
Bioinformatics of Metalloproteins and Metalloproteomes

Abstract
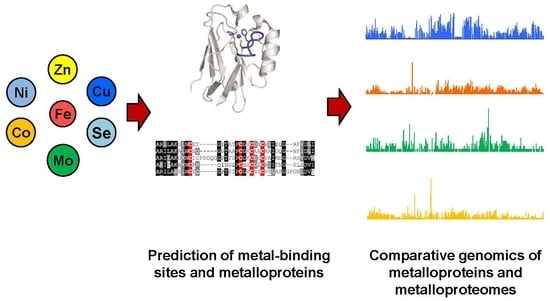
1. Introduction
2. Identification of Metalloprotein Genes and Related Resources
2.1. Homology-Based Identification of Known Metalloprotein Genes
2.2. Methods for Prediction of Metal-Binding Sites and Novel Metalloprotein Genes
2.3. Metalloprotein Databases
3. Comparative Genomics of Metalloproteins and Metalloproteomes
3.1. Zinc and Iron
3.2. Copper
3.3. Molybdenum and Tungsten
3.4. Nickel and Cobalt
3.5. Selenium
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zoroddu, M.A.; Aaseth, J.; Crisponi, G.; Medici, S.; Peana, M.; Nurchi, V.M. The essential metals for humans: A brief overview. J. Inorg. Biochem. 2019, 195, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Freeland-Graves, J.H.; Sanjeevi, N.; Lee, J.J. Global perspectives on trace element requirements. J. Trace Elem. Med. Biol. 2015, 31, 135–141. [Google Scholar] [CrossRef]
- Mertz, W. Review of the scientific basis for establishing the essentiality of trace elements. Biol. Trace Elem. Res. 1998, 66, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Nordberg, M.; Nordberg, G.F. Trace element research-historical and future aspects. J. Trace Elem. Med. Biol. 2016, 38, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Roman, M.; Jitaru, P.; Barbante, C. Selenium biochemistry and its role for human health. Metallomics 2014, 6, 25–54. [Google Scholar] [CrossRef]
- Van Gossum, A.; Neve, J. Trace element deficiency and toxicity. Curr. Opin. Clin. Nutr. Metab. Care 1998, 1, 499–507. [Google Scholar] [CrossRef]
- Sánchez, M.; Sabio, L.; Gálvez, N.; Capdevila, M.; Dominguez-Vera, J.M. Iron chemistry at the service of life. IUBMB Life 2017, 69, 382–388. [Google Scholar] [CrossRef]
- King, J.C. Zinc: An essential but elusive nutrient. Am. J. Clin. Nutr. 2011, 94, 679Ss–684Ss. [Google Scholar] [CrossRef]
- Magalon, A.; Mendel, R.R. Biosynthesis and insertion of the molybdenum cofactor. EcoSal Plus 2015. [Google Scholar] [CrossRef]
- Giedyk, M.; Goliszewska, K.; Gryko, D. Vitamin B12 catalysed reactions. Chem. Soc. Rev. 2015, 44, 3391–3404. [Google Scholar] [CrossRef]
- Degtyarenko, K. Bioinorganic motifs: Towards functional classification of metalloproteins. Bioinformatics 2000, 16, 851–864. [Google Scholar] [CrossRef] [PubMed]
- Waldron, K.J.; Rutherford, J.C.; Ford, D.; Robinson, N.J. Metalloproteins and metal sensing. Nature 2009, 460, 823–830. [Google Scholar] [CrossRef]
- Andreini, C.; Bertini, I.; Cavallaro, G.; Holliday, G.L.; Thornton, J.M. Metal ions in biological catalysis: From enzyme databases to general principles. J. Biol. Inorg. Chem. 2008, 13, 1205–1218. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Zinc and the zinc proteome. Met. Ions Life Sci. 2013, 12, 479–501. [Google Scholar] [PubMed]
- Alfano, M.; Cavazza, C. Structure, function, and biosynthesis of nickel-dependent enzymes. Protein Sci. 2020, 29, 1071–1089. [Google Scholar] [CrossRef]
- Zoidis, E.; Seremelis, I.; Kontopoulos, N.; Danezis, G.P. Selenium-dependent antioxidant enzymes: Actions and properties of selenoproteins. Antioxidants (Basel) 2018, 7, 66. [Google Scholar] [CrossRef]
- Chandrangsu, P.; Rensing, C.; Helmann, J.D. Metal homeostasis and resistance in bacteria. Nat. Rev. Microbiol. 2017, 15, 338–350. [Google Scholar] [CrossRef]
- Martinez-Finley, E.J.; Chakraborty, S.; Fretham, S.J.; Aschner, M. Cellular transport and homeostasis of essential and nonessential metals. Metallomics 2012, 4, 593–605. [Google Scholar] [CrossRef]
- Zhang, Y.; Gladyshev, V.N. Comparative genomics of trace element dependence in biology. J. Biol. Chem. 2011, 286, 23623–23629. [Google Scholar] [CrossRef]
- Sukdeo, N.; Clugston, S.L.; Daub, E.; Honek, J.F. Distinct classes of glyoxalase I: Metal specificity of the Yersinia pestis, Pseudomonas aeruginosa and Neisseria meningitidis enzymes. Biochem. J. 2004, 384, 111–117. [Google Scholar] [CrossRef]
- Bulteau, A.L.; Chavatte, L. Update on selenoprotein biosynthesis. Antioxid. Redox Signal. 2015, 23, 775–794. [Google Scholar] [CrossRef]
- Gonzalez-Flores, J.N.; Shetty, S.P.; Dubey, A.; Copeland, P.R. The molecular biology of selenocysteine. Biomol. Concepts 2013, 4, 349–365. [Google Scholar] [CrossRef]
- Zhang, Y.; Romero, H.; Salinas, G.; Gladyshev, V.N. Dynamic evolution of selenocysteine utilization in bacteria: A balance between selenoprotein loss and evolution of selenocysteine from redox active cysteine residues. Genome Biol. 2006, 7, R94. [Google Scholar] [CrossRef]
