
Noble Gas Bonding Interactions Involving Xenon Oxides and Fluorides


Abstract
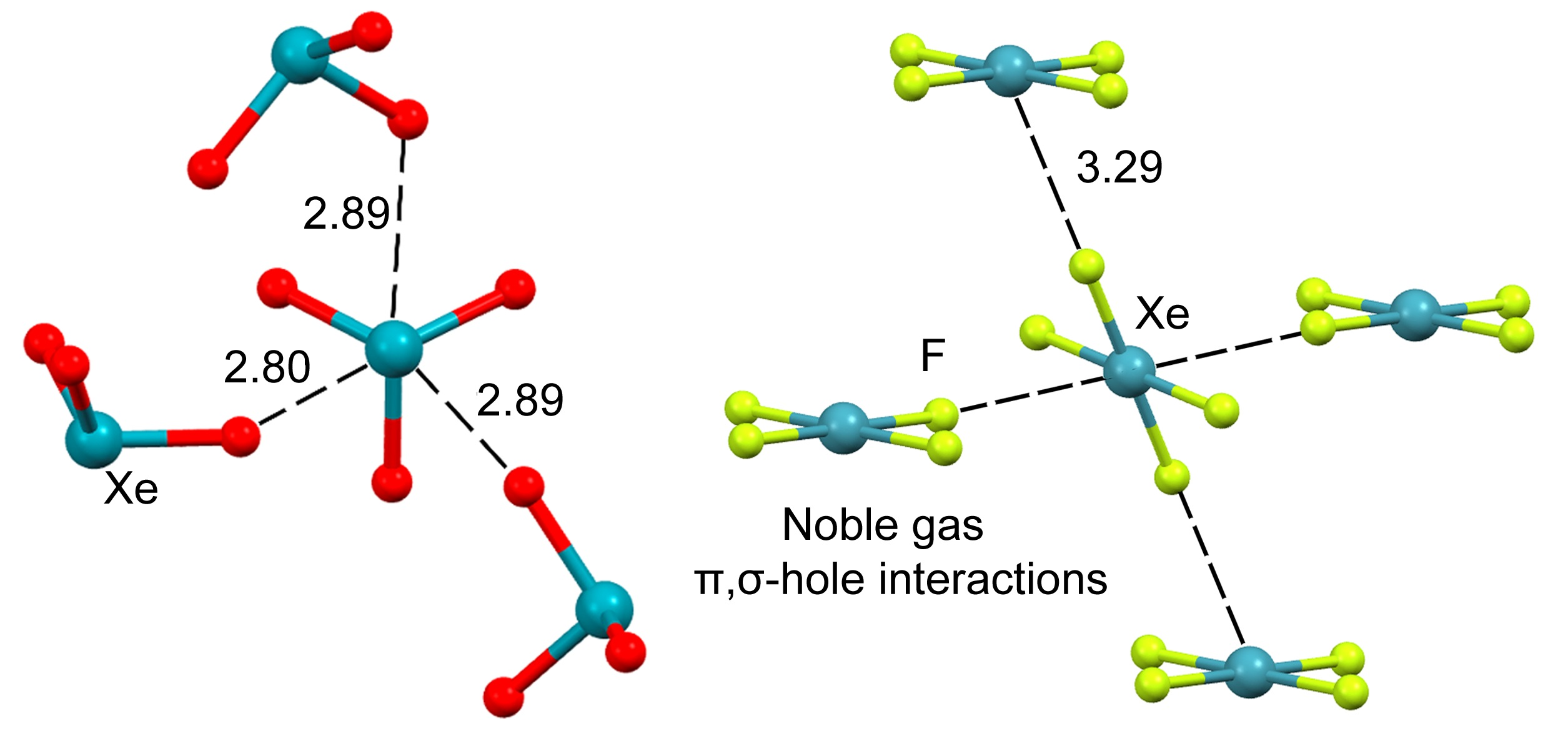
1. Introduction
2. Results
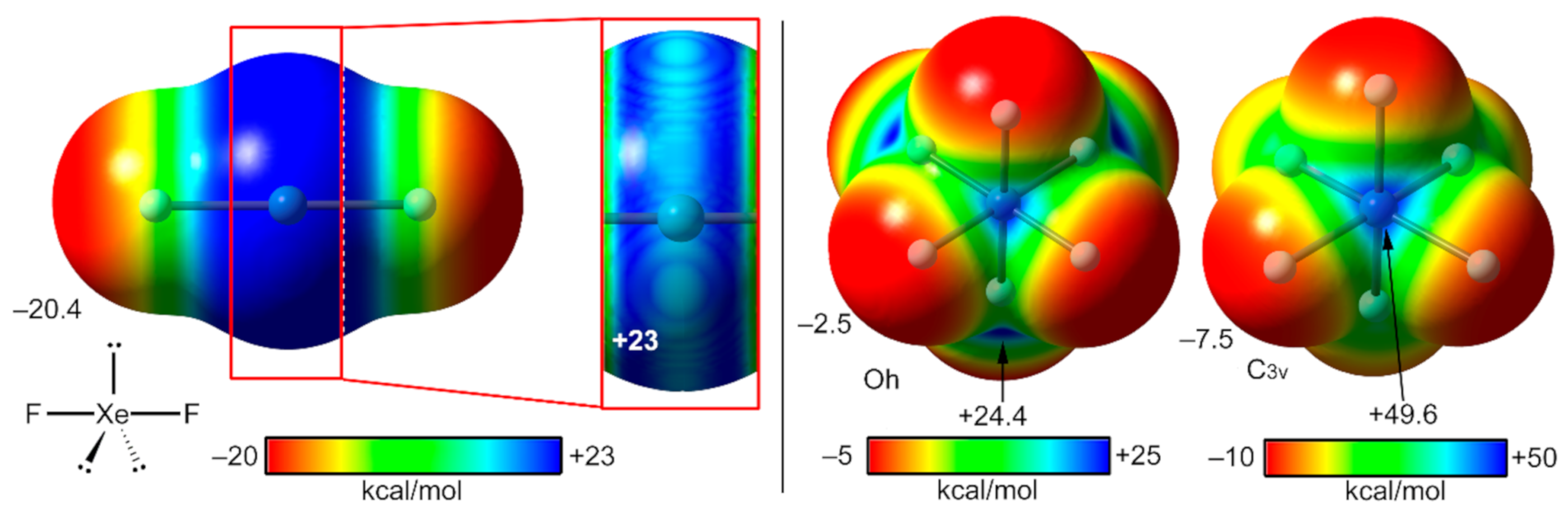
2.1. Pioneering Works and Physical Insights
2.2. Cooperativity
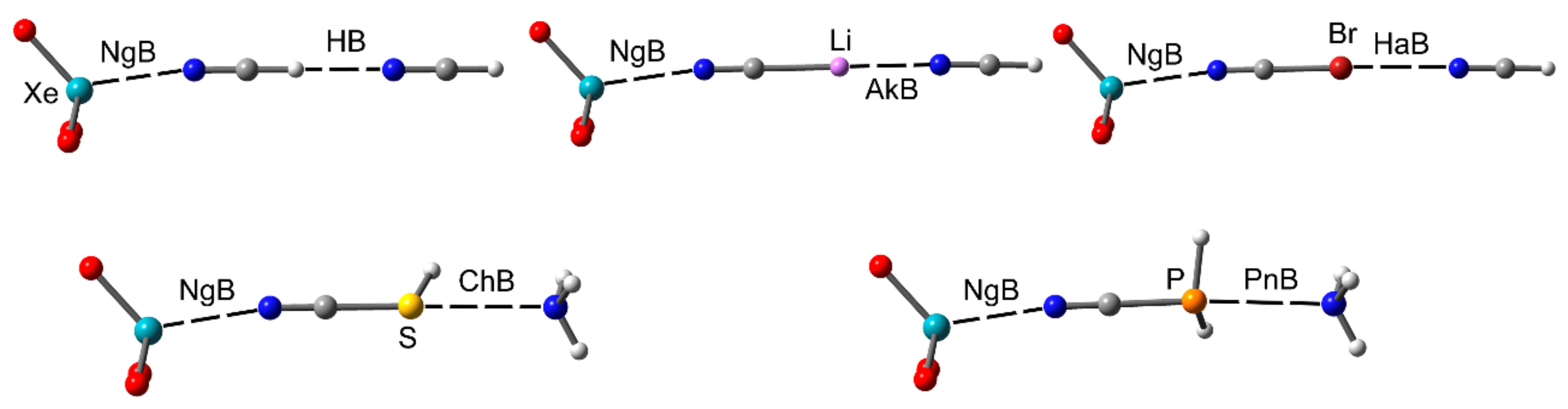
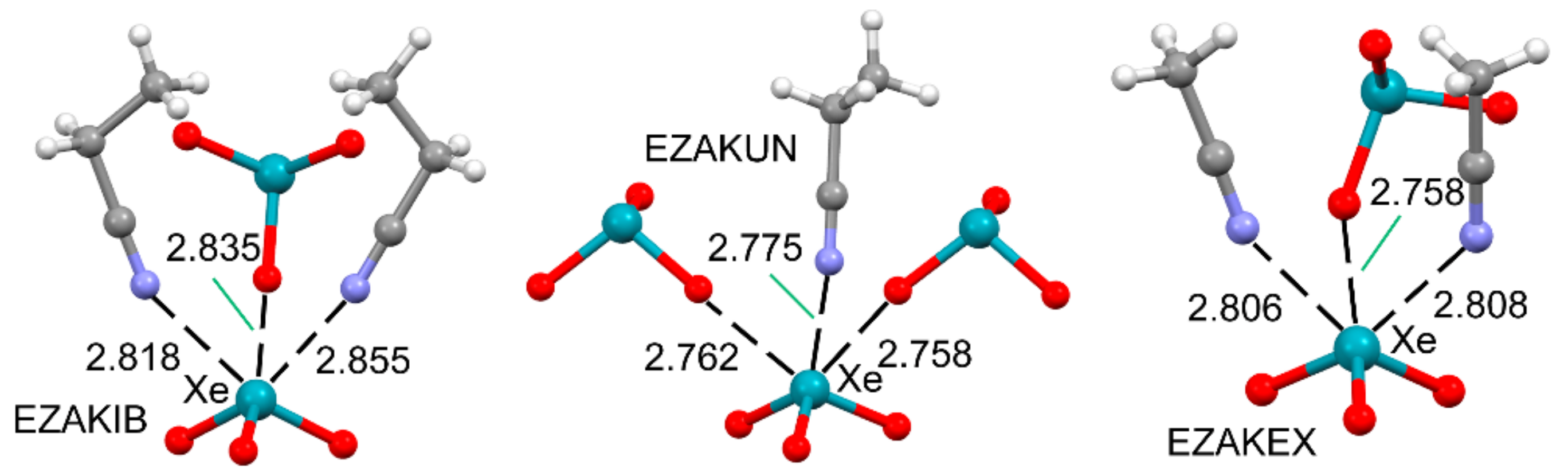
2.2.1. NgBs and H-bonds or Alkali (Lithium) Bonds
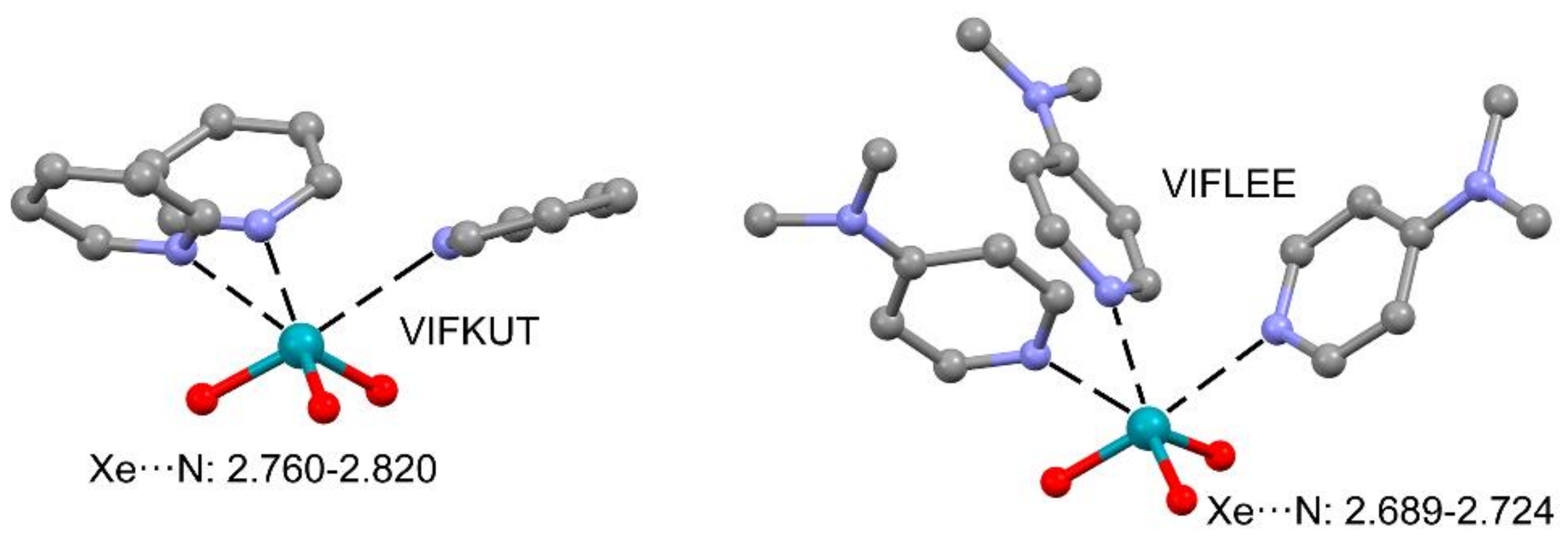
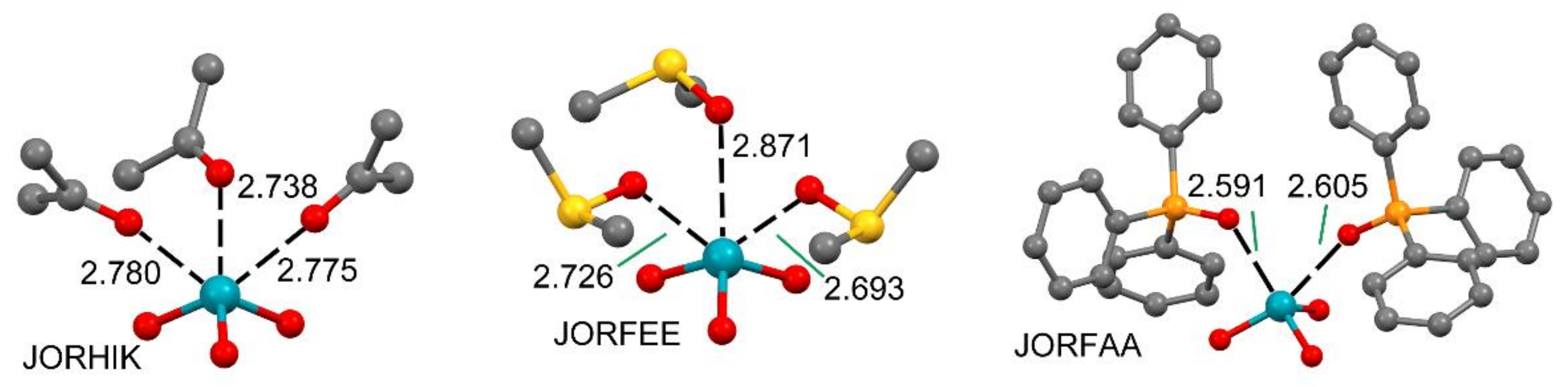
2.2.2. NgBs and ChB and PnB Interactions
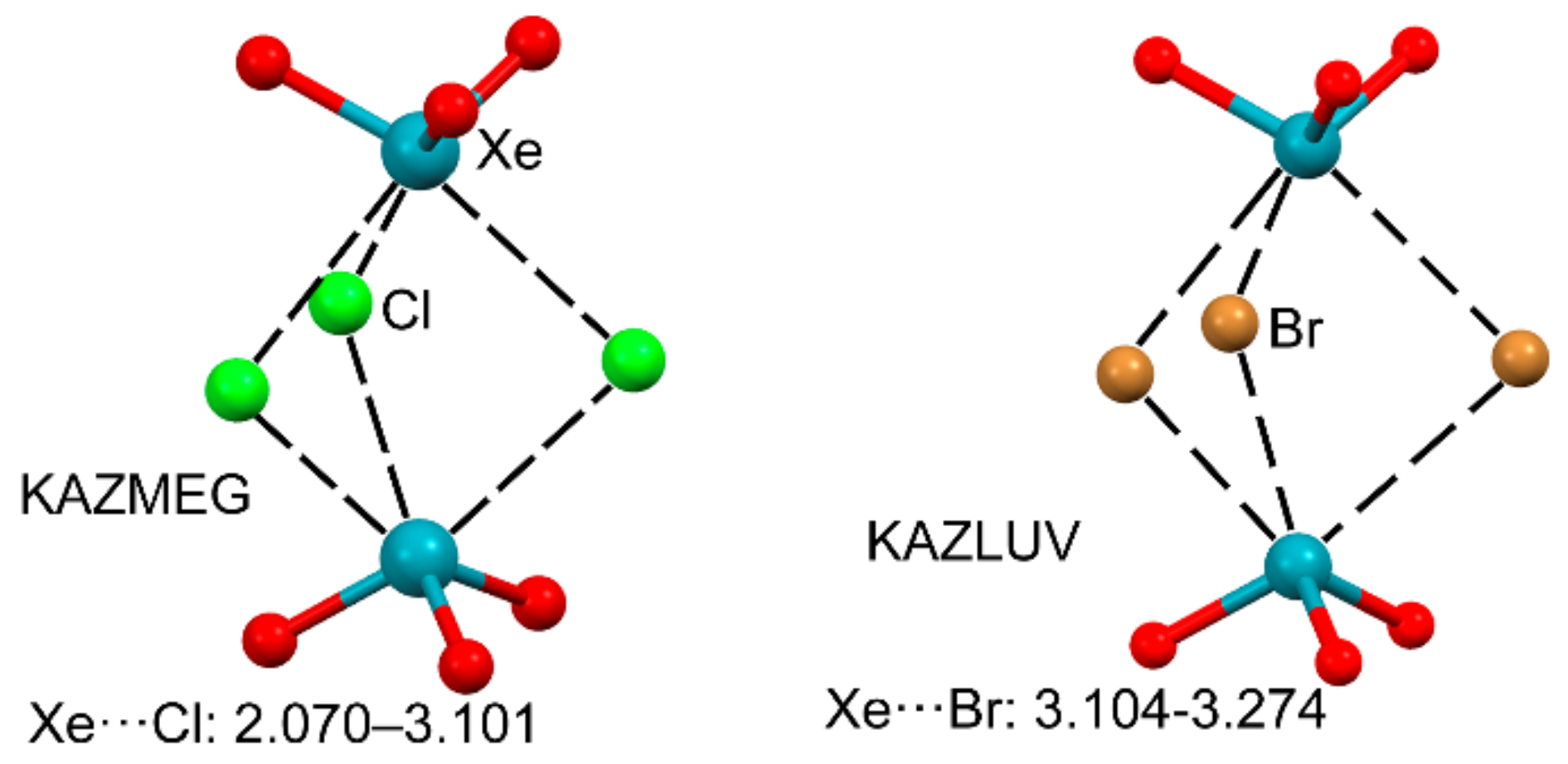
2.2.3. NgB and HaB Interactions
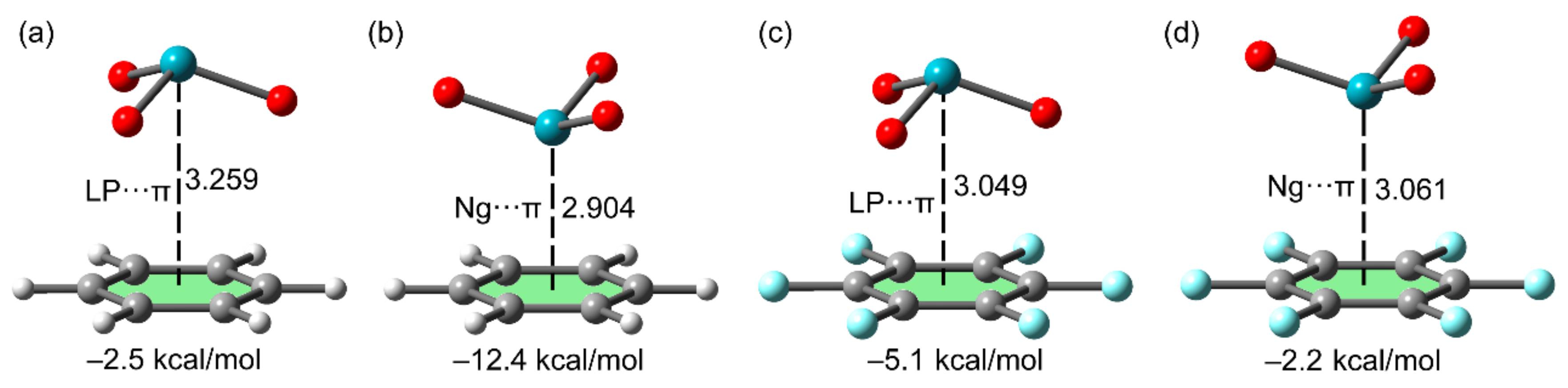
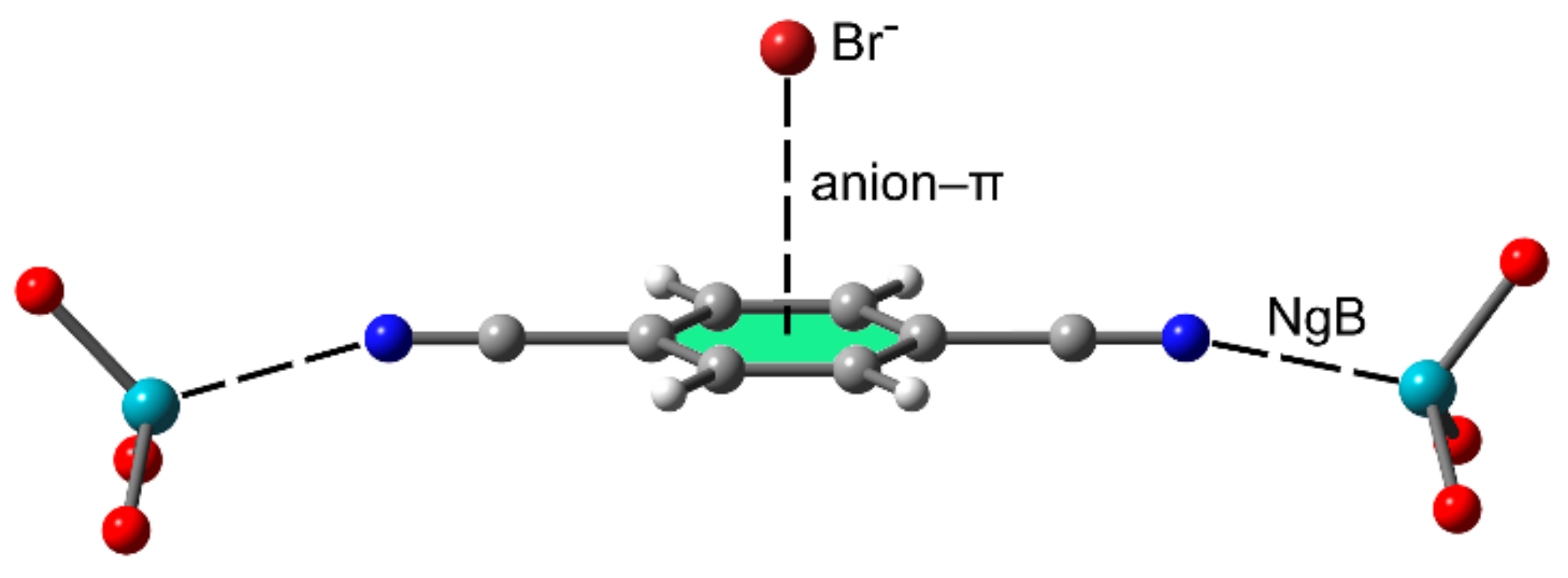
2.2.4. NgBs and Anion⋯π or Lone Pair⋯π Interactions
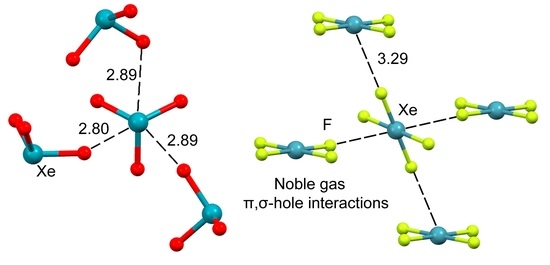
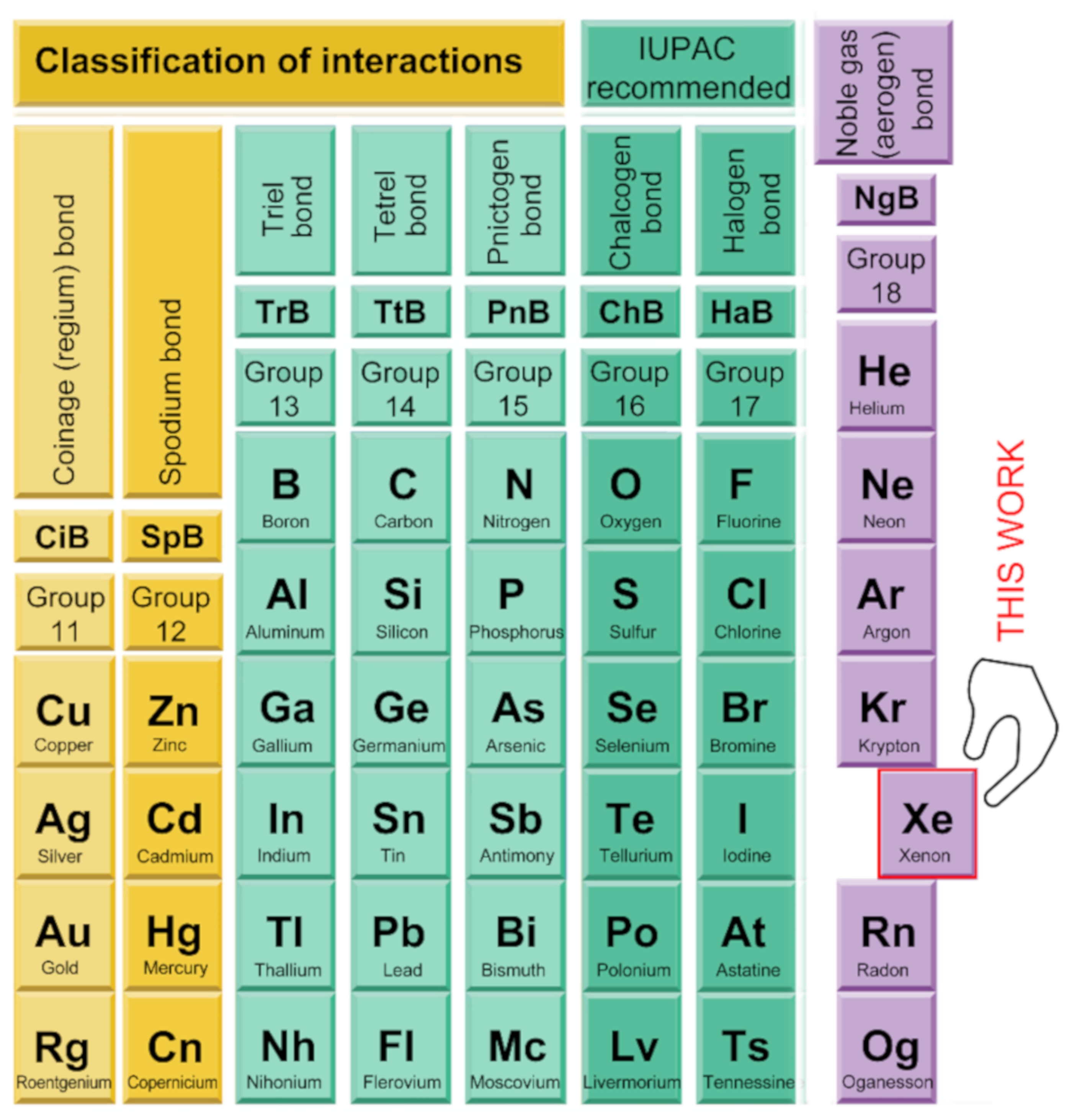
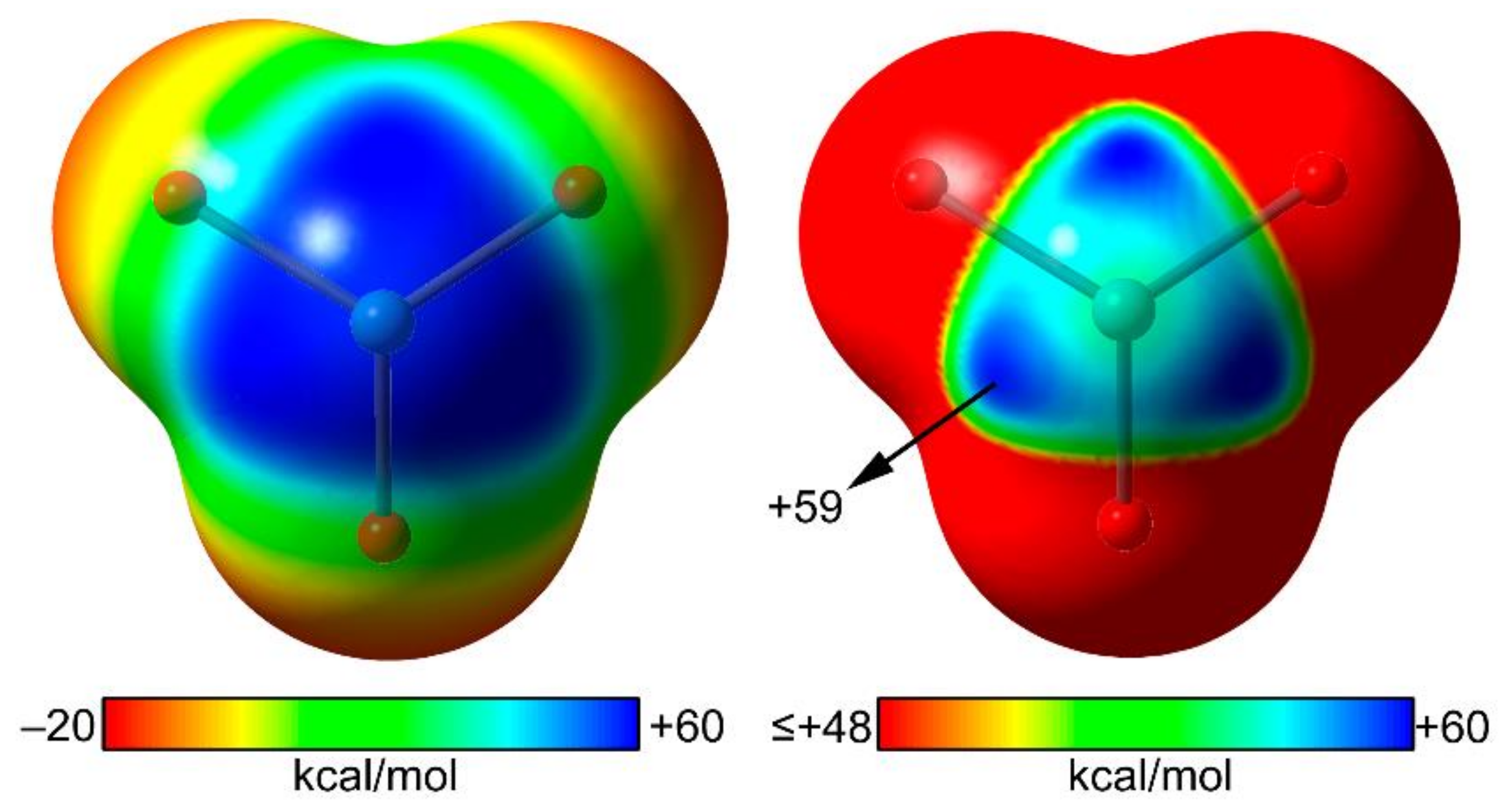
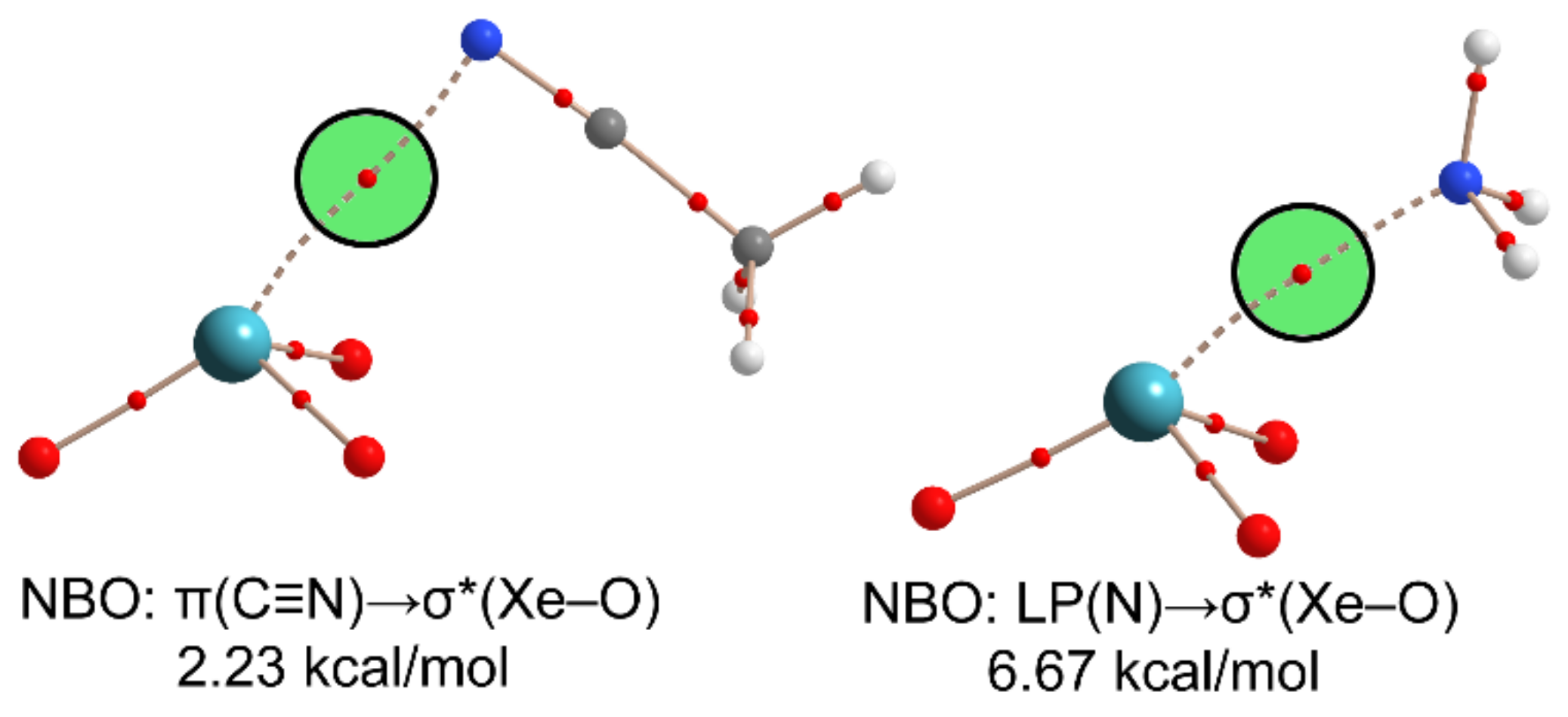
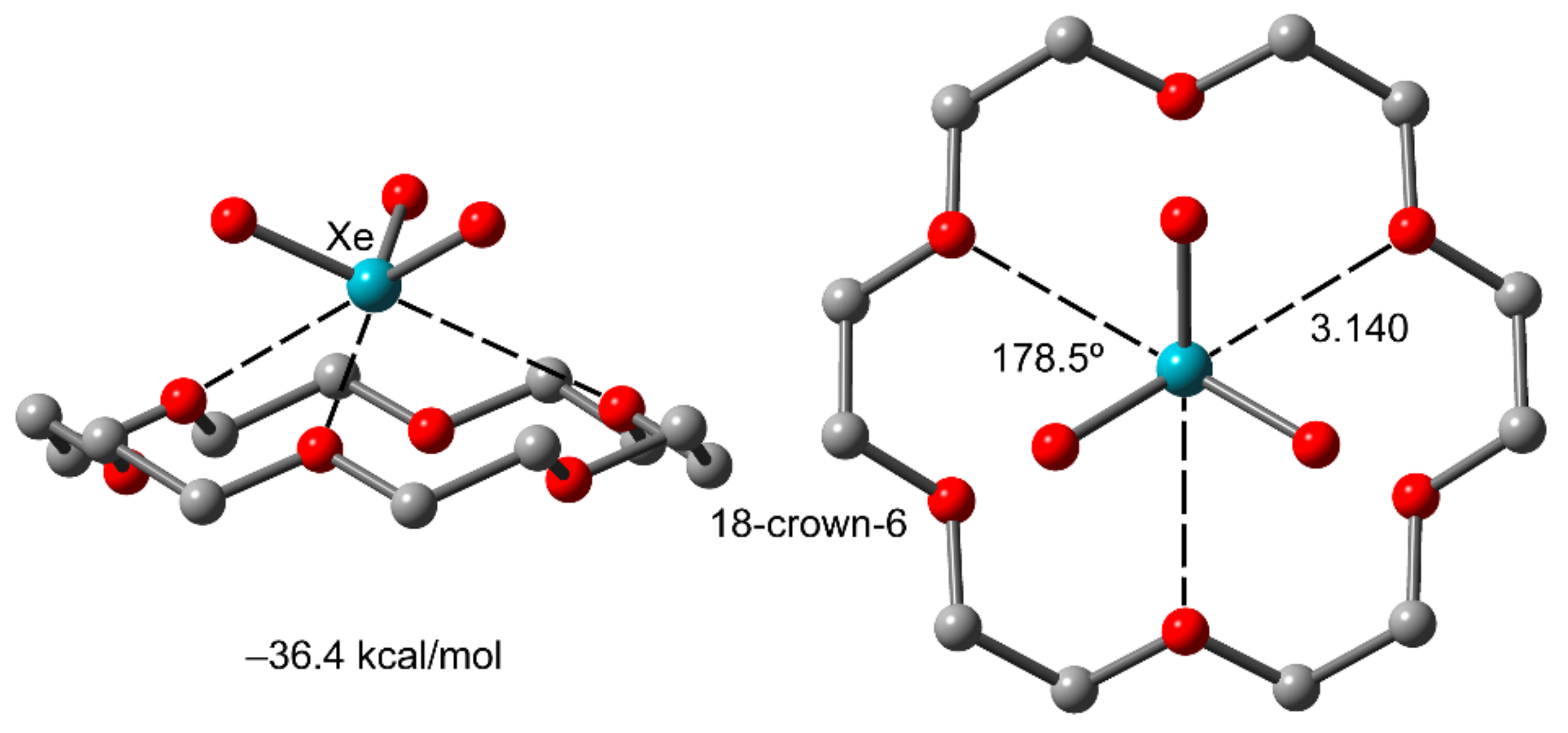
2.3. NgB in XeO3 Adducts
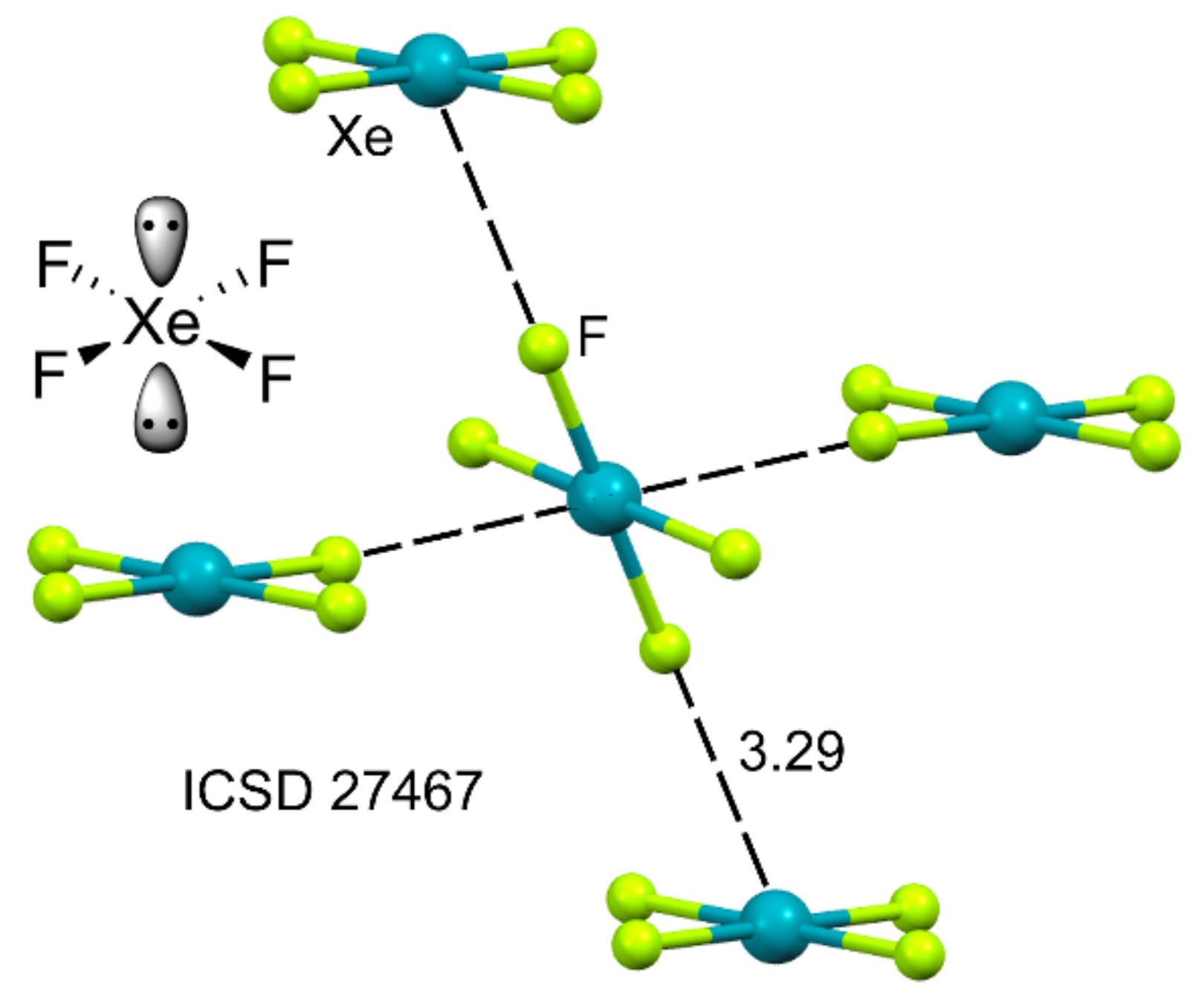
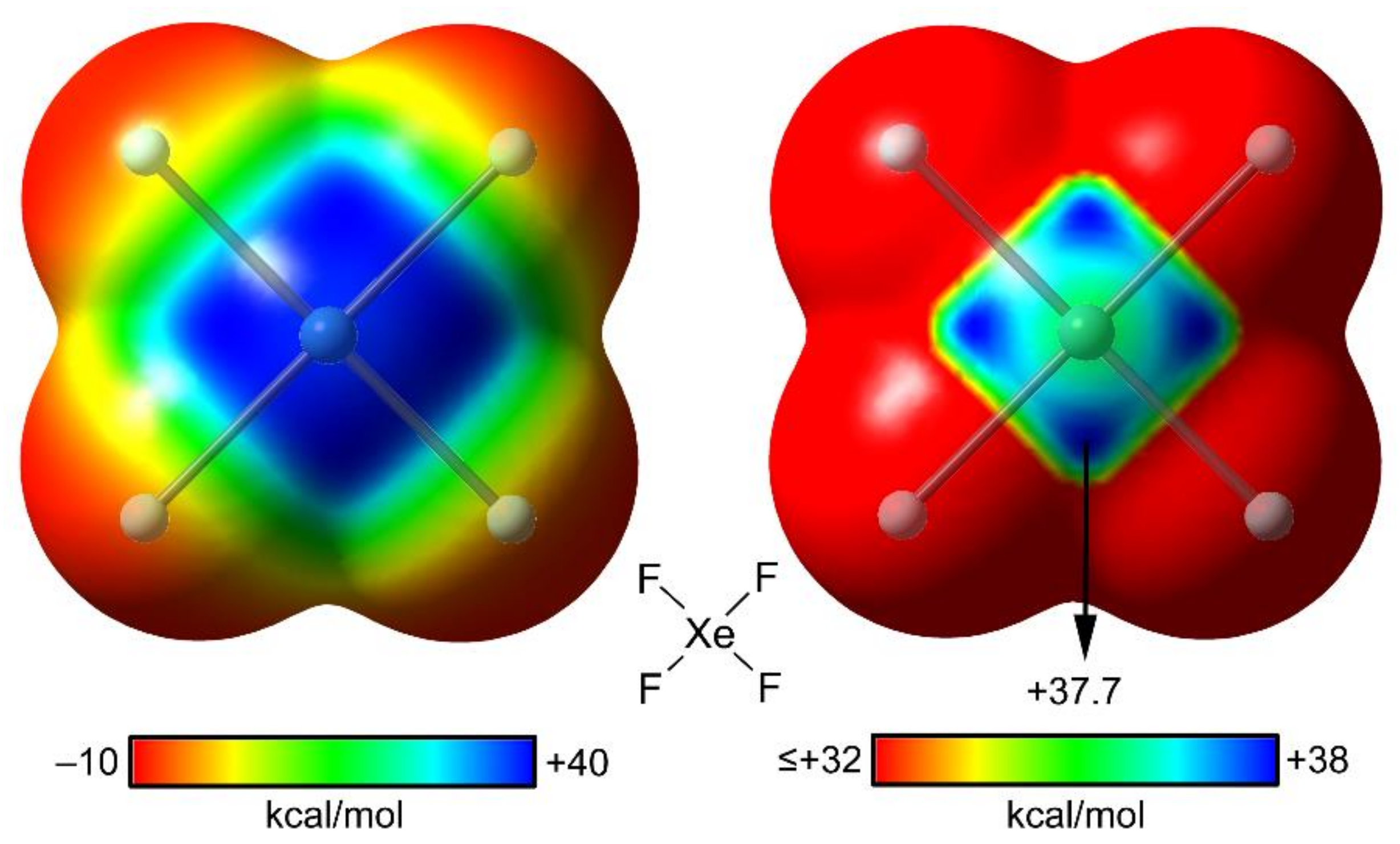
2.4. NgB in XeF2, XeF4, and XeF6
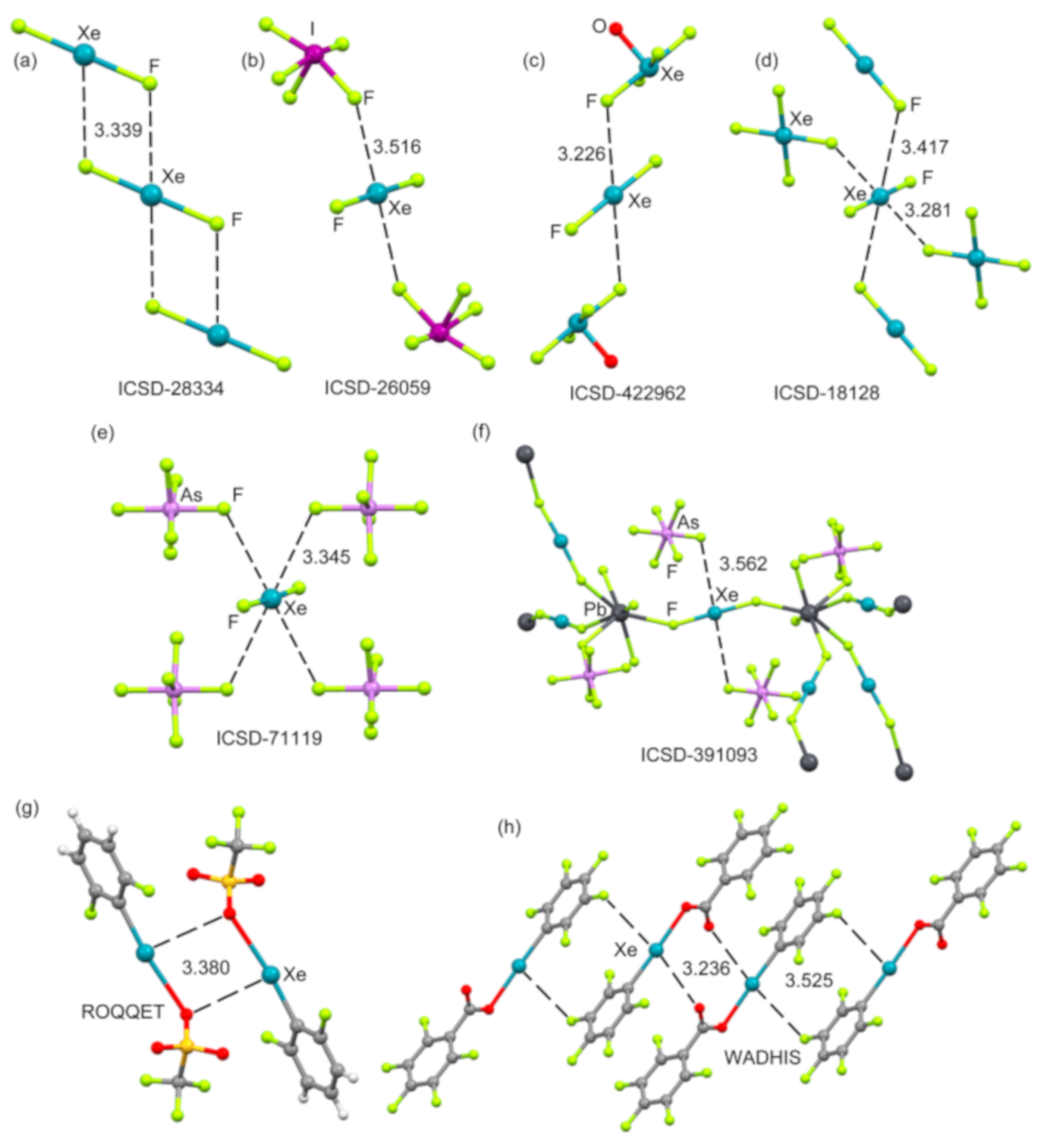
2.4.1. X-Ray Structures of XeF2
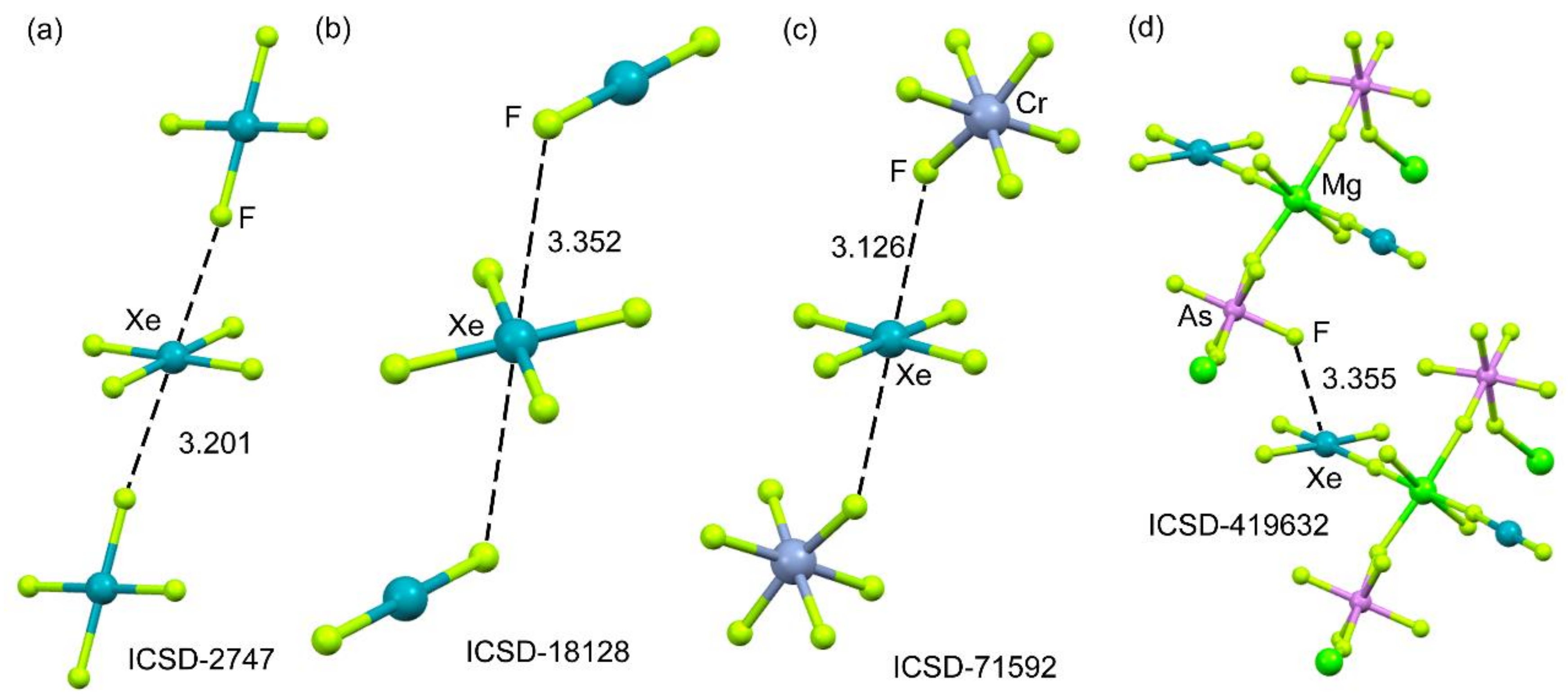
2.4.2. X-Ray Structures of XeF4
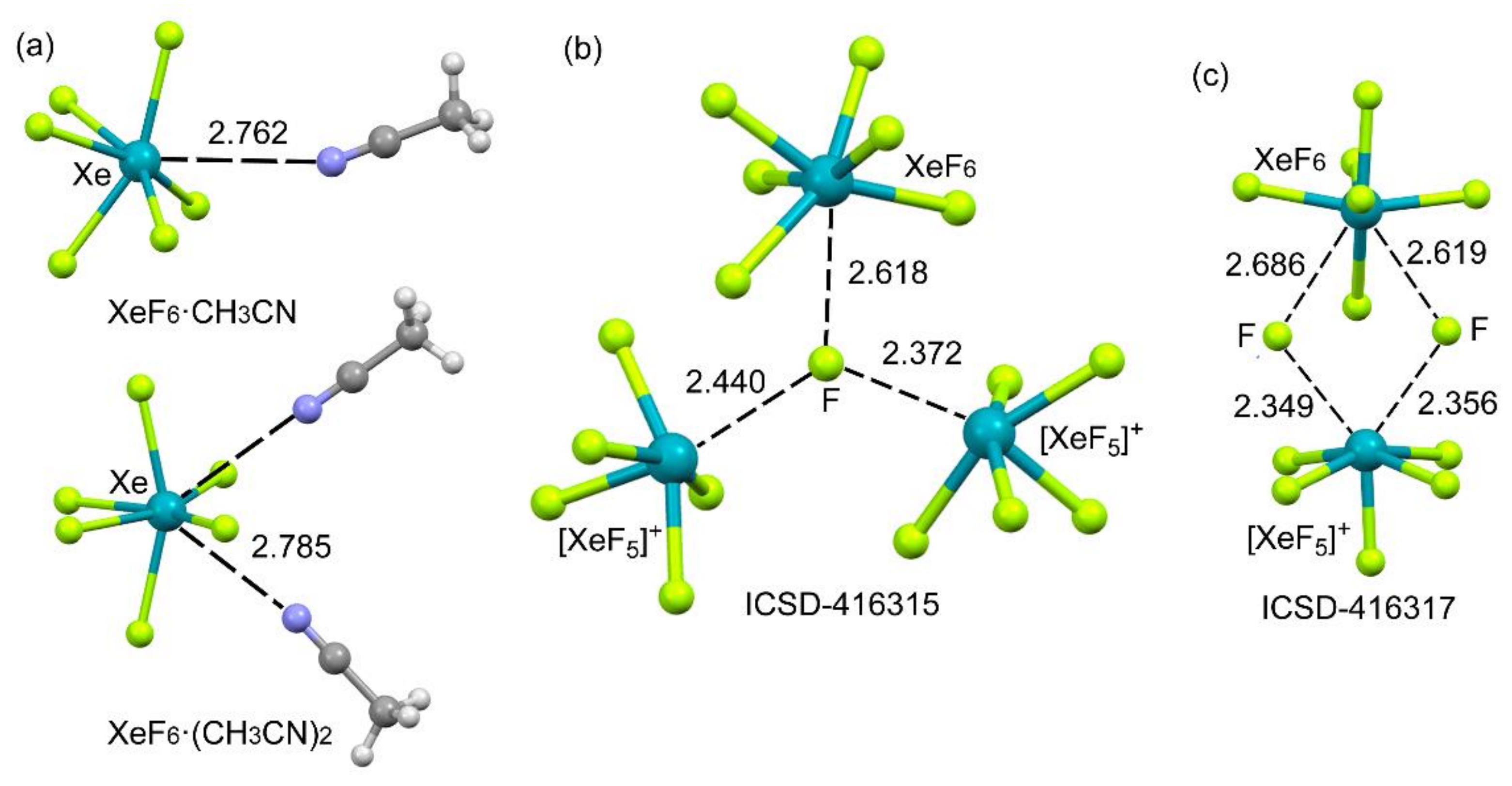
2.4.3. X-Ray Structures of XeF6
3. Concluding Remarks
Funding
Conflicts of Interest
References
- Busschaert, N.; Caltagirone, C.; Van Rossom, W.; Gale, P.A. Applications of Supramolecular Anion Recognition. Chem. Rev. 2015, 115, 8038–8155. [Google Scholar] [CrossRef] [PubMed]
- Evans, N.H.; Beer, P.D. Advances in Anion Supramolecular Chemistry: From Recognition to Chemical Applications. Angew. Chem. Int. Ed. 2014, 53, 11716–11754. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Noncovalent Forces; Springer: Cham, Switzerland, 2015. [Google Scholar]
- Alkorta, I.; Elguero, J.; Frontera, A. Not Only Hydrogen Bonds: Other Noncovalent Interactions. Crystals 2020, 10, 180. [Google Scholar] [CrossRef]
- Desiraju, G.R. Crystal Engineering: From Molecule to Crystal. J. Am. Chem. Soc. 2013, 135, 9952–9967. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.-J. Binding Mechanisms in Supramolecular Complexes. Angew. Chem. Int. Ed. 2009, 48, 3924–3977. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.-J.; Yatsimirski, A. Principles and Methods in Supramolecular Chemistry; Wiley: Chichester, UK, 2000. [Google Scholar]
- Desiraju, G.R.; Steiner, T. The weak hydrogen bond in structural chemistry and biology. In IUCr Monographs on Crystallography; Oxford University Press/International Union of Crystallography: Oxford, UK, 1999; Volume 9. [Google Scholar]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef]
- Tepper, R.; Schubert, U.S. Halogen Bonding in Solution: Anion Recognition, Templated Self-Assembly, and Organocatalysis. Angew. Chem. Int. Ed. 2018, 57, 6004–6016. [Google Scholar] [CrossRef]
- Frontera, A.; Gamez, P.; Mascal, M.; Mooibroek, T.J.; Reedijk, J. Putting Anion-π Interactions into Perspective. Angew. Chem. Int. Ed. 2011, 50, 9564–9583. [Google Scholar] [CrossRef]
- Gamez, P.; Mooibroek, T.J.; Teat, S.J.; Reedijk, J. Anion binding involving π-acidic heteroaromatic rings. Acc. Chem. Res. 2007, 40, 435–444. [Google Scholar] [CrossRef]
- Quiñonero, D.; Garau, C.; Rotger, C.; Frontera, A.; Ballester, P.; Costa, A.; Deyà, P.; Rotger, C. Anion–π Interactions: Do They Exist? Angew. Chem. Int. Ed. 2002, 41, 3389–3392. [Google Scholar] [CrossRef]
