
The Diverse World of Foldamers: Endless Possibilities of Self-Assembly †

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Definition and Overview
1.2. Organization of the Review
1.3. Aim, Scope and Exclusions
2. β-Peptide Foldamers
2.1. Discrete Self-Assembly in Solution: Up to Bundles
2.1.1. Nucleobase-Guided Pairing in Aqueous Solution
2.1.2. Bundles in Aqueous Solution
2.1.3. Dimers and Bundles in Methanol
2.1.4. Ion Channels in Phospholipid Bilayers
2.2. Extended Self-Assembly: Vesicles and Fibers
2.2.1. Vesicles in Protic Solvents
2.2.2. Fibers in Methanol
2.2.3. Fibers in Aqueous Solution
2.3. Self-Assembly into Solid Tridimensional Structures in Aqueous Environment
2.4. Challenges in Design and Structural Characterization of Self-Assembled β-Peptide Foldamers
3. Aromatic Foldamers
3.1. Discrete Self-Assembly in Solution: The Realm of Double Helices and Other Multiple Helices
3.1.1. Aggregates Different from Interpenetrating Multiple Helices in Nonpolar Solvents
3.1.2. Double, Triple and Quadruple Helices in Chloroform
3.1.3. Double Helices in Water
3.2. Extended Self-Assembly: Monolayers, Vesicles, and Fibers
3.2.1. Self-Assembled Monolayers
3.2.2. Vesicles: Effects of Varying the Solvent Composition
3.2.3. Vesicles in Water
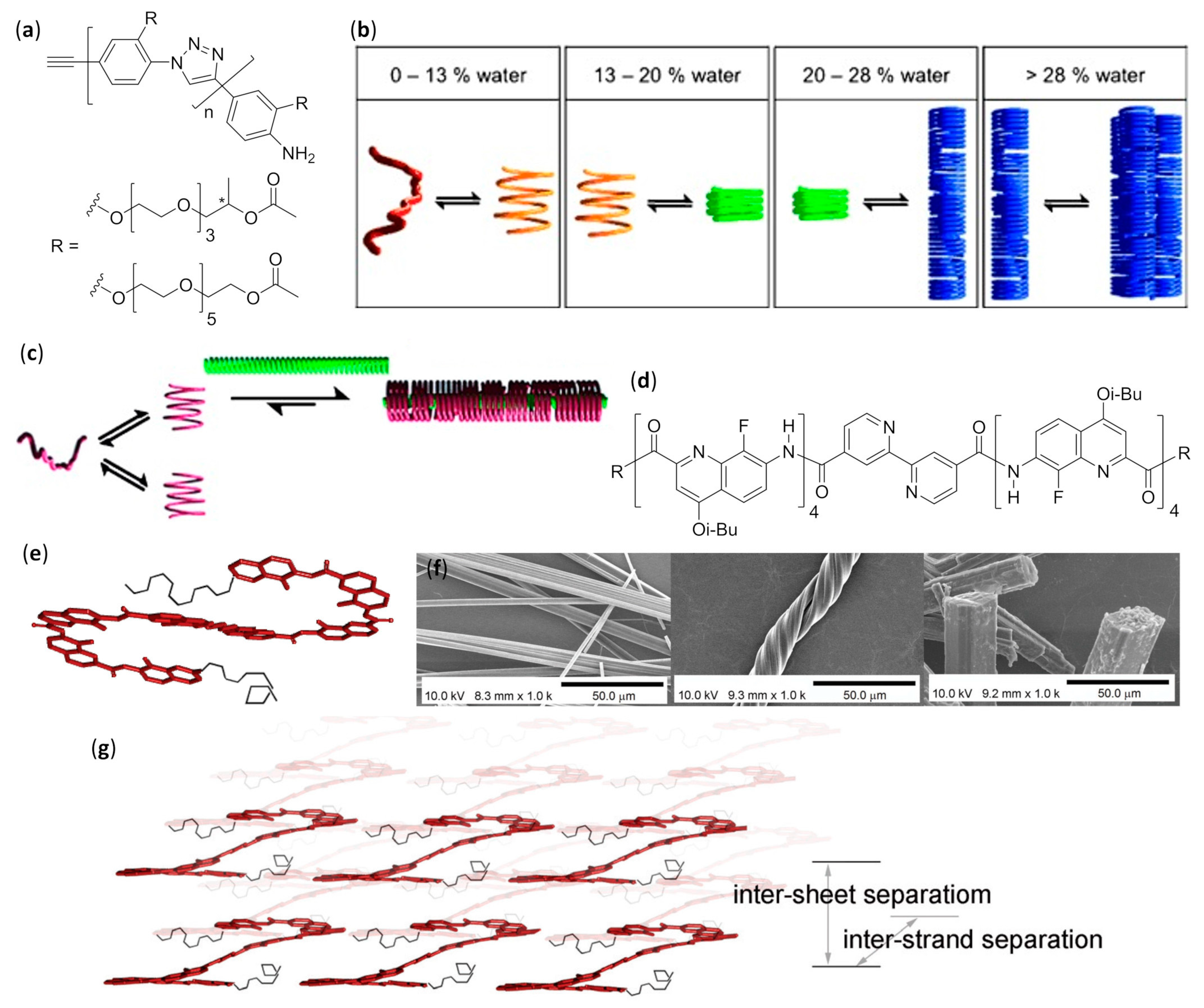
3.2.4. Fibers: Effect of Varying the Water Content
3.2.5. Fibers in Organic Solvents
3.3. Challenges in Design and Structural Characterization of Self-Assembled Aromatic Foldamers
4. Foldamers Based on Other Building Blocks
4.1. Discrete Self-Assembly in Solution: Up to Bundles
4.1.1. Duplexes, β-Sheets, and Double Helices
Aib Oligomers in Chloroform
Mixed Aliphatic/Aromatic Foldamers in Aqueous Solution
Mixed Aliphatic/Aromatic Foldamers in Chloroform
Mixed α/γ Aliphatic Foldamers in Chloroform
4.1.2. Bundles in Aqueous Solution
Mixed α/β Foldamers
Urea Foldamers
4.2. Extended Self-Assembly: Monolayers, Vesicles, and Fibers
4.2.1. Self-Assembled Monolayers
Mixed α/β Foldamers at the Air–Water Interface
Catechol-Based Foldamers at Different Interfaces
Urea Foldamers on Gold
4.2.2. Nanosheets
Peptoids in Aqueous Solutions
4.2.3. Microspheres
Peptoids in Aqueous Solutions
4.2.4. Vesicles
Peptoids in Aqueous Solutions
Mixed Aliphatic–Aromatic Foldamers in Methanolic Solutions
4.2.5. Fibers
Mixed α–Δβ Foldamers in Different Solvents
Mixed Aliphatic–Aromatic Foldamers in Aqueous Environment
Mixed Aliphatic–Aromatic Foldamers in Organic Solvents
Aib-Containing Aliphatic Oligomers in Organic Solvents