- Mariotti, M.; Guigó, R. Selenoprofiles: Profile-based scanning of eukaryotic genome sequences for selenoprotein genes. Bioinformatics 2010, 26, 2656–2663. [Google Scholar] [CrossRef] [PubMed]
- Andreini, C.; Banci, L.; Bertini, I.; Rosato, A. Zinc through the three domains of life. J. Proteome Res. 2006, 5, 3173–3178. [Google Scholar] [CrossRef] [PubMed]
- Andreini, C.; Banci, L.; Bertini, I.; Elmi, S.; Rosato, A. Non-heme iron through the three domains of life. Proteins 2007, 67, 317–324. [Google Scholar] [CrossRef]
- Andreini, C.; Banci, L.; Bertini, I.; Rosato, A. Occurrence of copper proteins through the three domains of life: A bioinformatic approach. J. Proteome Res. 2008, 7, 209–216. [Google Scholar] [CrossRef]
- Andreini, C.; Bertini, I.; Rosato, A. Metalloproteomes: A bioinformatic approach. Acc. Chem. Res. 2009, 42, 1471–1479. [Google Scholar] [CrossRef]
- Andreini, C.; Bertini, I. A bioinformatics view of zinc enzymes. J. Inorg. Biochem. 2012, 111, 150–156. [Google Scholar] [CrossRef]
- Andreini, C.; Putignano, V.; Rosato, A.; Banci, L. The human iron-proteome. Metallomics 2018, 10, 1223–1231. [Google Scholar] [CrossRef]
- Andreini, C.; Rosato, A.; Banci, L. The relationship between environmental dioxygen and iron-sulfur proteins explored at the genome level. PLoS ONE 2017, 12, e0171279. [Google Scholar] [CrossRef]
- Andreini, C.; Bertini, I.; Cavallaro, G.; Decaria, L.; Rosato, A. A simple protocol for the comparative analysis of the structure and occurrence of biochemical pathways across superkingdoms. J. Chem. Inf. Model. 2011, 51, 730–738. [Google Scholar] [CrossRef]
- Passerini, A.; Andreini, C.; Menchetti, S.; Rosato, A.; Frasconi, P. Predicting zinc binding at the proteome level. BMC Bioinform. 2007, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Shu, N.; Zhou, T.; Hovmöller, S. Prediction of zinc-binding sites in proteins from sequence. Bioinformatics 2008, 24, 775–782. [Google Scholar] [CrossRef]
- Zhao, W.; Xu, M.; Liang, Z.; Ding, B.; Niu, L.; Liu, H.; Teng, M. Structure-based de novo prediction of zinc-binding sites in proteins of unknown function. Bioinformatics 2011, 27, 1262–1268. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Wang, M.; Takemoto, K.; Akutsu, T.; Zhang, Z.; Song, J. An integrative computational framework based on a two-step random forest algorithm improves prediction of zinc-binding sites in proteins. PLoS ONE 2012, 7, e49716. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, Z.; Wang, Y.; Zhai, Y.F.; Song, J.; Zhang, Z. ZincExplorer: An accurate hybrid method to improve the prediction of zinc-binding sites from protein sequences. Mol. Biosyst. 2013, 9, 2213–2222. [Google Scholar] [CrossRef]
- Srivastava, A.; Kumar, M. Prediction of zinc binding sites in proteins using sequence derived information. J. Biomol. Struct. Dyn. 2018, 36, 4413–4423. [Google Scholar] [CrossRef]
- Ajitha, M.; Sundar, K.; Arul Mugilan, S.; Arumugam, S. Development of METAL-ACTIVE SITE and ZINCCLUSTER tool to predict active site pockets. Proteins 2018, 86, 322–331. [Google Scholar] [CrossRef]
- Yan, R.; Wang, X.; Tian, Y.; Xu, J.; Xu, X.; Lin, J. Prediction of zinc-binding sites using multiple sequence profiles and machine learning methods. Mol. Omics 2019, 15, 205–215. [Google Scholar] [CrossRef]
- Liu, R.; Hu, J. HemeBIND: A novel method for heme binding residue prediction by combining structural and sequence information. BMC Bioinform. 2011, 12, 207. [Google Scholar] [CrossRef] [PubMed]
- Liou, Y.F.; Charoenkwan, P.; Srinivasulu, Y.; Vasylenko, T.; Lai, S.C.; Lee, H.C.; Chen, Y.H.; Huang, H.L.; Ho, S.Y. SCMHBP: Prediction and analysis of heme binding proteins using propensity scores of dipeptides. BMC Bioinform. 2014, 15 (Suppl. 16), S4. [Google Scholar] [CrossRef]
- Estellon, J.; Ollagnier de Choudens, S.; Smadja, M.; Fontecave, M.; Vandenbrouck, Y. An integrative computational model for large-scale identification of metalloproteins in microbial genomes: A focus on iron-sulfur cluster proteins. Metallomics 2014, 6, 1913–1930. [Google Scholar] [CrossRef] [PubMed]
- Valasatava, Y.; Rosato, A.; Banci, L.; Andreini, C. MetalPredator: A web server to predict iron-sulfur cluster binding proteomes. Bioinformatics 2016, 32, 2850–2852. [Google Scholar] [CrossRef]
- Sodhi, J.S.; Bryson, K.; McGuffin, L.J.; Ward, J.J.; Wernisch, L.; Jones, D.T. Predicting metal-binding site residues in low-resolution structural models. J. Mol. Biol. 2004, 342, 307–320. [Google Scholar] [CrossRef]