- Meyer, E.A.; Castellano, R.K.; Diederich, F. Interactions with aromatic rings in chemical and biological recognition. Angew. Chem. Int. Ed. 2003, 42, 1210–1250. [Google Scholar] [CrossRef] [PubMed]
- Neel, A.J.; Hilton, M.J.; Sigman, M.S.; Toste, F.D. Exploiting non-covalent π interactions for catalyst design. Nature 2017, 543, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhao, C.; Huang, J.; Leung, L.R.; Qian, Y.; Yu, H.; Huang, L.; Kalashnikova, O.V. Trans-pacific transport and evolution of aerosols: Evaluation of quasi global WRF-Chem simulation with multiple observations. Eur. J. 2016, 22, 14434–14450. [Google Scholar] [CrossRef]
- Benz, S.; López-Andarias, J.; Mareda, J.; Sakai, N.; Matile, S. Catalysis with chalcogen bonds. Angew. Chem. Int. Ed. 2017, 56, 812–815. [Google Scholar] [CrossRef]
- Benz, S.; Poblador-Bahamonde, A.I.; Low-Ders, N.; Matile, S. Catalysis with pnictogen, chalcogen, and halogen bonds. Angew. Chem. Int. Ed. 2018, 57, 5408–5412. [Google Scholar] [CrossRef]
- Lee, L.M.; Tsemperouli, M.; Poblador-Bahamonde, A.I.; Benz, S.; Sakai, N.; Sugihara, K.; Matile, S.J. Anion Transport with Pnictogen Bonds in Direct Comparison with Chalcogen and Halogen Bonds. Am. Chem. Soc. 2019, 141, 810–814. [Google Scholar] [CrossRef]
- Macchione, M.; Goujon, A.; Strakova, K.; Humeniuk, H.V.; Licari, G.; Tajkhorshid, E.; Sakai, N.; Matile, S. A Chalcogen-Bonding Cascade Switch for Planarizable Push–Pull Probes. Angew. Chem. Int. Ed. 2019, 58, 15752–15756. [Google Scholar] [CrossRef]
- Legon, A.C. Tetrel, pnictogen and chalcogen bonds identified in the gas phase before they had names: A systematic look at non-covalent interactions. Phys. Chem. Chem. Phys. 2017, 19, 14884–14896. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. The Bright Future of Unconventional σ/π-Hole Interactions. ChemPhysChem 2015, 16, 2496–2517. [Google Scholar] [CrossRef]
- Gilday, L.C.; Robinson, S.W.; Barendt, T.A.; Langton, M.J.; Mullaney, B.R.; Beer, P.D. Halogen bonding in supramolecular chemistry. Chem. Rev. 2015, 115, 7118–7195. [Google Scholar] [CrossRef]
- Troff, R.W.; Makela, T.; Topic, F.; Valkonen, A.; Raatikainen, K.; Rissanen, K. Alternative motifs for halogen bonding. Eur. J. Org. Chem. 2013, 2013, 1617–1637. [Google Scholar] [CrossRef]
- Kolář, M.H.; Hobza, P. Computer Modeling of Halogen Bonds and Other σ-Hole Interactions. Chem. Rev. 2016, 116, 5155–5187. [Google Scholar] [CrossRef] [PubMed]
- Sure, R.; Grimme, S. Halogen bonded supramolecular capsules: A challenging test case for quantum chemical methods. Chem. Commun. 2016, 52, 9893–9896. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S. Halogen bonding: An interim discussion. ChemPhysChem 2013, 14, 278–294. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding: An electrostatically-driven highly directional noncovalent interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef]
- Bauza, A.; Mooibroek, T.J.; Frontera, A. Tetrel Bonding Interactions. Chem. Rec. 2016, 16, 473–487. [Google Scholar] [CrossRef]
- Grabowski, S.J. Tetrel bond–σ-hole bond as a preliminary stage of the SN2 reaction. Phys. Chem. Chem. Phys. 2014, 16, 1824–1834. [Google Scholar] [CrossRef]
- Daolio, A.; Scilabra, P.; Terraneo, G.; Resnati, G. C(sp3) atoms as tetrel bond donors: A crystallographic survey. Coord. Chem. Rev. 2020, 413, 213265. [Google Scholar] [CrossRef]