Aib-Containing Aliphatic Oligomers in Organic Solvent–Water Mixtures
γ-Foldamers in Different Solvents
Peptoid Foldamers in Aqueous Solution
4.2.6. Nanotubes
Peptoid Foldamers in Aqueous Solution
4.3. Self-Assembly into Solid Tridimensional Structures
Aliphatic Foldamers with Heterogeneous Backbones in Aqueous Environment
Mixed α–β Foldamers
Mixed α–γ Foldamers
4.4. Challenges in Design and Structural Characterization of Other Self-Assembled Foldamers
5. Conclusions
Funding
Conflicts of Interest
References
- Gellman, S.H. Foldamers: A manifesto. Acc. Chem. Res. 1998, 31, 173–180. [Google Scholar] [CrossRef]
- Hill, D.J.; Mio, M.J.; Prince, R.B.; Hughes, T.S.; Moore, J.S. A field guide to foldamers. Chem. Rev. 2001, 101, 3893–4012. [Google Scholar] [CrossRef] [PubMed]
- Seebach, D.; Matthews, J.L. β-Peptides: A surprise at every turn. J. Chem. Soc. Chem. Commun. 1997, 21, 2015–2022. [Google Scholar] [CrossRef]
- Cheng, R.P.; Gellman, S.H.; DeGrado, W.F. β-Peptides: From structure to function. Chem. Rev. 2001, 101, 3219–3232. [Google Scholar] [CrossRef]
- Fülöp, F.; Martinek, T.A.; Tóth, G.K. Application of alicyclic β-amino acids in peptide chemistry. Chem. Soc. Rev. 2006, 35, 323–334. [Google Scholar] [CrossRef]
- Hecht, S.; Huc, I. Foldamers: Structure, Properties, and Applications. Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- Goodman, C.M.; Choi, S.; Shandler, S.; DeGrado, W.F. Foldamers as versatile frameworks for the design and evolution of function. Nat. Chem. Biol. 2007, 3, 252–262. [Google Scholar] [CrossRef]
- Martinek, T.A.; Fülöp, F. Peptidic foldamers: Ramping up diversity. Chem. Soc. Rev. 2012, 41, 687–702. [Google Scholar] [CrossRef]
- Rudzińska-Szostak, E.; Berlicki, Ł. Sequence engineering to control the helix handedness of peptide foldamers. Chem. Eur. J. 2017, 23, 14980–14986. [Google Scholar] [CrossRef]
- Huc, I. Aromatic oligoamide foldamers. Eur. J. Org. Chem. 2004, 17–29. [Google Scholar] [CrossRef]
- Saraogi, I.; Hamilton, A.D. Recent advances in the development of aryl-based foldamers. Chem. Soc. Rev. 2009, 38, 1726–1743. [Google Scholar] [CrossRef]
- Li, Z.T.; Hou, J.L.; Li, C. Peptide mimics by linear arylamides: A structural and functional diversity test. Acc. Chem. Res. 2008, 41, 1343–1353. [Google Scholar] [CrossRef] [PubMed]
- Menegazzo, I.; Fries, A.; Mammi, S.; Galeazzi, R.; Martelli, G.; Orena, M.; Rinaldi, S. Synthesis and structural characterisation as 12-helix of the hexamer of a β-amino acid tethered to a pyrrolidin-2-one ring. Chem. Commun. 2006, 47, 4915–4917. [Google Scholar] [CrossRef] [PubMed]