- Brylinski, M.; Skolnick, J. FINDSITE-metal: Integrating evolutionary information and machine learning for structure-based metal-binding site prediction at the proteome level. Proteins 2011, 79, 735–751. [Google Scholar] [CrossRef] [PubMed]
- Levy, R.; Edelman, M.; Sobolev, V. Prediction of 3D metal binding sites from translated gene sequences based on remote-homology templates. Proteins 2009, 76, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Passerini, A.; Lippi, M.; Frasconi, P. MetalDetector v2.0: Predicting the geometry of metal binding sites from protein sequence. Nucleic Acids Res. 2011, 39, W288–W292. [Google Scholar] [CrossRef]
- Lin, Y.F.; Cheng, C.W.; Shih, C.S.; Hwang, J.K.; Yu, C.S.; Lu, C.H. MIB: Metal ion-binding site prediction and docking server. J. Chem. Inf. Model. 2016, 56, 2287–2291. [Google Scholar] [CrossRef]
- Mariotti, M. SECISearch3 and seblastian: In-silico tools to predict SECIS elements and selenoproteins. Methods Mol. Biol. 2018, 1661, 3–16. [Google Scholar]
- Jiang, L.; Liu, Q. SelGenAmic: An algorithm for selenoprotein gene assembly. Methods Mol. Biol. 2018, 1661, 29–39. [Google Scholar] [PubMed]
- Zhang, Y.; Gladyshev, V.N. An algorithm for identification of bacterial selenocysteine insertion sequence elements and selenoprotein genes. Bioinformatics 2005, 21, 2580–2589. [Google Scholar] [CrossRef] [PubMed]
- Haberal, İ.; Oğul, H. Prediction of protein metal binding sites using deep neural networks. Mol. Inform. 2019, 38, e1800169. [Google Scholar] [CrossRef] [PubMed]
- Cvetkovic, A.; Menon, A.L.; Thorgersen, M.P.; Scott, J.W.; Poole, F.L., 2nd; Jenney, F.E., Jr.; Lancaster, W.A.; Praissman, J.L.; Shanmukh, S.; Vaccaro, B.J.; et al. Microbial metalloproteomes are largely uncharacterized. Nature 2010, 466, 779–782. [Google Scholar] [CrossRef]
- Azia, A.; Levy, R.; Unger, R.; Edelman, M.; Sobolev, V. Genome-wide computational determination of the human metalloproteome. Proteins 2015, 83, 931–939. [Google Scholar] [CrossRef]
- Santesmasses, D.; Mariotti, M.; Gladyshev, V.N. Bioinformatics of selenoproteins. Antioxid. Redox Signal. 2020, in press. [Google Scholar] [CrossRef]
- Kryukov, G.V.; Gladyshev, V.N. The prokaryotic selenoproteome. EMBO Rep. 2004, 5, 538–543. [Google Scholar] [CrossRef]
- Castagnetto, J.M.; Hennessy, S.W.; Roberts, V.A.; Getzoff, E.D.; Tainer, J.A.; Pique, M.E. MDB: The metalloprotein database and browser at the scripps research institute. Nucleic Acids Res. 2002, 30, 379–382. [Google Scholar] [CrossRef]
- Andreini, C.; Bertini, I.; Cavallaro, G.; Holliday, G.L.; Thornton, J.M. Metal-MACiE: A database of metals involved in biological catalysis. Bioinformatics 2009, 25, 2088–2089. [Google Scholar] [CrossRef]
- Zhang, Y.; Gladyshev, V.N. dbTEU: A protein database of trace element utilization. Bioinformatics 2010, 26, 700–702. [Google Scholar] [CrossRef]
- Harding, M.M.; Hsin, K.Y. Mespeus—A database of metal interactions with proteins. Methods Mol. Biol. 2014, 1091, 333–342. [Google Scholar] [PubMed]
- Putignano, V.; Rosato, A.; Banci, L.; Andreini, C. MetalPDB in 2018: A database of metal sites in biological macromolecular structures. Nucleic Acids Res. 2018, 46, D459–D464. [Google Scholar] [CrossRef] [PubMed]
- Romagné, F.; Santesmasses, D.; White, L.; Sarangi, G.K.; Mariotti, M.; Hübler, R.; Weihmann, A.; Parra, G.; Gladyshev, V.N.; Guigó, R.; et al. SelenoDB 2.0: Annotation of selenoprotein genes in animals and their genetic diversity in humans. Nucleic Acids Res. 2014, 42, D437–D443. [Google Scholar] [CrossRef] [PubMed]
- Ireland, S.M.; Martin, A.C.R. ZincBind-the database of zinc binding sites. Database (Oxford) 2019, 2019, baz006. [Google Scholar] [CrossRef]
- Miller, W.; Makova, K.D.; Nekrutenko, A.; Hardison, R.C. Comparative genomics. Annu. Rev. Genomics Hum. Genet. 2004, 5, 15–56. [Google Scholar] [CrossRef]
- Maret, W. Metalloproteomics, metalloproteomes, and the annotation of metalloproteins. Metallomics 2010, 2, 117–125. [Google Scholar] [CrossRef]
- Zhang, Y.; Gladyshev, V.N. Comparative genomics of trace elements: Emerging dynamic view of trace element utilization and function. Chem. Rev. 2009, 109, 4828–4861. [Google Scholar] [CrossRef]
- Tzou, W.S.; Chu, Y.; Lin, T.Y.; Hu, C.H.; Pai, T.W.; Liu, H.F.; Lin, H.J.; Cases, I.; Rojas, A.; Sanchez, M.; et al. Molecular evolution of multiple-level control of heme biosynthesis pathway in animal kingdom. PLoS ONE 2014, 9, e86718. [Google Scholar] [CrossRef][Green Version]
- Zhang, Y.; Ying, H.; Xu, Y. Comparative genomics and metagenomics of the metallomes. Metallomics 2019, 11, 1026–1043. [Google Scholar] [CrossRef]
