides 2003, 24, 1669–1680. [Google Scholar] [CrossRef]
- Conlon, J.M.; Sonnevend, A.; Davidson, C.; Smith, D.D.; Nielsen, P.F. The ascaphins: A family of antimicrobial peptides from the skin secretions of the most primitive extant frog, Ascaphus truei. Biochem. Biophys. Res. Commun. 2004, 320, 170–175. [Google Scholar] [CrossRef]
- Conlon, J.M.; Kolodziejek, J.; Nowotny, N. Antimicrobial peptides from the skins of North American frogs. Biochim. Biophys. Acta 2009, 1788, 1556–1563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, P.; Xiao, K.Q.; Wang, H.J.; Xu, H.; Xu, P.P.; Jia, Y.; Häggblom, M.M.; Zhu, J.G. Characterization and Potential Applications of a Selenium Nanoparticle Producing and Nitrate Reducing Bacterium Bacillus oryziterrae sp. nov. Sci. Rep. 2016, 6, 34054. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B.; Lohner, K. Detergent-like actions of linear amphipathic cationic antimicrobial peptides. Biochim. Biophys. Acta 2006, 1758, 1529–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segrest, J.P.; De Loof, H.; Dohlman, J.G.; Brouillette, C.G.; Anantharamaiah, G. Amphipathic helix motif: Classes and properties. Proteins 1990, 8, 103–117. [Google Scholar] [CrossRef]
- Russ, W.P.; Engelman, D.M. The GxxxG motif: A framework for transmembrane helix-helix association. J. Mol. Biol. 2000, 296, 911–919. [Google Scholar] [CrossRef]
- Walters, R.F.; DeGrado, W.F. Helix-packing motifs in membrane proteins. Proc. Natl. Acad. Sci. USA 2006, 103, 13658–13663. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Anuwongcharoen, N.; Malik, A.A.; Prachayasittikul, V.; Wikberg, J.E.; Nantasenamat, C. Roles of d-Amino Acids on the Bioactivity of Host Defense Peptides. Int. J. Mol. Sci. 2016, 17, 1023. [Google Scholar] [CrossRef] [Green Version]
- Langel, U. Cell-Penetrating Peptides: Processes and Applications; CRC Press: Boca Raton, FL, USA, 2002. [Google Scholar]
- Milletti, F. Cell-penetrating peptides: Classes, origin, and current landscape. Drug Discov. Today 2012, 17, 850–860. [Google Scholar] [CrossRef]
- Oehlke, J.; Scheller, A.; Wiesner, B.; Krause, E.; Beyermann, M.; Klauschenz, E.; Melzig, M.; Bienert, M. Cellular uptake of an alpha-helical amphipathic model peptide with the potential to deliver polar compounds into the cell interior non-endocytically. Biochim. Biophys. Acta 1998, 1414, 127–139. [Google Scholar] [CrossRef] [Green Version]