- Galeazzi, R.; Martelli, G.; Mazzanti, A.; Orena, M.; Rinaldi, S. Quaternary centres as a tool for modulating foldamer conformation. Chem. Eur. J. 2011, 17, 12564–12568. [Google Scholar] [CrossRef] [PubMed]
- Amabili, P.; Calvaresi, M.; Martelli, G.; Orena, M.; Rinaldi, S.; Sgolastra, F. Imidazolidinone-tethered α-hydrazidopeptides—Synthesis and conformational investigation. Eur. J. Org. Chem. 2019, 907–917. [Google Scholar] [CrossRef]
- Horne, W.S.; Gellman, S.H. Foldamers with heterogeneous backbones. Acc. Chem. Res. 2008, 41, 1399–1408. [Google Scholar] [CrossRef]
- Nair, R.V.; Vijayadas, K.N.; Roy, A.; Sanjayan, G.J. Heterogeneous foldamers from aliphatic–aromatic amino acid building blocks: Current trends and future prospects. Eur. J. Org. Chem. 2014, 7763–7780. [Google Scholar] [CrossRef]
- Gan, Q.; Wang, Y.; Jiang, H. Aromatic oligoamide foldamers: A paradigm for structure-property relationship. Curr. Org. Chem. 2011, 15, 1293–1301. [Google Scholar] [CrossRef]
- Zhang, D.W.; Zhao, X.; Hou, J.L.; Li, Z.T. Aromatic amide foldamers: Structures, properties, and functions. Chem. Rev. 2012, 112, 5271–5316. [Google Scholar] [CrossRef]
- Clerici, F.; Erba, E.; Gelmi, M.L.; Pellegrino, S. Non-standard amino acids and peptides: From self-assembly to nanomaterials. Tetrahedron Lett. 2016, 57, 5540–5550. [Google Scholar] [CrossRef]
- Gopalan, R.D.; Del Borgo, M.P.; Mechler, A.I.; Perlmutter, P.; Aguilar, M.-I. Geometrically precise building blocks: The self-assembly of β-peptides. Chem. Biol. 2015, 22, 1417–1423. [Google Scholar] [CrossRef]
- Wang, P.S.P.; Schepartz, A. β-Peptide bundles: Design. Build. Analyze. Biosynthesize. Chem. Commun. 2016, 52, 7420–7432. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.H.; Lee, H.S. Foldectures: 3D molecular architectures from self-assembly of peptide foldamers. Acc. Chem. Res. 2017, 50, 832–841. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, K.; Habila, N.; Del Borgo, M.P.; Aguilar, M.-I. Novel materials from the supramolecular self-assembly of short helical β3-peptide foldamers. Front. Chem. 2019, 7, 70. [Google Scholar] [CrossRef] [PubMed]
- Yamato, K.; Kline, M.; Gong, B. Cavity-containing, backbone-rigidified foldamers and macrocycles. Chem. Commun. 2012, 48, 12142–12158. [Google Scholar] [CrossRef]