- Bauza, A.; Mooibroek, T.J.; Frontera, A. Small Cycloalkane (CN)2C–C(CN)2 Structures Are Highly Directional Non-covalent Carbon-Bond Donors. Chem. A Eur. J. 2014, 20, 10245–10248. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel-Bonding Interaction: Rediscovered Supramolecular Force. Angew. Chem. Int. Ed. 2013, 52, 12317–12321. [Google Scholar] [CrossRef] [PubMed]
- Southern, S.A.; Errulat, D.; Frost, J.M.; Gabidullin, B.; Bryce, D.L. Prospects for 207Pb solid-state NMR studies of lead tetrel bonds. Faraday Discuss. 2017, 203, 165–186. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Seth, S.K.; Frontera, A. Tetrel bonding interactions at work: Impact on tin and lead coordination compounds. Coord. Chem. Rev. 2019, 384, 107–125. [Google Scholar] [CrossRef]
- Roeleveld, J.J.; Deprez, S.J.L.; Verhoofstad, A.; Frontera, A.; Van Der Vlugt, I.J.I.; Mooibroek, T.J. Engineering Crystals Using sp3-C Centred Tetrel Bonding Interactions. Chem. A Eur. J. 2020, 26. [Google Scholar] [CrossRef]
- Bauza, A.; Mooibroek, T.J.; Frontera, A. Non-covalent sp3 carbon bonding with ArCF3 is analogous to CH–π interactions. Chem. Commun. 2014, 50, 12626–12629. [Google Scholar] [CrossRef] [PubMed]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. The Pnicogen Bond in Review: Structures, Binding Energies, Bonding Properties, and Spin-Spin Coupling Constants of Complexes Stabilized by Pnicogen Bonds. In Noncovalent Forces: Challenges and Advances in Computational Chemistry and Physics, 1st ed.; Scheiner, S., Ed.; Springer: New York, NY, USA, 2015; Volume 19, pp. 191–263. [Google Scholar]
- Setiawan, D.; Kraka, E.; Cremer, D. Strength of the Pnicogen Bond in Complexes Involving Group Va Elements N, P, and As. J. Phys. Chem. A 2014, 119, 1642–1656. [Google Scholar] [CrossRef]
- Sarkar, S.; Pavan, M.S.; Row, T.N.G. Experimental validation of ‘pnicogen bonding’ in nitrogen by charge density analysis. Phys. Chem. Chem. Phys. 2015, 17, 2330–2334. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Gurbanov, A.V.; Aliyeva, V.A.; Resnati, G.; Pombeiro, A.J.L. Pnictogen bonding in coordination chemistry. Coord. Chem. Rev. 2020, 418, 213381. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. Pnicogen-Bonded Anionic Complexes. J. Phys. Chem. A 2014, 118, 3386–3392. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Janjić, G.V.; Zarić, S. σ-Hole Interactions of Covalently-Bonded Nitrogen, Phosphorus and Arsenic: A Survey of Crystal Structures. Crystals 2014, 4, 12–31. [Google Scholar] [CrossRef]
- Eskandari, K.; Mahmoodabadi, N. Pnicogen bonds: A theoretical study based on the Laplacian of electron density. J. Phys. Chem. A 2013, 117, 13018–13024. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. The Pnicogen Bond: Its Relation to Hydrogen, Halogen, and Other Noncovalent Bonds. Acc. Chem. Res. 2012, 46, 280–288. [Google Scholar] [CrossRef]
- Scheiner, S. Detailed comparison of the pnicogen bond with chalcogen, halogen, and hydrogen bonds. Int. J. Quantum Chem. 2012, 113, 1609–1620. [Google Scholar] [CrossRef]
- Adhikari, U.; Scheiner, S. Comparison of P⋯D (D = P,N) with other noncovalent bonds in molecular aggregates. J. Chem. Phys. 2011, 135, 184306. [Google Scholar] [CrossRef]
- Zahn, S.; Frank, R.; Hey-Hawkins, E.; Kirchner, B. Pnicogen Bonds: A New Molecular Linker? Chem. A Eur. J. 2011, 17, 6034–6038. [Google Scholar] [CrossRef] [PubMed]
- Aakeroy, C.B.; Bryce, D.L.; Desiraju, G.R.; Frontera, A.; Legon, A.C.; Nicotra, F.; Rissanen, K.; Scheiner, S.; Terraneo, G.; Metrangolo, P.; et al. Definition of the chalcogen bond (IUPAC Recommendations 2019). Pure Appl. Chem. 2019, 91, 1889–1892. [Google Scholar] [CrossRef]