- Bertini, I.; Decaria, L.; Rosato, A. The annotation of full zinc proteomes. J. Biol. Inorg. Chem. 2010, 15, 1071–1078. [Google Scholar] [CrossRef]
- Decaria, L.; Bertini, I.; Williams, R.J. Zinc proteomes, phylogenetics and evolution. Metallomics 2010, 2, 706–709. [Google Scholar] [CrossRef] [PubMed]
- Aruga, J.; Hatayama, M. Comparative genomics of the Zic family genes. Adv. Exp. Med. Biol. 2018, 1046, 3–26. [Google Scholar] [PubMed]
- Vervoort, M.; Meulemeester, D.; Béhague, J.; Kerner, P. Evolution of Prdm genes in animals: Insights from comparative genomics. Mol. Biol. Evol. 2016, 33, 679–696. [Google Scholar] [CrossRef] [PubMed]
- Najafabadi, H.S.; Garton, M.; Weirauch, M.T.; Mnaimneh, S.; Yang, A.; Kim, P.M.; Hughes, T.R. Non-base-contacting residues enable kaleidoscopic evolution of metazoan C2H2 zinc finger DNA binding. Genome Biol. 2017, 18, 167. [Google Scholar] [CrossRef] [PubMed]
- Salih, H.; Odongo, M.R.; Gong, W.; He, S.; Du, X. Genome-wide analysis of cotton C2H2-zinc finger transcription factor family and their expression analysis during fiber development. BMC Plant. Biol. 2019, 19, 400. [Google Scholar] [CrossRef] [PubMed]
- Tsaousis, A.D.; Gentekaki, E.; Eme, L.; Gaston, D.; Roger, A.J. Evolution of the cytosolic iron-sulfur cluster assembly machinery in Blastocystis species and other microbial eukaryotes. Eukaryot. Cell 2014, 13, 143–153. [Google Scholar] [CrossRef]
- Cavallaro, G.; Decaria, L.; Rosato, A. Genome-based analysis of heme biosynthesis and uptake in prokaryotic systems. J. Proteome Res. 2008, 7, 4946–4954. [Google Scholar] [CrossRef]
- Hayrapetyan, H.; Siezen, R.; Abee, T.; Nierop Groot, M. Comparative genomics of iron-transporting systems in bacillus cereus strains and impact of iron sources on growth and biofilm formation. Front. Microbiol. 2016, 7, 842. [Google Scholar] [CrossRef]
- Decaria, L.; Bertini, I.; Williams, R.J. Copper proteomes, phylogenetics and evolution. Metallomics 2011, 3, 56–60. [Google Scholar] [CrossRef]
- Ridge, P.G.; Zhang, Y.; Gladyshev, V.N. Comparative genomic analyses of copper transporters and cuproproteomes reveal evolutionary dynamics of copper utilization and its link to oxygen. PLoS ONE 2008, 3, e1378. [Google Scholar] [CrossRef]
- Zhang, Y.; Gladyshev, V.N. General trends in trace element utilization revealed by comparative genomic analyses of Co, Cu, Mo, Ni, and Se. J. Biol. Chem. 2010, 285, 3393–3405. [Google Scholar] [CrossRef] [PubMed]
- Scherbaum, S.; Hellmann, N.; Fernández, R.; Pick, C.; Burmester, T. Diversity, evolution, and function of myriapod hemocyanins. BMC Evol. Biol. 2018, 18, 107. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Xu, Y.; Zhang, Y. Comparative genomics of molybdenum utilization in prokaryotes and eukaryotes. BMC Genomics 2018, 19, 691. [Google Scholar] [CrossRef] [PubMed]
- Mendel, R.R. The molybdenum cofactor. J. Biol. Chem. 2013, 288, 13165–13172. [Google Scholar] [CrossRef] [PubMed]
- Moura, J.J.; Bernhardt, P.V.; Maia, L.B.; Gonzalez, P.J. Molybdenum and tungsten enzymes: From biology to chemistry and back. J. Biol. Inorg. Chem. 2015, 20, 181–182. [Google Scholar] [CrossRef] [PubMed]
- Hille, R.; Hall, J.; Basu, P. The mononuclear molybdenum enzymes. Chem. Rev. 2014, 114, 3963–4038. [Google Scholar] [CrossRef] [PubMed]
- Leimkühler, S.; Iobbi-Nivol, C. Bacterial molybdoenzymes: Old enzymes for new purposes. FEMS Microbiol. Rev. 2016, 40, 1–18. [Google Scholar] [CrossRef]
- Gladyshev, V.N.; Zhang, Y. Abundance, ubiquity and evolution of molybdoenzymes. In Molybdenum and Tungsten Enzymes: Biochemistry, 1st ed.; Hille, R., Schulzke, C., Kirk, M.L., Eds.; The Royal Society of Chemistry: Cambridge, UK, 2016; pp. 81–99. [Google Scholar]
- Llamas, A.; Tejada-Jiménez, M.; Fernández, E.; Galván, A. Molybdenum metabolism in the alga Chlamydomonas stands at the crossroad of those in Arabidopsis and humans. Metallomics 2011, 3, 578–590. [Google Scholar] [CrossRef]
- Zhang, Y.; Rump, S.; Gladyshev, V.N. Comparative genomics and evolution of molybdenum utilization. Coord. Chem. Rev. 2011, 255, 1206–1217. [Google Scholar] [CrossRef]
- Bevers, L.; Hagedoorn, P.; Hagen, W. The bioinorganic chemistry of tungsten. Coord. Chem. Rev. 2009, 253, 269–290. [Google Scholar] [CrossRef]