- Gong, B.; Shao, Z. Self-assembling organic nanotubes with precisely defined, sub-nanometer pores: Formation and mass transport characteristics. Acc. Chem. Res. 2013, 46, 2856–2866. [Google Scholar] [CrossRef]
- Das, A.; Ghosh, S. H-bonding directed programmed supramolecular assembly of naphthalene-diimide (NDI) derivatives. Chem. Commun. 2016, 52, 6860–6872. [Google Scholar] [CrossRef]
- Ikkanda, B.A.; Iverson, B.L. Exploiting the interactions of aromatic units for folding and assembly in aqueous environments. Chem. Commun. 2016, 52, 7752–7759. [Google Scholar] [CrossRef]
- Yashima, E.; Ousaka, N.; Taura, D.; Shimomura, K.; Ikai, T.; Maeda, K. Supramolecular helical systems: Helical assemblies of small molecules, foldamers, and polymers with chiral amplification and their functions. Chem. Rev. 2016, 116, 13752–13990. [Google Scholar] [CrossRef]
- Ferrand, Y.; Huc, I. Designing helical molecular capsules based on folded aromatic amide oligomers. Acc. Chem. Res. 2018, 51, 970–977. [Google Scholar] [CrossRef]
- Shen, Y.; Chen, C.F. Helicenes: Synthesis and applications. Chem. Rev. 2012, 112, 1463–1535. [Google Scholar] [CrossRef]
- Gange, D.; Magnus, P.; Bass, L.; Arnold, E.V.; Clardy, J. Helixanes. The first primary helical molecules: Polyoxapolyspiroalkanones. J. Am. Chem. Soc. 1980, 102, 2134–2135. [Google Scholar] [CrossRef][Green Version]
- Tsubaki, K.; Tanaka, H.; Takaishi, K.; Miura, M.; Morikawa, H.; Furuta, T.; Tanaka, K.; Fuji, K.; Sasamori, T.; Tokitoh, N.; et al. Bottom-up synthesis of optically active oligonaphthalenes: Three different pathways for controlling axial chirality. J. Org. Chem. 2006, 71, 6579–6587. [Google Scholar] [CrossRef] [PubMed]
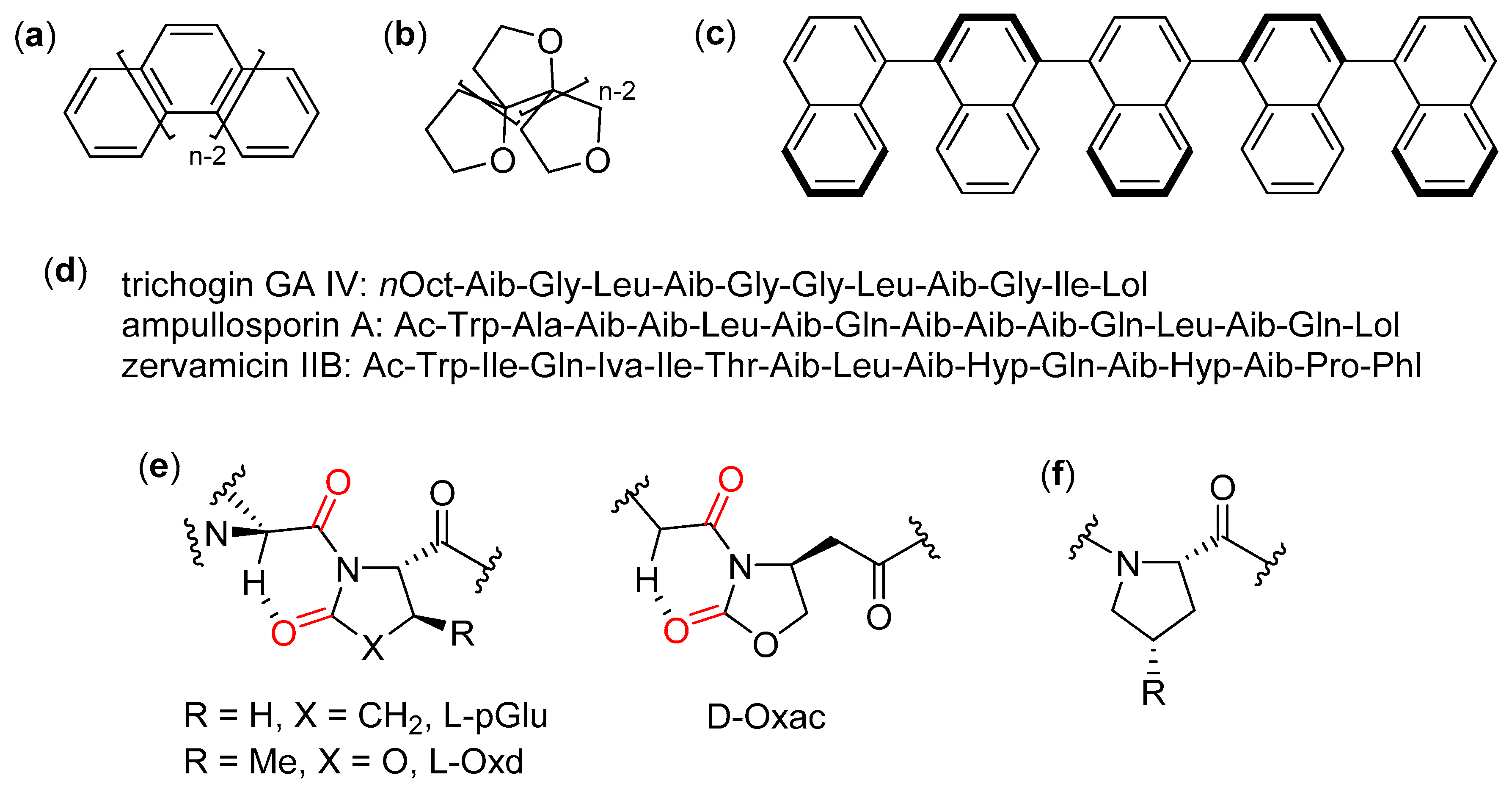
- Venanzi, M.; Gatto, E.; Formaggio, F.; Toniolo, C. The importance of being Aib. Aggregation and self-assembly studies on conformationally constrained oligopeptides. J. Pept. Sci. 2017, 23, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Toniolo, C.; Breckner, H. Peptaibiotics: Fungal Peptides Containing α-Dialkyl α-Amino Acids; Wiley-VCH: Weinheim, Germany, 2009. [Google Scholar]
- Zeilinger, S. Peptaibiotics and peptaibols from fungi. In Fungal Biomolecules: Sources, Applications and Recent Developments; Gupta, V.K., Mach, R.L., Sreenivasaprasad, S., Eds.; Wiley-Blackwell: Hoboken, NJ, USA, 2015; pp. 101–113. [Google Scholar]
- Salnikov, E.S.; De Zotti, M.; Bobone, S.; Mazzuca, C.; Raya, J.; Siano, A.S.; Peggion, C.; Toniolo, C.; Stella, L.; Bechinger, B. Trichogin GA IV alignment and oligomerization in phospholipid bilayers. ChemBioChem 2019, 20, 2141–2150. [Google Scholar] [CrossRef]