- Scilabra, P.; Terraneo, G.; Resnati, G. The Chalcogen Bond in Crystalline Solids: A World Parallel to Halogen Bond. Acc. Chem. Res. 2019, 52, 1313–1324. [Google Scholar] [CrossRef] [PubMed]
- Vogel, L.; Wonner, P.; Huber, S.M. Chalcogen Bonding: An Overview. Angew. Chem. Int. Ed. 2018, 58, 1880–1891. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhu, H.; Liu, S.; Zhao, Z.; Zhang, L.; Hao, J.; Wang, Y. Chalcogen–Chalcogen Bonding Catalysis Enables Assembly of Discrete Molecules. J. Am. Chem. Soc. 2019, 141, 9175–9179. [Google Scholar] [CrossRef]
- Borissov, A.; Marques, I.; Lim, J.Y.C.; Félix, V.; Smith, M.D.; Beer, P.D. Anion Recognition in Water by Charge-Neutral Halogen and Chalcogen Bonding Foldamer Receptors. J. Am. Chem. Soc. 2019, 141, 4119–4129. [Google Scholar] [CrossRef]
- Shukla, R.; Chopra, D. “Pnicogen bonds” or “chalcogen bonds”: Exploiting the effect of substitution on the formation of P⋯Se noncovalent bonds. Phys. Chem. Chem. Phys. 2016, 18, 13820–13829. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.S. Anion recognition based on halogen, chalcogen, pnictogen and tetrel bonding. Coord. Chem. Rev. 2020, 413, 213270. [Google Scholar] [CrossRef]
- Azofra, L.M.; Alkorta, I.; Scheiner, S. Chalcogen Bonds in Complexes of SOXY (X, Y = F, Cl) with Nitrogen Bases. J. Phys. Chem. A 2015, 119, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Biot, N.; Bonifazi, D. Chalcogen-bond driven molecular recognition at work. Coord. Chem. Rev. 2020, 413, 213243. [Google Scholar] [CrossRef]
- Fourmigue, M.; Dhaka, A. Chalcogen bonding in crystalline diselenides and selenocyanates: From molecules of pharmaceutical interest to conducting materials. Coord. Chem. Rev. 2020, 403, 213084. [Google Scholar] [CrossRef]
- Nziko, V.D.P.N.; Scheiner, S. Intramolecular S⋯O Chalcogen Bond as Stabilizing Factor in Geometry of Substituted Phenyl-SF3 Molecules. J. Org. Chem. 2015, 80, 2356–2363. [Google Scholar] [CrossRef]
- Garrett, G.E.; Gibson, G.L.; Straus, R.N.; Seferos, D.S.; Taylor, M.S. Chalcogen Bonding in Solution: Interactions of Benzotelluradiazoles with Anionic and Uncharged Lewis Bases. J. Am. Chem. Soc. 2015, 137, 4126–4133. [Google Scholar] [CrossRef]
- Thomas, S.P.; Satheeshkumar, K.; Mugesh, G.; Row, T.N.G. Unusually Short Chalcogen Bonds Involving Organoselenium: Insights into the Se-N Bond Cleavage Mechanism of the Antioxidant Ebselen and Analogues. Chem. A Eur. J. 2015, 21, 6793–6800. [Google Scholar] [CrossRef]
- Shukla, R.; Chopra, D. Exploring the Role of Substitution on the Formation of Se⋯O/N Noncovalent Bonds. J. Phys. Chem. B 2015, 119, 14857–14870. [Google Scholar] [CrossRef]
- Adhikari, U.; Scheiner, S. Effects of Charge and Substituent on the S⋯N Chalcogen Bond. J. Phys. Chem. A 2014, 118, 3183–3192. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Sato, N. Origin of Attraction in Chalgogen–Nitrogen Interaction of 1,2,5-Chalcogenadiazole Dimers. J. Phys. Chem. B 2013, 117, 6849–6855. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Lane, P.; Politzer, P. Simultaneous σ-hole and hydrogen bonding by sulfur- and selenium-containing heterocycles. Int. J. Quantum Chem. 2008, 108, 2770–2781. [Google Scholar] [CrossRef]
- Wang, W.; Ji, B.; Zhang, Y. Chalcogen Bond: A Sister Noncovalent Bond to Halogen Bond. J. Phys. Chem. A 2009, 113, 8132–8135. [Google Scholar] [CrossRef] [PubMed]