- Pushie, M.J.; Cotelesage, J.J.; George, G.N. Molybdenum and tungsten oxygen transferases--and functional diversity within a common active site motif. Metallomics 2014, 6, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Niks, D.; Hille, R. Molybdenum- and tungsten-containing formate dehydrogenases and formylmethanofuran dehydrogenases: Structure, mechanism, and cofactor insertion. Protein Sci. 2019, 28, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Boll, M.; Einsle, O.; Ermler, U.; Kroneck, P.M.; Ullmann, G.M. Structure and function of the unusual tungsten enzymes acetylene hydratase and class II benzoyl-coenzyme a reductase. J. Mol. Microbiol. Biotechnol. 2016, 26, 119–137. [Google Scholar] [CrossRef]
- Boer, J.L.; Mulrooney, S.B.; Hausinger, R.P. Nickel-dependent metalloenzymes. Arch. Biochem. Biophys. 2014, 544, 142–152. [Google Scholar] [CrossRef]
- Kräutler, B. Vitamin B12: Chemistry and biochemistry. Biochem. Soc. Trans. 2005, 33, 806–810. [Google Scholar] [CrossRef]
- Smith, A.D.; Warren, M.J.; Refsum, H. Vitamin B12. Adv. Food Nutr. Res. 2018, 83, 215–279. [Google Scholar]
- Takano, H.; Mise, K.; Hagiwara, K.; Hirata, N.; Watanabe, S.; Toriyabe, M.; Shiratori-Takano, H.; Ueda, K. Role and function of LitR, an adenosyl B12-bound light-sensitive regulator of bacillus megaterium QM B1551, in regulation of carotenoid production. J. Bacteriol. 2015, 197, 2301–2315. [Google Scholar] [CrossRef]
- Kobayashi, M.; Shimizu, S. Cobalt proteins. Eur. J. Biochem. 1999, 261, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Rodionov, D.A.; Gelfand, M.S.; Gladyshev, V.N. Comparative genomic analyses of nickel, cobalt and vitamin B12 utilization. BMC Genomics 2009, 10, 78. [Google Scholar] [CrossRef]
- Zheng, K.; Ngo, P.D.; Owens, V.L.; Yang, X.P.; Mansoorabadi, S.O. The biosynthetic pathway of coenzyme F430 in methanogenic and methanotrophic archaea. Science 2016, 354, 339–342. [Google Scholar] [CrossRef]
- Mulrooney, S.B.; Hausinger, R.P. Nickel uptake and utilization by microorganisms. FEMS Microbiol. Rev. 2003, 27, 239–261. [Google Scholar] [CrossRef]
- Eitinger, T.; Suhr, J.; Moore, L.; Smith, J.A. Secondary transporters for nickel and cobalt ions: Theme and variations. BioMetals 2005, 18, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Shelton, A.N.; Seth, E.C.; Mok, K.C.; Han, A.W.; Jackson, S.N.; Haft, D.R.; Taga, M.E. Uneven distribution of cobamide biosynthesis and dependence in bacteria predicted by comparative genomics. ISME J. 2019, 13, 789–804. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Zhang, Y. Systems biology of selenium and complex disease. Biol. Trace Elem. Res. 2019, 192, 38–50. [Google Scholar] [CrossRef]
- Steinbrenner, H.; Speckmann, B.; Klotz, L.O. Selenoproteins: Antioxidant selenoenzymes and beyond. Arch. Biochem. Biophys. 2016, 595, 113–119. [Google Scholar] [CrossRef]
- Hariharan, S.; Dharmaraj, S. Selenium and selenoproteins: It’s role in regulation of inflammation. Inflammopharmacology 2020, 28, 667–695. [Google Scholar] [CrossRef]
- Li, M.; Huang, Y.; Xiao, Y. A method for identification of selenoprotein genes in archaeal genomes. Genom. Proteom. Bioinform. 2009, 7, 62–70. [Google Scholar] [CrossRef]
- Santesmasses, D.; Mariotti, M.; Guigó, R. Selenoprofiles: A computational pipeline for annotation of selenoproteins. Methods Mol. Biol. 2018, 1661, 17–28. [Google Scholar]
- Castellano, S.; Lobanov, A.V.; Chapple, C.; Novoselov, S.V.; Albrecht, M.; Hua, D.; Lescure, A.; Lengauer, T.; Krol, A.; Gladyshev, V.N.; et al. Diversity and functional plasticity of eukaryotic selenoproteins: Identification and characterization of the SelJ family. Proc. Natl. Acad. Sci. USA 2005, 102, 16188–16193. [Google Scholar] [CrossRef]
- Zhang, Y.; Gladyshev, V.N. Trends in selenium utilization in marine microbial world revealed through the analysis of the global ocean sampling (GOS) project. PLoS Genet. 2008, 4, e1000095. [Google Scholar] [CrossRef]
- Lin, J.; Peng, T.; Jiang, L.; Ni, J.Z.; Liu, Q.; Chen, L.; Zhang, Y. Comparative genomics reveals new candidate genes involved in selenium metabolism in prokaryotes. Genome Biol. Evol. 2015, 7, 664–676. [Google Scholar] [CrossRef]
- Cravedi, P.; Mori, G.; Fischer, F.; Percudani, R. Evolution of the selenoproteome in helicobacter pylori and epsilonproteobacteria. Genome Biol. Evol. 2015, 7, 2692–2704. [Google Scholar] [PubMed]
- Peng, T.; Lin, J.; Xu, Y.Z.; Zhang, Y. Comparative genomics reveals new evolutionary and ecological patterns of selenium utilization in bacteria. ISME J. 2016, 10, 2048–2059. [Google Scholar] [CrossRef]
- Miller, W.G.; Yee, E.; Lopes, B.S.; Chapman, M.H.; Huynh, S.; Bono, J.L.; Parker, C.T.; Strachan, N.J.C.; Forbes, K.J. Comparative genomic analysis identifies a campylobacter clade deficient in selenium metabolism. Genome Biol. Evol. 2017, 9, 1843–1858. [Google Scholar] [CrossRef] [PubMed]