- Strandberg, E.; Tiltak, D.; Ieronimo, M.; Kanithasen, N.; Wadhwani, P.; Ulrich, A.S. Influence of C-terminal amidation on the antimicrobial and hemolytic activities of cationic alpha-helical peptides. Pure Appl. Chem. 2007, 79, 717–728. [Google Scholar] [CrossRef]
- Juretić, D.; Jerončić, A.; Zucić, D. Sequence Analysis of Membrane Proteins with the Web Server SPLIT. Croat. Chem. Acta 1999, 72, 975–997. [Google Scholar]
- Sani, M.A.; Saenger, C.; Juretic, D.; Separovic, F. Glycine Substitution Reduces Antimicrobial Activity and Helical Stretch of diPGLa-H in Lipid Micelles. J. Phys. Chem. B 2017, 121, 4817–4822. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, M.P.D.; Costa, S.T.B.; de Souza, B.M.; Palma, M.S.; Ruggiero, J.R.; Neto, J.R. Selectivity in the mechanism of action of antimicrobial mastoparan peptide Polybia-MP1. Eur. Biophys. J. 2008, 37, 879–891. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, J.; Liu, L.W.; Wang, K.R.; Song, J.J.; Yan, J.X.; Li, T.Y.; Zhang, B.Z.; Wang, R. A novel analog of antimicrobial peptide Polybia-MPI, with thioamide bond substitution, exhibits increased therapeutic efficacy against cancer and diminished toxicity in mice. Peptides 2010, 31, 1832–1838. [Google Scholar] [CrossRef] [PubMed]
- Luong, H.X.; Kim, D.H.; Lee, B.J.; Kim, Y.W. Antimicrobial activity and stability of stapled helices of polybia-MP1. Arch. Pharm. Res. 2017, 40, 1414–1419. [Google Scholar] [CrossRef] [PubMed]
- de Souza, B.M.; da Silva, A.V.R.; Resende, V.M.F.; Arcuri, H.A.; Cabrera, M.P.D.; Neto, J.R.; Palma, M.S. Characterization of two novel polyfunctional mastoparan peptides from the venom of the social wasp Polybia paulista. Peptides 2009, 30, 1387–1395. [Google Scholar] [CrossRef]
- Wang, K.R.; Yan, J.X.; Dang, W.; Xie, J.Q.; Yan, B.; Yan, W.J.; Sun, M.; Zhang, B.; Ma, M.; Zhao, Y. Dual antifungal properties of cationic antimicrobial peptides polybia-MPI: Membrane integrity disruption and inhibition of biofilm formation. Peptides 2014, 56, 22–29. [Google Scholar] [CrossRef]