- Franconetti, A.; Quiñonero, D.; Frontera, A.; Resnati, G. Unexpected chalcogen bonds in tetravalent sulfur compounds. Phys. Chem. Chem. Phys. 2019, 21, 11313–11319. [Google Scholar] [CrossRef]
- Tellinghuisen, J. Spectroscopic studies of diatomic noble gas halides. II. Analysis of bound-free emission from XeBr, XeI, and KrF. J. Chem. Phys. 1976, 65, 4473. [Google Scholar] [CrossRef]
- Tellinghuisen, J.; Tellinghuisen, P.C.; Tisone, G.C.; Hoffman, J.M.; Hays, A.K. Spectroscopic studies of diatomic noble gas halides. III. Analysis of XeF 3500 Å band system. J. Chem. Phys. 1978, 68, 5177. [Google Scholar] [CrossRef]
- Tellinghuisen, P.C.; Tellinghuisen, J.; Coxon, J.A.; Velazco, J.E.; Setser, D.W. Spectroscopic studies of diatomic noble gas halides. IV. Vibrational and rotational constants for the X, B, and D states of XeF. J. Chem. Phys. 1978, 68, 5187–5198. [Google Scholar] [CrossRef]
- Kolts, J.H.; Setser, D.W. Rate constants for argon fluoride (ArF*) formation from reactions of argon(3P2,0) with fluorine-containing molecules and the pressure dependence of the C to B state ratios for argon fluoride (ArF*) krypton fluoride (KrF*), and xenon fluoride (XeF*). J. Phys. Chem. 1978, 82, 1766–1768. [Google Scholar] [CrossRef]
- Becker, C.H.; Casavecchia, P.; Lee, Y.T. Crossed molecular beam studies on the interaction potential for F(2P)+Xe(1S). J. Chem. Phys. 1978, 69, 2377–2381. [Google Scholar] [CrossRef]
- Becker, C.H.; Casavecchia, P.; Lee, Y.T. Crossed molecular beam studies on the interaction potentials for F(2P) + Ne,Ar,Kr(1S). J. Chem. Phys. 1979, 70, 2986. [Google Scholar] [CrossRef]
- Krauss, M. The electronic structure of rare gas halide excimers. J. Chem. Phys. 1977, 67, 1712. [Google Scholar] [CrossRef]
- Dunning, T.H.; Hay, P.J. Low-lying electronic states of the rare gas oxides. J. Chem. Phys. 1977, 66, 3767. [Google Scholar] [CrossRef]
- Dunning, T.H.; Hay, P.J. The covalent and ionic states of the rare gas monofluorides. J. Chem. Phys. 1978, 69, 134. [Google Scholar] [CrossRef]
- Aquilanti, V.; Candori, R.; Pirani, F. Molecular beam studies of weak interactions for open-shell systems: The ground and lowest excited states of rare gas oxides. J. Chem. Phys. 1988, 89, 6157–6164. [Google Scholar] [CrossRef]
- Aquilanti, V.; Luzzatti, E.; Pirani, F.; Volpi, G.G. Molecular beam studies of weak interactions for open-shell systems: The ground and lowest excited states of ArF, KrF, and XeF. J. Chem. Phys. 1988, 89, 6165–6175. [Google Scholar] [CrossRef]
- Pirani, F.; Maciel, G.S.; Cappelletti, D.M.; Aquilanti, V. Experimental benchmarks and phenomenology of interatomic forces: Open-shell and electronic anisotropy effects. Int. Rev. Phys. Chem. 2006, 25, 165–199. [Google Scholar] [CrossRef]
- Bauzá, A.; Frontera, A. Aerogen bonding interaction: A new supramolecular force? Angew. Chem. Int. Ed. 2015, 54, 7340–7343. [Google Scholar] [CrossRef]
- Goettel, J.T.; Matsumoto, K.; Mercier, H.P.A.; Schrobilgen, G.J. Syntheses and Structures of Xenon Trioxide Alkylnitrile Adducts. Angew. Chem. Int. Ed. 2016, 55, 13780–13783. [Google Scholar] [CrossRef]
- Britvin, S.N.; Kashtanov, S.A.; Krivovichev, S.V.; Chukanov, N.V. Xenon in Rigid Oxide Frameworks: Structure, Bonding and Explosive Properties of Layered Perovskite K4Xe3O12. J. Am. Chem. Soc. 2016, 138, 13838–13841. [Google Scholar] [CrossRef]
- Britvin, S.N.; Kashtanov, S.A.; Krzhizhanovskaya, M.; Gurinov, A.A.; Glumov, O.V.; Strekopytov, S.; Kretser, Y.L.; Zaitsev, A.; Chukanov, N.V.; Krivovichev, S.V. Perovskites with the Framework-Forming Xenon. Angew. Chem. Int. Ed. 2015, 54, 14340–14344. [Google Scholar] [CrossRef]