- Santesmasses, D.; Mariotti, M.; Guigó, R. Computational identification of the selenocysteine tRNA (tRNASec) in genomes. PLoS Comput. Biol. 2017, 13, e1005383. [Google Scholar] [CrossRef] [PubMed]
- Rother, M.; Quitzke, V. Selenoprotein synthesis and regulation in Archaea. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2451–2462. [Google Scholar] [CrossRef] [PubMed]
- Mariotti, M.; Lobanov, A.V.; Manta, B.; Santesmasses, D.; Bofill, A.; Guigó, R.; Gabaldón, T.; Gladyshev, V.N. Lokiarchaeota marks the transition between the archaeal and eukaryotic selenocysteine encoding systems. Mol. Biol. Evol. 2016, 33, 2441–2453. [Google Scholar] [CrossRef] [PubMed]
- Lobanov, A.V.; Fomenko, D.E.; Zhang, Y.; Sengupta, A.; Hatfield, D.L.; Gladyshev, V.N. Evolutionary dynamics of eukaryotic selenoproteomes: Large selenoproteomes may associate with aquatic life and small with terrestrial life. Genome Biol. 2007, 8, R198. [Google Scholar] [CrossRef]
- Mariotti, M.; Ridge, P.G.; Zhang, Y.; Lobanov, A.V.; Pringle, T.H.; Guigo, R.; Hatfield, D.L.; Gladyshev, V.N. Composition and evolution of the vertebrate and mammalian selenoproteomes. PLoS ONE 2012, 7, e33066. [Google Scholar] [CrossRef]
- Jiang, L.; Ni, J.; Liu, Q. Evolution of selenoproteins in the metazoan. BMC Genomics 2012, 13, 446. [Google Scholar] [CrossRef]
- Liang, H.; Wei, T.; Xu, Y.; Li, L.; Kumar Sahu, S.; Wang, H.; Li, H.; Fu, X.; Zhang, G.; Melkonian, M.; et al. Phylogenomics provides new insights into gains and losses of selenoproteins among archaeplastida. Int. J. Mol. Sci. 2019, 20, 3020. [Google Scholar] [CrossRef]
- Mariotti, M.; Santesmasses, D.; Capella-Gutierrez, S.; Mateo, A.; Arnan, C.; Johnson, R.; D’Aniello, S.; Yim, S.H.; Gladyshev, V.N.; Serras, F.; et al. Evolution of selenophosphate synthetases: Emergence and relocation of function through independent duplications and recurrent subfunctionalization. Genome Res. 2015, 25, 1256–1267. [Google Scholar] [CrossRef] [PubMed]
- Sarangi, G.K.; Romagné, F.; Castellano, S. Distinct patterns of selection in selenium-dependent genes between land and aquatic vertebrates. Mol. Biol. Evol. 2018, 35, 1744–1756. [Google Scholar] [CrossRef] [PubMed]
- Gobler, C.J.; Berry, D.L.; Dyhrman, S.T.; Wilhelm, S.W.; Salamov, A.; Lobanov, A.V.; Zhang, Y.; Collier, J.L.; Wurch, L.L.; Kustka, A.B.; et al. Niche of harmful alga Aureococcus anophagefferens revealed through ecogenomics. Proc. Natl. Acad. Sci. USA 2011, 108, 4352–4357. [Google Scholar] [CrossRef]
- Lobanov, A.V.; Hatfield, D.L.; Gladyshev, V.N. Reduced reliance on the trace element selenium during evolution of mammals. Genome Biol. 2008, 9, R62. [Google Scholar] [CrossRef]
- Baclaocos, J.; Santesmasses, D.; Mariotti, M.; Bierła, K.; Vetick, M.B.; Lynch, S.; McAllen, R.; Mackrill, J.J.; Loughran, G.; Guigó, R.; et al. Processive recoding and metazoan evolution of selenoprotein P: Up to 132 UGAs in molluscs. J. Mol. Biol. 2019, 431, 4381–4407. [Google Scholar] [CrossRef]
- Mariotti, M.; Salinas, G.; Gabaldón, T.; Gladyshev, V.N. Utilization of selenocysteine in early-branching fungal phyla. Nat. Microbiol. 2019, 4, 759–765. [Google Scholar] [CrossRef]
- Khrustalev, V.V.; Barkovsky, E.V.; Khrustaleva, T.A. Magnesium and manganese binding sites on proteins have the same predominant motif of secondary structure. J. Theor. Biol. 2016, 395, 174–185. [Google Scholar] [CrossRef]
- Vincent, J.B. The biochemistry of chromium. J. Nutr. 2000, 130, 715–718. [Google Scholar] [CrossRef]
- Ueki, T.; Adachi, T.; Kawano, S.; Aoshima, M.; Yamaguchi, N.; Kanamori, K.; Michibata, H. Vanadium-binding proteins (vanabins) from a vanadium-rich ascidian Ascidia sydneiensis samea. Biochim. Biophys. Acta 2003, 1626, 43–50. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Website | Related Metals | Main Algorithm | Reported Performance | Ref. |
|---|---|---|---|---|---|
| RDGB | http://www.cerm.unifi.it/home/research/genomebrowsing.html | Zn, Cu, Fe and other metals | Integration of tools for retrieval of protein domains and genome analysis | Accuracy: 89.6%, precision: 85.9% | [32] |
| Zincfinder | http://zincfinder.dsi.unifi.it | Zn | a SVM | b AURPC: 0.590 (local predictor) and 0.633 (gating network) | [33] |
| Zincpred | http://www.fos.su.se/~nanjiang/zincpred/download/ | Zn | SVM- and homology-based algorithm | AURPC: 0.723 (local predictor) and 0.701 (gating network) | [34] |
| TEMSP | http://netalign.ustc.edu.cn/temsp/ | Zn | Structure-based algorithm with a range of geometric criteria | c AUC: 0.945 | [35] |