- Vinhote, J.F.C.; Lima, D.B.; de Menezes, R.; Mello, C.P.; de Souza, B.M.; Havt, A.; Palma, M.S.; dos Santos, R.P.; de Albuquerque, E.L.; Freire, V.N.; et al. Trypanocidal activity of mastoparan from Polybia paulista wasp venom by interaction with TcGAPDH. Toxicon 2017, 137, 168–172. [Google Scholar] [CrossRef]
- Wang, K.R.; Zhang, B.Z.; Zhang, W.; Yan, J.X.; Li, J.; Wang, R. Antitumor effects, cell selectivity and structure-activity relationship of a novel antimicrobial peptide polybia-MPI. Peptides 2008, 29, 963–968. [Google Scholar] [CrossRef]
- Wang, K.R.; Yan, J.X.; Zhang, B.Z.; Song, J.J.; Jia, P.F.; Wang, R. Novel mode of action of polybia-MPI, a novel antimicrobial peptide, in multi-drug resistant leukemic cells. Cancer Lett. 2009, 278, 65–72. [Google Scholar] [CrossRef]
- Cabrera, M.P.D.; Arcisio-Miranda, M.; Gorjao, R.; Leite, N.B.; de Souza, B.M.; Curi, R.; Procopio, J.; Neto, J.R.; Palma, M.S. Influence of the Bilayer Composition on the Binding and Membrane Disrupting Effect of Polybia-MP1, an Antimicrobial Mastoparan Peptide with Leukemic T-Lymphocyte Cell Selectivity. Biochemistry 2012, 51, 4898–4908. [Google Scholar] [CrossRef]
- Chen, X.L.; Zhang, L.Y.; Ma, C.B.; Zhang, Y.Q.; Xi, X.P.; Wang, L.; Zhou, M.; Burrows, J.F.; Chen, T. A novel antimicrobial peptide, Ranatuerin-2PLx, showing therapeutic potential in inhibiting proliferation of cancer cells. Biosci. Rep. 2018, 38, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leite, N.B.; Aufderhorst-Roberts, A.; Palma, M.S.; Connell, S.D.; Neto, J.R.; Beales, P.A. PE and PS Lipids Synergistically Enhance Membrane Poration by a Peptide with Anticancer Properties. Biophys. J. 2015, 109, 936–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvares, D.S.; Fanani, M.L.; Neto, J.R.; Wilke, N. The interfacial properties of the peptide Polybia-MP1 and its interaction with DPPC are modulated by lateral electrostatic attractions. Biochim. Biophys. Acta 2016, 1858, 393–402. [Google Scholar] [CrossRef]
- Jiang, Z.Q.; Mant, C.T.; Vasil, M.; Hodges, R.S. Role of positively charged residues on the polar and non-polar faces of amphipathic alpha-helical antimicrobial peptides on specificity and selectivity for Gram-negative pathogens. Chem. Biol. Drug Des. 2018, 91, 75–92. [Google Scholar] [CrossRef]