- Syryamina, V.N.; Samoilova, R.I.; Tsvetkov, Y.D.; Ischenko, A.V.; De Zotti, M.; Gobbo, M.; Toniolo, C.; Formaggio, F.; Dzuba, S.A. Peptides on the surface: Spin-label EPR and PELDOR study of adsorption of the antimicrobial peptides Trichogin GA IV and Ampullosporin A on the silica nanoparticles. Appl. Magn. Reson. 2016, 47, 309–320. [Google Scholar] [CrossRef]
- De Zotti, M.; Muzzi, B.; Gatto, E.; Di Napoli, B.; Mazzuca, C.; Palleschi, A.; Placidi, E.; Formaggio, F.; Toniolo, C.; Venanzi, M. Tuning the morphology of nanostructured peptide films by introduction of a secondary structure conformational constraint: A case-study of hierarchical self-assembly. J. Phys. Chem. B 2018, 122, 6305–6313. [Google Scholar] [CrossRef]
- De Zotti, M.; Corvi, G.; Gatto, E.; Di Napoli, B.; Mazzuca, C.; Palleschi, A.; Placidi, E.; Biondi, B.; Crisma, M.; Formaggio, F.; et al. Controlling the formation of peptide films: Fully-developed helical peptides are required to obtain a homogenous coating over a large area. ChemPlusChem 2019, 84, 1688–1696. [Google Scholar] [CrossRef]
- Afanasyeva, E.F.; Syryamina, V.N.; De Zotti, M.; Formaggio, F.; Toniolo, C.; Dzuba, S.A. Peptide antibiotic trichogin in model membranes: Self-association and capture of fatty acids. Biochim. Biophys. Acta Biomembr. 2019, 1861, 524–531. [Google Scholar] [CrossRef]
- Martí-Centelles, R.; Escuder, B. Morphology diversity of L-phenylalanine-based short peptide supramolecular aggregates and hydrogels. ChemNanoMat 2018, 4, 796–800. [Google Scholar] [CrossRef]
- Das, T.; Häring, M.; Haldar, D.; Díaz Díaz, D. Phenylalanine and derivatives as versatile low-molecular-weight gelators: Design, structure and tailored function. Biomater. Sci. 2018, 6, 38–59. [Google Scholar] [CrossRef]
- Aldilla, V.R.; Chen, R.; Martin, A.D.; Marjo, C.E.; Rich, A.M.; Black, D.S.; Thordarson, P.; Kumar, N. Anthranilamide-based short peptides self-assembled hydrogels as antibacterial agents. Sci. Rep. 2020, 10, 770. [Google Scholar] [CrossRef] [PubMed]
- Zanna, N.; Tomasini, C. Peptide-based physical gels endowed with thixotropic behaviour. Gels 2017, 3, 39. [Google Scholar] [CrossRef] [PubMed]
- Tomasini, C.; Zanna, N. Oxazolidinone-containing pseudopeptides: Supramolecular materials, fibers, crystals, and gels. Biopolymers 2017, 108, e22898. [Google Scholar] [CrossRef] [PubMed]
- Tomasini, C.; Angelici, G.; Castellucci, N. Foldamers based on oxazolidin-2-ones. Eur. J. Org. Chem. 2011, 3648–3669. [Google Scholar] [CrossRef]