- Makarewicz, E.; Lundell, J.; Gordon, A.J.; Berski, S. Perovskites with the framework-forming xenon. J. Comput. Chem. 2016, 37, 1876–1886. [Google Scholar] [CrossRef]
- Miao, J.; Xiong, Z.; Gao, Y. The effects of aerogen-bonding on the geometries and spectral properties of several small molecular clusters containing XeO3. J. Phys. Cond. Mat. 2018, 30, 44. [Google Scholar] [CrossRef]
- Borocci, S.; Grandinetti, F.; Sanna, N.; Antoniotti, P.; Nunzi, F. Noncovalent Complexes of the Noble-Gas Atoms: Analyzing the Transition from Physical to Chemical Interactions. J. Comput. Chem. 2019, 40, 2318–2328. [Google Scholar] [CrossRef] [PubMed]
- Gomila, R.M.; Frontera, A. Covalent and Non-covalent Noble Gas Bonding Interactions in XeFn Derivatives (n = 2–6): A Combined Theoretical and ICSD Analysis. Front. Chem. 2020, 8, 395. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Frontera, A. σ/π-Hole noble gas bonding interactions: Insights from theory and experiment. Coord. Chem. Rev. 2020, 404, 213112. [Google Scholar] [CrossRef]
- Templeton, D.H.; Zalkin, A.; Forrester, J.D.; Williamson, S.M. Crystal and Molecular Structure of Xenon Trioxide. J. Am. Chem. Soc. 1963, 85, 817. [Google Scholar] [CrossRef]
- Goettel, J.T.; Schrobilgen, G.J. Solid-State Structures of XeO3. Inorg. Chem. 2016, 55, 12975–12981. [Google Scholar] [CrossRef]
- Hou, C.J.; Wang, X.; Botana, J.; Miao, M.-S. Noble gas bond and the behaviour of XeO3 under pressure. Phys. Chem. Chem. Phys. 2017, 19, 27463–27467. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R. Valency and Bonding: A Natural Bond Orbital Donor–Acceptor Perspective; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Bauzá, A.; Frontera, A. π-Hole aerogen bonding interactions. Phys. Chem. Chem. Phys. 2015, 17, 24748–24753. [Google Scholar] [CrossRef]
- Bauza, A.; Frontera, A. Theoretical Study on the Dual Behavior of XeO3 and XeF4 toward Aromatic Rings: Lone Pair-π versus Aerogen-π Interactions. ChemPhysChem 2015, 16, 3625–3630. [Google Scholar] [CrossRef]
- Miao, J.; Song, B.; Gao, Y. Is aerogen–π interaction capable of initiating the noncovalent chemistry of group 18? Chem. Asian J. 2015, 10, 2615–2618. [Google Scholar] [CrossRef] [PubMed]
- Bavafa, S.; Nowroozi, A.; Ebrahimi, A. Ab initio study of aerogen-bonds between some heterocyclic compounds of benzene with the noble gas elements (Ne, Ar, and Kr). Struct. Chem. 2019, 31, 435–445. [Google Scholar] [CrossRef]
- Gao, M.; Cheng, J.; Li, W.-Z.; Xiao, B.; Li, Q. The aerogen–π bonds involving π systems. Chem. Phys. Lett. 2016, 651, 50–55. [Google Scholar] [CrossRef]
- Zierkiewicz, W.; Michalczyk, M.; Scheiner, S. Aerogen bonds formed between AeOF2 (Ae = Kr, Xe) and diazines: Comparisons between σ-hole and π-hole complexes. Phys. Chem. Chem. Phys. 2018, 20, 4676–4687. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Asadollahi, S.; Vakili, M. Investigation of substituent effects in aerogen-bonding interaction between ZO3 (Z=Kr, Xe) and nitrogen bases. Int. J. Quantum Chem. 2016, 116, 1254–1260. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Mohammadian-Sabet, F.; Solimannejad, M. Single-electron aerogen bonds: Do they exist? Chem. Phys. Lett. 2016, 659, 196–202. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Mohammadian-Sabet, F. An ab initio study on anionic aerogen bonds. Chem. Phys. Lett. 2017, 667, 337–344. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Sabouri, A. Carbene–aerogen bonds: Anab initiostudy. Mol. Phys. 2017, 115, 971–980. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Sadr-Mousavi, A. A computational study on the strength and nature of bifurcated aerogen bonds. Chem. Phys. Lett. 2018, 698, 1–6. [Google Scholar] [CrossRef]