- Jiang, Z.Q.; Vasil, A.I.; Vasil, M.L.; Hodges, R.S. “Specificity Determinants” Improve Therapeutic Indices of Two Antimicrobial Peptides Piscidin 1 and Dermaseptin S4 against the Gram-negative Pathogens Acinetobacter baumannii and Pseudomonas aeruginosa. Pharmaceuticals 2014, 7, 366–391. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, N.; Tamura, A. Designed low amphipathic peptides with alpha-helical propensity exhibiting antimicrobial activity via a lipid domain formation mechanism. Peptides 2010, 31, 794–805. [Google Scholar] [CrossRef]
- Ter-Avetisyan, G.; Tuennemann, G.; Nowak, D.; Nitschke, M.; Herrmann, A.; Drab, M.; Cardoso, M.C. Cell Entry of Arginine-rich Peptides Is Independent of Endocytosis. J. Biol. Chem. 2009, 284, 3370–3378. [Google Scholar] [CrossRef] [Green Version]
- Derossi, D.; Joliot, A.H.; Chassaing, G.; Prochiantz, A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 1994, 269, 10444–10450. [Google Scholar]
- Bahnsen, J.S.; Franzyk, H.; Sandberg-Schaal, A.; Nielsen, H.M. Antimicrobial and cell-penetrating properties of penetratin analogs: Effect of sequence and secondary structure. Biochim. Biophys. Acta 2013, 1828, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.L.; Shin, S.Y. Antimicrobial and cytolytic activities and plausible mode of bactericidal action of the cell penetrating peptide penetratin and its Lys-linked two-stranded peptide. Chem. Biol. Drug Des. 2009, 73, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Sugita, T.; Yoshikawa, T.; Mukai, Y.; Yamanada, N.; Imai, S.; Nagano, K.; Yoshida, Y.; Shibata, H.; Yoshioka, Y.; Nakagawa, S. Comparative study on transduction and toxicity of protein transduction domains. Br. J. Pharmacol. 2008, 153, 1143–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porto, W.F.; Irazazabal, L.; Alves, E.S.F.; Ribeiro, S.M.; Matos, C.O.; Pires, A.S.; Fensterseifer, I.C.M.; Miranda, V.J.; Haney, E.F.; Humblot, V.; et al. In silico optimization of a guava antimicrobial peptide enables combinatorial exploration for peptide design. Nat. Commun. 2018, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Bibow, S.; Polyhach, Y.; Eichmann, C.; Chi, C.N.; Kowal, J.; Albiez, S.; McLeod, R.A.; Stahlberg, H.; Jeschke, G.; Güntert, P.; et al. Solution structure of discoidal high-density lipoprotein particles with a shortened apolipoprotein AI. Nat. Struct. Mol. Biol. 2017, 24, 187. [Google Scholar] [CrossRef]