- Dobitz, S.; Aronoff, M.R.; Helma Wennemers, H. Oligoprolines as molecular entities for controlling distance in biological and material sciences. Acc. Chem. Res. 2017, 50, 2420–2428. [Google Scholar] [CrossRef] [PubMed]
- Siebler, C.; Erdmann, R.S.; Wennemers, H. From azidoproline to functionalizable collagen. Chimia 2013, 67, 891–895. [Google Scholar] [CrossRef]
- Kirshenbaum, K.; Barron, A.E.; Goldsmith, R.A.; Armand, P.; Bradley, E.K.; Truong, K.T.V.; Dill, K.A.; Cohen, F.E.; Zuckermann, R.N. Sequence-specific polypeptoids: A diverse family of heteropolymers with stable secondary structure. Proc. Natl. Acad. Sci. USA 1998, 95, 4303–4308. [Google Scholar] [CrossRef]
- Wetzler, M.; Barron, A.E. Progress in the de novo design of structured peptoid protein mimics. Biopolymers 2011, 96, 556–560. [Google Scholar] [CrossRef]
- Szekely, T.; Caumes, C.; Roy, O.; Faure, S.; Taillefumier, C. α-Peptoids and related compounds: Synthesis and control of the conformation. C. R. Chim. 2013, 16, 318–330. [Google Scholar] [CrossRef]
- Weiser, L.J.; Santiso, E.E. Molecular modeling studies of peptoid polymers. AIMS Mater. Sci. 2017, 4, 1029–1051. [Google Scholar] [CrossRef]
- Culf, A.S. Peptoids as tools and sensors. Biopolymers 2019, 110, e23285. [Google Scholar] [CrossRef]
- De Riccardis, F. The challenge of conformational isomerism in cyclic peptoids. Eur. J. Org. Chem. 2020, 2981–2994. [Google Scholar] [CrossRef]
- Huang, K.; Wu, C.W.; Sanborn, T.J.; Patch, J.A.; Kirshenbaum, K.; Zuckermann, R.N.; Barron, A.E.; Radhakrishnan, I. A threaded loop conformation adopted by a family of peptoid nonamers. J. Am. Chem. Soc. 2006, 128, 1733–1738. [Google Scholar] [CrossRef]
- Stringer, J.R.; Crapster, J.A.; Guzei, I.A.; Blackwell, H.E. Construction of peptoids with all trans-amide backbones and peptoid reverse turns via the tactical incorporation of N-aryl side chains capable of hydrogen bonding. J. Org. Chem. 2010, 75, 6068–6078. [Google Scholar] [CrossRef]
- Yoo, B.; Kirshenbaum, K. Peptoid architectures: Elaboration, actuation, and application. Curr. Opin. Chem. Biol. 2008, 12, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Drexler, K.E. Peptoids at the 7th summit: Toward macromolecular systems engineering. Biopolymers 2011, 96, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zuckermann, R.N. Peptoid polymers: A highly designable bioinspired material. ACS Nano 2013, 7, 4715–4732. [Google Scholar] [CrossRef] [PubMed]