| Zincidentifier | http://protein.cau.edu.cn/zincidentifier/ | Zn | A two-step feature selection method based on random forest algorithm | AUC: 0.955, AURPC: 0.829 | [36] |
| ZincExplorer | http://protein.cau.edu.cn/ZincExplorer/ | Zn | A combination of SVM-, cluster- and template-based predictors | AURPC: 0.907 | [37] |
| ZincBinder | http://proteininformatics.org/mkumar/znbinder/ | Zn | SVM model trained on PSSM-based input feature | AUC: 0.91 | [38] |
| ZINCCLUSTER | http://www.metalactive.in | Zn | SVM-based Ligand Finder and Cluster Finder algorithms | d MCC: 0.798, F1-score: 0.801 | [39] |
| ZnMachine | http://bioinformatics.fzu.edu.cn/znMachine.html | Zn | A combination of several intensively-trained machine learning models | AUC: 0.933 (SVM) and 0.910 (neural network) | [40] |
| HemeBIND | http://mleg.cse.sc.edu/hemeBIND/ | Fe (heme) | SVM | MCC: 0.504, F1-score: 56.87% | [41] |
| SCMHBP | http://iclab.life.nctu.edu.tw/SCMHBP/ | Fe (heme) | Based on a newly-developed scoring card method for predicting heme-binding proteins | Accuracy: 85.90% | [42] |
| Isph | http://biodev.extra.cea.fr/isph | Fe (Fe-S) | A penalized linear model based on machine learning approach | Precision: 87.9%, recall: 80.1% (extended model) | [43] |
| MetalPredator | http://metalweb.cerm.unifi.it/tools/metalpredator/ | Fe (Fe-S) | Integration of existing domain-based methodology with a new approach for discovering metal-binding motifs | Precision: 85.2%, recall: 88.6% | [44] |
| MetSite | N/A | Fe, Zn, Cu, Mn, Ca, Mg | Artificial neural network | Mean accuracy: 94.5% | [45] |
| FINDSITE-metal | http://cssb.biology.gatech.edu/findsite-metal/ | Fe, Zn, Cu, Mn, Ni, Co, Ca, Mg | Integration of structure/evolutionary information and machine learning approach (SVM) | Overall accuracy: 70–90% | [46] |
| SeqCHED | http://ligin.weizmann.ac.il/seqched | Fe, Zn, Cu, Mn, Ni, Co, Ca, Mg | A modification of the CHED algorithm and machine learning filters (decision tree classifier and SVM) | Sensitivity: 84–85%, selectivity: 82–93% (stringent filtration) | [47] |
| MetalDetector | http://metaldetector.dsi.unifi.it/v2.0/ | Transition metals that use cysteine and histidine as ligands | A combination of different machine learning algorithms (SVM-HMM, structured-output SVM) | Precision: 60–79%, recall: 71–88% | [48] |
| MIB | http://bioinfo.cmu.edu.tw/MIB/ | Ca, Cu, Fe, Mg, Mn, Zn, Cd, Ni, Hg, Co | Fragment transformation method | Overall accuracy: 92.9–95.1% | [49] |
| SECISearch3 and Seblastian | http://seblastian.crg.eu/, http://gladyshevlab.org/SelenoproteinPredictionServer | Se | Homology-based RNA motif finding and selenoprotein gene detection approach | Precision: 81.48–100%, recall: 33.33–100% | [50] |
| SelGenAmic | N/A | Se | Selenoprotein gene assembly algorithm based on the GenAmic approach used by geneid | N/A | [51] |
| bSECISearch | http://genomics.unl.edu/bSECISearch/ | Se | An algorithm for prediction of bacterial selenoprotein genes based on a concensus RNA structural model | True positive rate: 96.5% | [52] |
| Name | Website | Main Content | Ref. |
|---|---|---|---|
| MDB | http://metallo.scripps.edu | Metalloproteins and metal-binding sites in protein structures | [58] |
| Metal-MACiE | http://www.ebi.ac.uk/thornton-srv/databases/Metal_MACiE/home.html | All metalloenzymes annotated in the MACiE database | [59] |
| dbTEU | http://gladyshevlab.bwh.harvard.edu/trace_element/ | Transporters and metalloproteins for Cu, Mo, Co, Ni, and Se in more than 700 organisms | [60] |
| Mespeus | http://mespeus.bch.ed.ac.uk/MESPEUS_10/ | Experimentally established geometry of metal and protein interactions | [61] |
| MetalPDB | http://metalweb.cerm.unifi.it | Metal-binding sites detected in the 3D structures of biological macromolecules | [62] |
| SelenoDB | http://www.selenodb.org | Selenoprotein genes in at least 58 animal genomes | [63] |
| ZincBind | http://zincbind.bioinf.org.uk | All known Zn-binding sites from PDB | [64] |
| Metal | Prokaryotes | Eukaryotes |
|---|---|---|
| Cu | Cytochrome c oxidase subunit I | Cytochrome c oxidase subunit I |
| Cytochrome c oxidase subunit II | Cytochrome c oxidase subunit II | |
| Plastocyanin family | Plastocyanin family | |
| Cu amine oxidase | Cu amine oxidase | |
| Cu-Zn superoxide dismutase | Cu-Zn superoxide dismutase | |
| Multicopper oxidase family | Multicopper oxidase family | |
| Tyrosinase | Tyrosinase | |
| Azurin | Galactose oxidase | |
| Rusticyanin | Hemocyanin | |