- Kapoor, P.; Singh, H.; Gautam, A.; Chaudhary, K.; Kumar, R.; Raghava, G.P. TumorHoPe: A database of tumor homing peptides. PLoS ONE 2012, 7, e35187. [Google Scholar] [CrossRef] [Green Version]
- Borrelli, A.; Tornesello, A.L.; Tornesello, M.L.; Buonaguro, F.M. Cell Penetrating Peptides as Molecular Carriers for Anti-Cancer Agents. Molecules 2018, 23, 295. [Google Scholar] [CrossRef] [Green Version]
- Prive, G.G.; Melnick, A. Specific peptides for the therapeutic targeting of oncogenes. Curr. Opin. Genet. Dev. 2006, 16, 71–77. [Google Scholar] [CrossRef]
- Koszalka, P.; Kamysz, E.; Wejda, M.; Kamysz, W.; Bigda, J. Antitumor activity of antimicrobial peptides against U937 histiocytic cell line. Acta Biochim. Pol. 2011, 58, 111–117. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Chen, C.X.; Zhang, S.Z.; Zhao, X.C.; Xu, H.; Zhao, X.B.; Lu, J.R. Designed Antimicrobial and Antitumor Peptides with High Selectivity. Biomacromolecules 2011, 12, 3839–3843. [Google Scholar] [CrossRef]
- Chen, C.; Hu, J.; Zeng, P.; Chen, Y.; Xu, H.; Lu, J.R. High cell selectivity and low-level antibacterial resistance of designed amphiphilic peptide G(IIKK)(3)I-NH(2). ACS Appl. Mater. Interfaces 2014, 6, 16529–16536. [Google Scholar] [CrossRef]
- Westerhoff, H.V.; Juretic, D.; Hendler, R.W.; Zasloff, M. Magainins and the disruption of membrane-linked free-energy transduction. Proc. Natl. Acad. Sci. USA 1989, 86, 6597–6601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennison, S.R.; Whittaker, M.; Harris, F.; Phoenix, D.A. Anticancer alpha-helical peptides and structure/function relationships underpinning their interactions with tumour cell membranes. Curr. Protein Pept. Sci. 2006, 7, 487–499. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name [Abbreviation] | A Sequence with Added or Substituted Residues in Bold Font and Underlined | Reference * | Amphipathic Helix Pred. ** | SMIC ¶ Template/Peptide | Anticancer Prediction & | CAMPR3 AMP Pred. All AI Classifiers $ | CPP Prediction # |
|---|---|---|---|---|---|---|---|
| Trichoplaxin-2 [T2] | HHWRRYARIGFRAVRTVIGK-NH2 | This work | 75% | 138 | 0.65/0.98/0.96 | >0.85 Yes | 0.73/0.88 |