- Gangloff, N.; Luxenhofer, R. Peptoids for biomimetic hierarchical structures. In Hierarchical Macromolecular Structures: 60 Years after the Staudinger Nobel Prize II; Percec, V., Ed.; Springer: Cham, Switzerland, 2013; Volume 262, pp. 389–413. [Google Scholar]
- Rosales, A.M.; Segalman, R.A.; Zuckermann, R.N. Polypeptoids: A model system to study the effect of monomer sequence on polymer properties and self-assembly. Soft Matter 2013, 9, 8400–8414. [Google Scholar] [CrossRef]
- Sun, J.; Proulx, C.; Zuckermann, R.N. Precision sequence control in bioinspired peptoid polymers. In Sequence-Controlled Polymers: Synthesis, Self-Assembly, and Properties; Lutz, J.-F., Meyer, T.Y., Ouchi, M., Sawamoto, M., Eds.; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2014; Volume 1170, pp. 35–53. [Google Scholar]
- Lau, K.H.A. Peptoids for biomaterials science. Biomater. Sci. 2014, 2, 627–633. [Google Scholar] [CrossRef]
- Knight, A.S.; Zhou, E.F.; Francis, M.B.; Zuckermann, R.N. Sequence programmable peptoid polymers for diverse materials applications. Adv. Mater. 2015, 27, 5665–5691. [Google Scholar] [CrossRef]
- Secker, C.; Brosnan, S.M.; Luxenhofer, R.; Schlaad, H. Poly(α-peptoid)s revisited: Synthesis, properties, and use as biomaterial. Macromol. Biosci. 2015, 15, 881–891. [Google Scholar] [CrossRef]
- Cai, S.L.; Zhang, W.G.; Zuckermann, R.N.; Li, Z.T.; Zhao, X.; Liu, Y. The organic flatland—Recent advances in synthetic 2D organic layers. Adv. Mater. 2015, 27, 5762–5770. [Google Scholar] [CrossRef]
- Robertson, E.J.; Battigelli, A.; Proulx, C.; Mannige, R.V.; Haxton, T.K.; Yun, L.; Whitelam, S.; Zuckermann, R.N. Design, synthesis, assembly, and engineering of peptoid nanosheets. Acc. Chem. Res. 2016, 49, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, S.D.; Saha, N.; Zandraa, O.; Zuckermann, R.N.; Sáha, P. Peptoids and polypeptoids: Biomimetic and bioinspired materials for biomedical applications. Polym. Bull. 2017, 74, 3455–3466. [Google Scholar] [CrossRef]
- Liu, L.; Klausen, L.H.; Dong, M. Two-dimensional peptide based functional nanomaterials. Nano Today 2018, 23, 40–58. [Google Scholar] [CrossRef]
- Battigelli, A. Design and preparation of organic nanomaterials using self-assembled peptoids. Biopolymers 2019, 110, e23265. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cai, B.; Cui, L.; Chen, C.L. Peptoid-based hierarchically-structured biomimetic nanomaterials: Synthesis, characterization and applications. Sci. China Mater. 2020, 63, 1099–1112. [Google Scholar] [CrossRef]