| Nitrosocyanin | Plantacyanin family | |
| Nitrous oxide reductase | Peptidylglycine α-hydroxylating monooxygenase | |
| Nitrite reductase | Dopamine β-monooxygenase | |
| NADH dehydrogenase 2 | Cnx1G | |
| Particulate methane monooxygenase | ||
| Mo | Sulfite oxidase | Sulfite oxidase |
| Xanthine oxidase | Xanthine oxidase | |
| Dimethylsulfoxide reductase | MOSC-containing protein (mARC) | |
| MOSC-containing protein | ||
| Fe-Mo-containing nitrogenase | ||
| W | Aldehyde:ferredoxin oxidoreductase | N/A |
| Certain members of dimethylsulfoxide reductase: | ||
| Formate dehydrogenase and acetylene hydratase (obligately anaerobic bacteria) | ||
| Formylmethanofuran dehydrogenase (methanogenic archaea) | ||
| Ni | Urease | Urease |
| Ni-Fe hydrogenase | ||
| Carbon monoxide dehydrogenase | ||
| Superoxide dismutase SodN | ||
| Acetyl-coenzyme A synthase/decarbonylase | ||
| Methyl-coenzyme M reductase | ||
| Lactate racemase | ||
| Co | Methylmalonyl-CoA mutase | Methylmalonyl-CoA mutase |
| Isobutyryl-CoA mutase | B12-dependent ribonucleotide reductase class II | |
| Ethylmalonyl-CoA mutase | Methionine synthase | |
| Glutamate mutase | ||
| Methyleneglutarate mutase | ||
| D-lysine 5,6-aminomutase | ||
| Diol dehydratase | ||
| Glycerol dehydratase | ||
| Ethanolamine ammonia lyase | ||
| B12-dependent ribonucleotide reductase class II | ||
| Methionine synthase | ||
| Methyltetrahydromethanopterin:coenzyme M methyltransferase subunit A | ||
| Other methyltransferases | ||
| B12-dependent reductive dehalogenase PceA/CprA | ||
| LitR/CarH/CarA | ||
| PpaA | ||
| Epoxyqueuosine reductase |
| Prokaryotes | Eukaryotes |
|---|---|
| Known selenoproteins | Known selenoproteins in mammals |
| Formate dehydrogenase alpha subunit | Deiodinase (DIO) family: DIO1, DIO2, and DIO3 |
| Selenophosphate synthetase | Glutathione peroxidase (GPX) family: GPX1, GPX2, GPX3, GPX4, and GPX6 |
| Coenzyme F420-reducing hydrogenase alpha subunit | Thioredoxin reductase (TXNRD) family: TXNRD1, TXNRD2, and TXNRD3 |
| Coenzyme F420-reducing hydrogenase delta subunit | Methionine sulfoxide reductase B1 |
| Methylviologen-reducing hydrogenase alpha subunit | Selenoprotein F |
| Glycine reductase selenoprotein A | Selenoprotein H |
| Glycine reductase selenoprotein B | Selenoprotein I |
| Proline reductase | Selenoprotein K |
| Heterodisulfide reductase alpha subunit | Selenoprotein M |
| Methionine-S-sulfoxide reductase | Selenoprotein N |
| Peroxiredoxin (Prx)Thioredoxin (Trx) | Selenoprotein O |
| Glutaredoxin (Grx) | Selenoprotein P |
| Arsenite S-adenosylmethyltransferase | Selenoprotein S |
| Selenoprotein T | |
| Predicted selenoproteins: | Selenoprotein V |
| Thiol:disulfide isomerase-like protein | Selenoprotein W |
| Thiol:disulfide interchange protein | Selenophosphate synthetase 2 |
| HesB-like | |
| Deiodinase-like | Other known selenoproteins: |
| Glutathione peroxidase-like | Methionine-S-sulfoxide reductase |
| Selenoprotein W-like | Protein disulfide isomerase |
| Fe-S oxidoreductase | Selenoprotein J |
| DsbA-like | Selenoprotein L |
| DsrE-like | Selenoprotein U |
| DsbG-like | Selenoprotein E |
| AhpD-like | SAM-dependent methyltransferase |
| Arsenate reductase | |
| Molybdopterin biosynthesis protein MoeB | Predicted selenoproteins: |
| Glutathione S-transferase | Prx-like protein |
| COG0737 UshA | Trx-fold protein |
| OsmC-like | Membrane selenoprotein MSP |
| Rhodanase-related protein | SelTryp |
| Sulfurtransferase COG2897 | Other hypothetical proteins |
| Cation-transporting ATPase, E1-E2 family | |
| Methylated-DNA-protein-cysteine methyltransferase | |
| UGSC-containing protein | |
| CMD domain containing protein | |
| Organic mercuric lyase MerB2 | |
| Predicted redox-active disulfide protein 2 | |
| Prx-like/Trx-like/Grx-like and Trx-fold proteins | |
| Other hypothetical selenoproteins |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Zheng, J. Bioinformatics of Metalloproteins and Metalloproteomes. Molecules 2020, 25, 3366. https://doi.org/10.3390/molecules25153366
Zhang Y, Zheng J. Bioinformatics of Metalloproteins and Metalloproteomes. Molecules. 2020; 25(15):3366. https://doi.org/10.3390/molecules25153366
Chicago/Turabian StyleZhang, Yan, and Junge Zheng. 2020. "Bioinformatics of Metalloproteins and Metalloproteomes" Molecules 25, no. 15: 3366. https://doi.org/10.3390/molecules25153366
APA StyleZhang, Y., & Zheng, J. (2020). Bioinformatics of Metalloproteins and Metalloproteomes. Molecules, 25(15), 3366. https://doi.org/10.3390/molecules25153366