| Trichoplaxin-2A [T2R1] | RHHWRRYARIGFRAVRTVIGK-NH2 | This work | 72% | 138/58 | 0.85/0.98/0.96 | >0.87 Yes | 0.73/0.91 |
| Adepantin-1A [A1A] | GIKKAVGKALKGLKGLLKALGES-NH2 | This work | 78% | 513/517 | 0.24/0.98/1.0 | >0.91 Yes | 0.93/0.39 |
| Pexiganan-L18 [PEXA] | GIGKFLKKAKKFGKAFVLILKK-NH2 | [23] | 77% | 352/435 | 0.82/0.98/1.0 | >0.99 Yes | 0.91/0.75 |
| Flexampin [FLEX] | GIKKWVKGVAKGVAKDLAKKIL-NH2 | [22] | 82% | 170/1460 | 0.29/0.98/1.0 | ≥0.99 Yes | 0.74/0.56 |
| Zyk-1 [ZYK1] | GIGREIIKKIIKKIGKKIGRII-NH2 | This work | 86% | 180/1098 | 0.43/0.97/0.99 | >0.97 Yes | 0.83/0.64 |
| DiPGLa-H [PG2] | KIAKVALKALKIAKVALKAL-NH2 | [24] | 75% | 16/204 | 0.61/0.98/0.99 | >0.48 | 0.94/0.85 |
| Kiadin-1 [KIA1] | KIAKVALKALKIAKGALKAL-NH2 | [24] | 80% | 16/251 | 0.61/0.98/0.99 | >0.48 | 0.97/0.86 |
| Mapegin [MAPA] | KIGKKILKALKGALKELA-NH2 | This work | 78% | 92/253 | 0.74/0.98/1.0 | >0.59 Yes | 0.95/0.71 |
| Polybia-MP1 [MP1] | IDWKKLLDAAKQIL-NH2 | [45] | 50% | 25 | 0.91/0.98/0.95 | >0.78 Yes | 0.57/0.53 |
| T2 | T2R1 | A1A | PEXA | FLEX | ZYK1 | PG2 | KIA1 | MAPA | MP1 | |
|---|---|---|---|---|---|---|---|---|---|---|
| MIC (E. coli ATCC 25922) | 0.5–1 | 1 | 1 | 0.5–1 | 0.25 | 1 | 1.5 | 0.75 | 1–2 | |
| MIC (E. coli MG1655) | >32 | 4 | 2 | 4 | 16 | 4 | 16 | 8 | 4 | >32 |
| MIC (E. coli clin. isolate) | 8 | 4 | 32 | 4 | 0.5 | 2 | 6 | 12 | 8–16 | |
| MIC (P. aerug. ATCC 27853) | 4 | 1 | 64 | 4 | 2 | 16 | 6 | 6 | 32–64 | |
| MIC (P. aerug. clin. isolate) | 32 | 8 | >64 | 16 | 2–4 | 16 | 6 | 3 | 32 | |
| MIC (K. pneum. ATCC 13883) | 4 | 2 | 4 | 2 | 0.5–1 | 2 | 3 | 3 | 8 | |
| MIC (K. pneum. clin. isolate) | 8 | 2–4 | 8 | 4 | 2–4 | 4 | 12 | 12 | 8 | |
| MIC (A. baum. ATCC 19606) | 1 | 2 | 2 | 1–2 | 0.5–1 | 2 | 1.5 | 1.5 | 1–2 | |
| MIC (A. baum. clin. isolate) | 8 | 8 | 4–8 | 1–2 | 1 | 2 | 1.5–3 | 1.5 | 4 | |
| MIC (S. aureus ATCC 29213) | 1 | 0.5 | 1 | 0.5 | 0.25 | 1 | 0.75 | 1 | 0.5 | |
| MIC (S. aureus clin. isolate) | 4 | 4 | 4 | 2 | 4 | 2 | 1.5 | 3 | 8 | |
| HC10 | 1.4 | 3 | 25 | 1.6 | 6 | 3 | 3 | 3 | 1.7 | 20 |
| HC20 | 3 | 25 | 80 | 7 | 25 | 8 | 14 | 6 | 3 | 37 |
| HC50 * | 28 | 7000 * | 125 * | 520 * | 1600 * | 29 | 18 | 20 | 20 | 170 |
| SIc = HC20/MIC(coli) & | 4 | 25 | 80 | 9.3 | 100 | 8 | 9.3 | 8 | 2 | |
| SIa = HC20/MIC(aureus) & | 3 | 50 | 80 | 14 | 100 | 8 | 18.7 | 6 | 6 | |