- Wang, R.; Liu, H.; Li, Q.; Scheiner, S. Xe⋯chalcogen aerogen bond. Effect of substituents and size of chalcogen atom. Phys. Chem. Chem. Phys. 2020, 22, 4115–4121. [Google Scholar] [CrossRef]
- Miao, J.; Xiong, Z.; Gao, Y. Unexpectedly strong Xe binding by host-guest interaction. Phys. Chem. Chem. Phys. 2019, 21, 26232–26236. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Vessally, E. The strengthening effect of a hydrogen or lithium bond on the Z⋯N aerogen bond (Z = Ar, Kr and Xe): A comparative study. Mol. Phys. 2016, 114, 1–12. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Mousavian, P.; Mohammadian-Sabet, F. Tuning of pnicogen and chalcogen bonds by an aerogen-bonding interaction: A comparative ab initio study. Mol. Phys. 2018, 117, 1–9. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Asadollahi, S. Strengthening of the halogen-bonding by an aerogen bond interaction: Substitution and cooperative effects in O3Z⋯NCX⋯NCY (Z = Ar, Kr, Xe; X = Cl, Br, I; Y = H, F, OH) complexes. Mol. Phys. 2016, 114, 1–10. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Qasemsolb, S. Tuning aerogen bonds via anion-π or lone pair-π interaction: A comparative ab initio study. Struct. Chem. 2017, 100, 143–1264. [Google Scholar] [CrossRef]
- Goettel, J.T.; Haensch, V.G.; Schrobilgen, G.J. Stable Chloro- and Bromoxenate Cage Anions; [X3(XeO3)3]3− and [X4(XeO3)4]4− (X = Cl or Br). J. Am. Chem. Soc. 2017, 139, 8725–8733. [Google Scholar] [CrossRef] [PubMed]
- Goettel, J.T.; Mercier, H.P.; Schrobilgen, G.J. XeO3 adducts of pyridine, 4-dimethylaminopyridine, and their pyridinium salts. J. Fluor. Chem. 2018, 211, 60–69. [Google Scholar] [CrossRef]
- Marczenko, K.M.; Goettel, J.T.; Mercier, H.P.A.; Schrobilgen, G.J. Xenon Trioxide Adducts of O-Donor Ligands; [(CH3)2CO]3XeO3, [(CH3)2SO]3(XeO3)2, (C5H5NO)3(XeO3)2, and [(C6H5)3PO]2XeO3. Chem. A Eur. J. 2019, 25, 12357–12366. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Pan, S.; Holzmann, N.; Schwerdtfeger, P.; Frenking, G. Chemical Bonding and Bonding Models of Main-Group Compounds. Chem. Rev. 2019, 119, 8781–8845. [Google Scholar] [CrossRef] [PubMed]
- Seppelt, K. Molecular Hexafluorides. Chem. Rev. 2014, 115, 1296–1306. [Google Scholar] [CrossRef]
- Gawrilow, M.; Beckers, H.; Hasenstab-Riedel, S.; Cheng, L. Matrix-Isolation and Quantum-Chemical Analysis of the C3v Conformer of XeF6, XeOF4, and Their Acetonitrile Adducts. J. Phys. Chem. A 2017, 122, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Dixon, D.A.; De Jong, W.A.; Peterson, K.A.; Christe, K.O.; Schrobilgen, G.J. Heats of Formation of Xenon Fluorides and the Fluxionality of XeF6 from High Level Electronic Structure Calculations. J. Am. Chem. Soc. 2005, 127, 8627–8634. [Google Scholar] [CrossRef] [PubMed]
- Levy, H.A.; Agron, P.A. The Crystal and Molecular Structure of Xenon Difluoride by Neutron Diffraction. J. Am. Chem. Soc. 1963, 85, 2. [Google Scholar] [CrossRef]
- Tavčar, G.; Tramsek, M. XeF2 as a ligand to a metal center, an interesting field of noble gas chemistry. J. Fluor. Chem. 2015, 174, 14–21. [Google Scholar] [CrossRef]