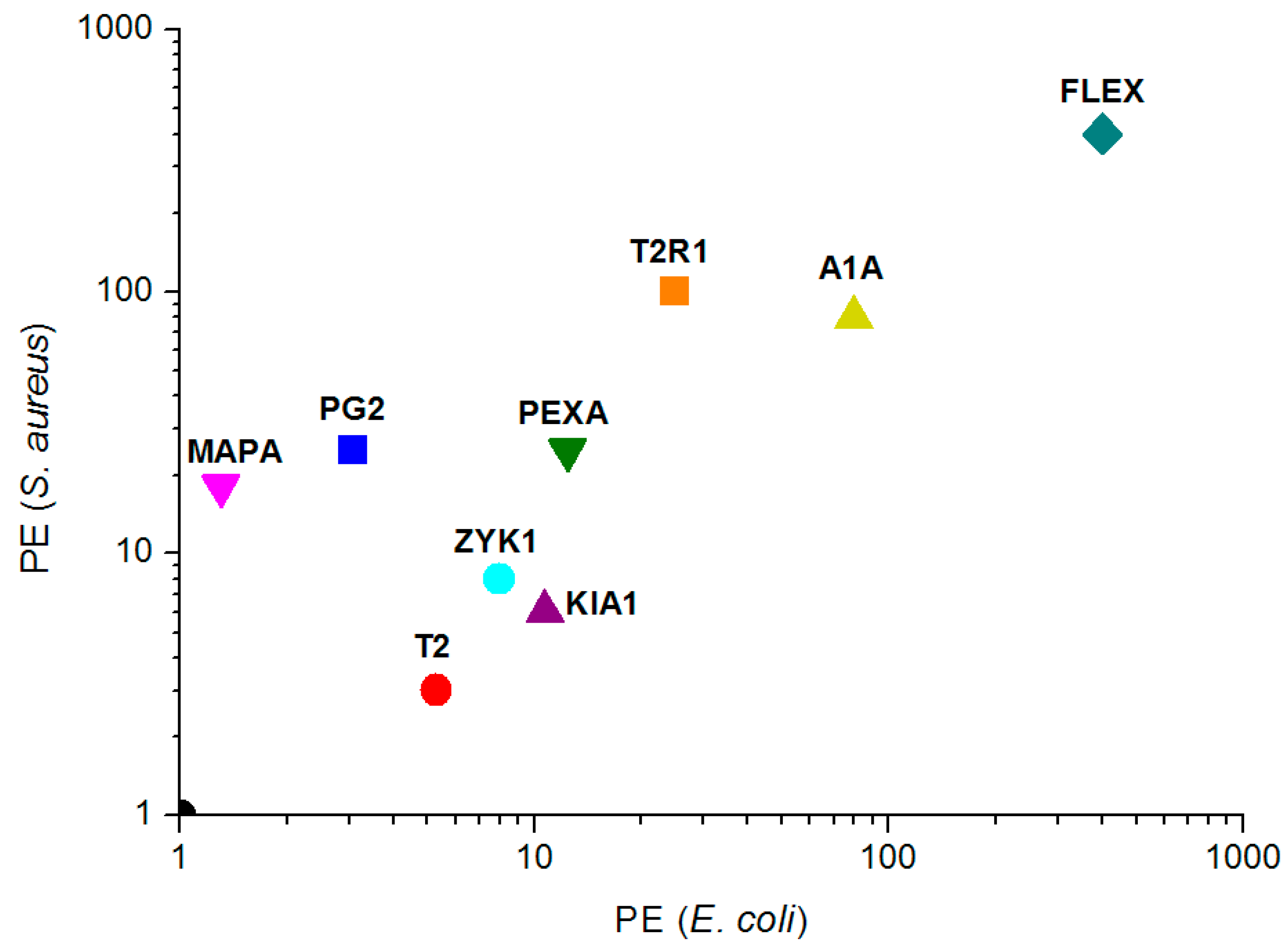
| PEc(20) = SIc/MIC(coli) $ | 5.3 | 25 | 80 | 12.4 | 400 | 8 | 3.1 | 10.7 | 1.3 | |
| PEa(20) = SI/MIC(aureus) $ | 3 | 100 | 80 | 24.9 | 400 | 8 | 24.9 | 6 | 12 |
| T2 | T2R1 | A1A | PEXA | FLEX | ZYK1 | PG2 | KIA1 | MAPA | MP1 | |
|---|---|---|---|---|---|---|---|---|---|---|
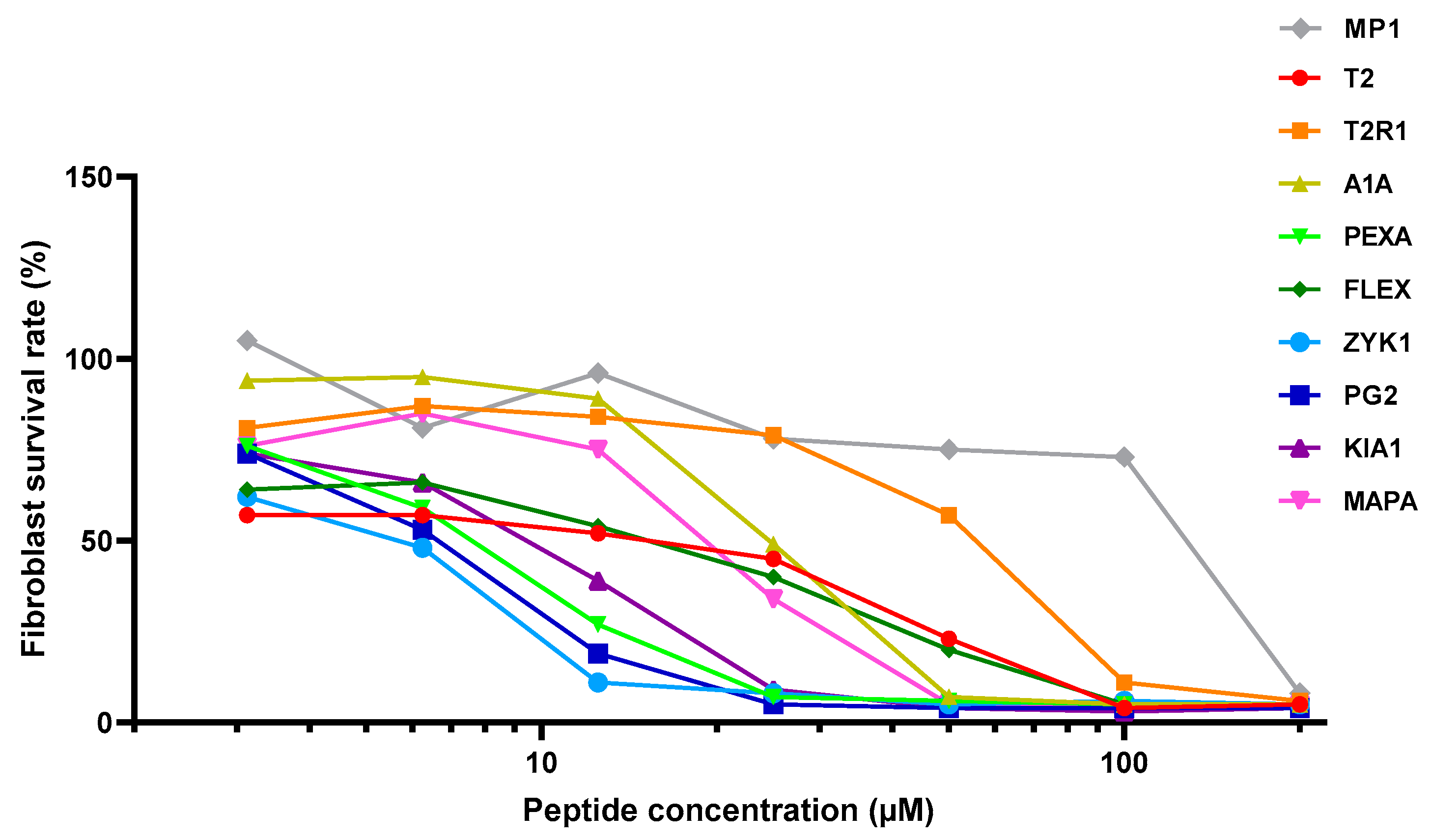
| IC50F (Fibroblasts) | 35 | 80 | 30 | 12 | 30 | 10 | 10 | 15 | 25 | 150 |
| IC50C (PC-3) | 10 | 8 | 12 | 4 | 6.25 | 1.5 | 6 | 15 | 8 | 60 |
| TI (IC50F/IC50C) | 3.5 | 10 | 2.5 | 3 | 4.8 | 6.7 | 1.7 | 1.0 | 3.1 | 2.5 |
| TI (HC20/IC20C) | 1 | 5 | 27 | 6.7 | 25 | 8.6 | 6 | 1 | 1 | 1 |
| TI/ IC50C | 0.35 | 1.25 | 0.21 | 0.75 | 0.77 | 4.47 | 0.28 | 0.07 | 0.39 | 0.04 |
| (TI/ IC50C) vs. MP1 * | 8.4 | 30 | 5 | 18 | 18.5 | 107 | 6.8 | 1.6 | 9.3 | 1.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juretić, D.; Golemac, A.; Strand, D.E.; Chung, K.; Ilić, N.; Goić-Barišić, I.; Pellay, F.-X. The Spectrum of Design Solutions for Improving the Activity-Selectivity Product of Peptide Antibiotics against Multidrug-Resistant Bacteria and Prostate Cancer PC-3 Cells. Molecules 2020, 25, 3526. https://doi.org/10.3390/molecules25153526
Juretić D, Golemac A, Strand DE, Chung K, Ilić N, Goić-Barišić I, Pellay F-X. The Spectrum of Design Solutions for Improving the Activity-Selectivity Product of Peptide Antibiotics against Multidrug-Resistant Bacteria and Prostate Cancer PC-3 Cells. Molecules. 2020; 25(15):3526. https://doi.org/10.3390/molecules25153526
Chicago/Turabian StyleJuretić, Davor, Anja Golemac, Denise E. Strand, Keshi Chung, Nada Ilić, Ivana Goić-Barišić, and François-Xavier Pellay. 2020. "The Spectrum of Design Solutions for Improving the Activity-Selectivity Product of Peptide Antibiotics against Multidrug-Resistant Bacteria and Prostate Cancer PC-3 Cells" Molecules 25, no. 15: 3526. https://doi.org/10.3390/molecules25153526
APA StyleJuretić, D., Golemac, A., Strand, D. E., Chung, K., Ilić, N., Goić-Barišić, I., & Pellay, F.-X. (2020). The Spectrum of Design Solutions for Improving the Activity-Selectivity Product of Peptide Antibiotics against Multidrug-Resistant Bacteria and Prostate Cancer PC-3 Cells. Molecules, 25(15), 3526. https://doi.org/10.3390/molecules25153526