- Tavčar, G.; Tramsek, M.; Bunič, T.; Benkic, P.; Žemva, B. New class of coordination compounds with noble gas fluorides as ligands to metal ions. J. Fluor. Chem. 2004, 125, 1579–1584. [Google Scholar] [CrossRef]
- Tramsek, M.; Žemva, B. Synthesis of novel salts with HF, AsF3 and XeF2 as ligands to metal cations. J. Fluor. Chem. 2006, 127, 1275–1284. [Google Scholar] [CrossRef]
- Hagiwara, R.; Hollander, F.; Maines, C.; Bartlett, N. The crystal–structure of [Ag(XeF2)2]AsF6 formed in the oxidation of Xe by AgFAsF6. Eur. J. Solid State Chem. 1991, 28, 855–866. [Google Scholar]
- Tramšek, M.; Benkič, P.; Žemva, B. [M(XeF2)3](AsF6)2 (M=Pb, Sr): The first coordination compounds of M2+ with XeF2 ligand. Solid State Sci. 2002, 4, 9–14. [Google Scholar] [CrossRef]
- Naumann, D.; Tyrra, W.; Gnann, R.; Pfolk, D.; Gilles, T.; Tebbe, K.-F. The Direct Synthesis of Arylxenon Trifluoromethanesulfonates via electrophilic substitution. Z. Anorg. Allg. Chem. 1997, 623, 1821–1834. [Google Scholar] [CrossRef]
- Frohn, H.J.; Klose, A.; Henkel, G. Pentafluorophenylxenon(II) Pentafluorobenzoate: The First Preparative Synthesis and Structural Characterization of an Acyloxy Compound of Xenon(II). Angew. Chem. Int. Ed. 1993, 32, 99–100. [Google Scholar] [CrossRef]
- Haner, J.; Schrobilgen, G.J. The Chemistry of Xenon(IV). Chem. Rev. 2015, 115, 1255–1295. [Google Scholar] [CrossRef] [PubMed]
- Ibers, J.A.; Hamilton, W.C. Xenon Tetrafluoride: Crystal Structure. Science 1963, 139, 106–107. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.H.; Ellison, R.D.; Levy, H.A. The crystal structure of the molecular addition compound xenon difluoride–xenon tetrafluoride. Acta Crystallogr. 1965, 18, 11–16. [Google Scholar] [CrossRef]
- Lutar, K.; Leban, I.; Ogrin, T.; Žemva, B. XeF2×2CrF4 and XeF5+CrF5−: Syntheses, Crystal Structures, and Some Properties. Eur. J. Solid State Inorg. Chem. 1992, 129, 713. [Google Scholar]
- Tavčar, G.; Žemva, B. XeF4 as a Ligand for a Metal Ion. Angew. Chem. Int. Ed. 2009, 48, 1432–1434. [Google Scholar] [CrossRef]
- Matsumoto, K.; Haner, J.; Mercier, H.P.A.; Schrobilgen, G.J. Syntheses and Structures of F6XeNCCH3 and F6Xe(NCCH3)2. Angew. Chem. Int. Ed. 2015, 54, 14169–14173. [Google Scholar] [CrossRef]
- Brock, D.S.; Bilir, V.; Mercier, H.P.A.; Schrobilgen, G.J. XeOF2, F2OXeN≡CCH3, and XeOF2·nHF: Rare Examples of Xe(IV) Oxide Fluorides. J. Am. Chem. Soc. 2007, 129, 3598–3611. [Google Scholar] [CrossRef]
- Hoyer, S.; Emmler, T.; Seppelt, K. The structure of xenon hexafluoride in the solid state. J. Fluor. Chem. 2006, 127, 1415–1422. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | ΔE | d |
|---|---|---|
| XeO3⋯NCCH3 | −9.5 | 3.142 |
| XeO3⋯NH3 | −9.0 | 2.779 |
| XeO3⋯Cl− | −37.2 | 2.784 |
| XeO3⋯Br− | −32.6 | 2.983 |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frontera, A. Noble Gas Bonding Interactions Involving Xenon Oxides and Fluorides. Molecules 2020, 25, 3419. https://doi.org/10.3390/molecules25153419
Frontera A. Noble Gas Bonding Interactions Involving Xenon Oxides and Fluorides. Molecules. 2020; 25(15):3419. https://doi.org/10.3390/molecules25153419
Chicago/Turabian StyleFrontera, Antonio. 2020. "Noble Gas Bonding Interactions Involving Xenon Oxides and Fluorides" Molecules 25, no. 15: 3419. https://doi.org/10.3390/molecules25153419
APA StyleFrontera, A. (2020). Noble Gas Bonding Interactions Involving Xenon Oxides and Fluorides. Molecules, 25(15), 3419. https://doi.org/10.3390/molecules25153419