- Xuan, S.; Zuckermann, R.N. Diblock copolypeptoids: A review of phase separation, self-assembly and biological applications. J. Mater. Chem. B 2020, 8, 5380–5394. [Google Scholar] [CrossRef]
- Campbell, V.E.; De Hatten, X.; Delsuc, N.; Kauffmann, B.; Huc, I.; Nitschke, J.R. Cascading transformations within a dynamic self-assembled system. Nat. Chem. 2010, 2, 684–687. [Google Scholar] [CrossRef]
- Jeon, H.G.; Lee, H.K.; Lee, S.; Jeong, K.S. Foldamer-based helicate displaying reversible switching between two distinct conformers. Chem. Commun. 2018, 54, 5740–5743. [Google Scholar] [CrossRef]
- Liu, C.Z.; Koppireddi, S.; Wang, H.; Zhang, D.W.; Li, Z.T. Halogen bonding-directed supramolecular quadruple and double helices from hydrogen bonded arylamide foldamers. Angew. Chem. Int. Ed. 2019, 58, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Parks, F.C.; Zhao, W.; Flood, A.H. Sequence-controlled stimuli-responsive single-double helix conversion between 1:1 and 2:2 chloride-foldamer complexes. J. Am. Chem. Soc. 2018, 140, 15477–15486. [Google Scholar] [CrossRef] [PubMed]
- Borissov, A.; Marques, I.; Lim, J.Y.C.; Félix, V.; Smith, M.D.; Beer, P.D. Anion recognition in water by charge-neutral halogen and chalcogen bonding foldamer receptors. J. Am. Chem. Soc. 2019, 141, 4119–4129. [Google Scholar] [CrossRef]
- Shi, Z.M.; Wu, C.F.; Zhou, T.Y.; Zhang, D.W.; Zhao, X.; Li, Z.T. Foldamer-based chiral supramolecular alternate block copolymers tuned by ion-pair binding. Chem. Commun. 2013, 49, 2673–2675. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.H.Y.; Ng, M.; Leung, S.Y.L.; Lam, W.H.; Yam, V.W.W. Synthesis of luminescent Platinum(II) 2,6-bis(N-dodecylbenzimidazol-2-yl)pyridine foldamers and their supramolecular assembly and metallogel formation. J. Am. Chem. Soc. 2017, 139, 8639–8645. [Google Scholar] [CrossRef]
- Li, C.; Zhu, Y.Y.; Yi, H.P.; Li, C.Z.; Jiang, X.K.; Li, Z.T.; Yu, Y.H. Strong Stacking between F···H-N hydrogen-bonded foldamers and fullerenes: Formation of supramolecular nano networks. Chem. Eur. J. 2007, 13, 9990–9998. [Google Scholar] [CrossRef]
- Ghosh, T.; Fridman, N.; Kosa, M.; Maayan, G. Self-assembled cyclic structures from copper(II) peptoids. Angew. Chem. Int. Ed. 2018, 57, 7703–7708. [Google Scholar] [CrossRef]
- Tigger-Zaborov, H.; Maayan, G. Aggregation of inorganic nanoparticles mediated by biomimetic oligomers. Org. Biomol. Chem. 2015, 13, 8978–8992. [Google Scholar] [CrossRef]
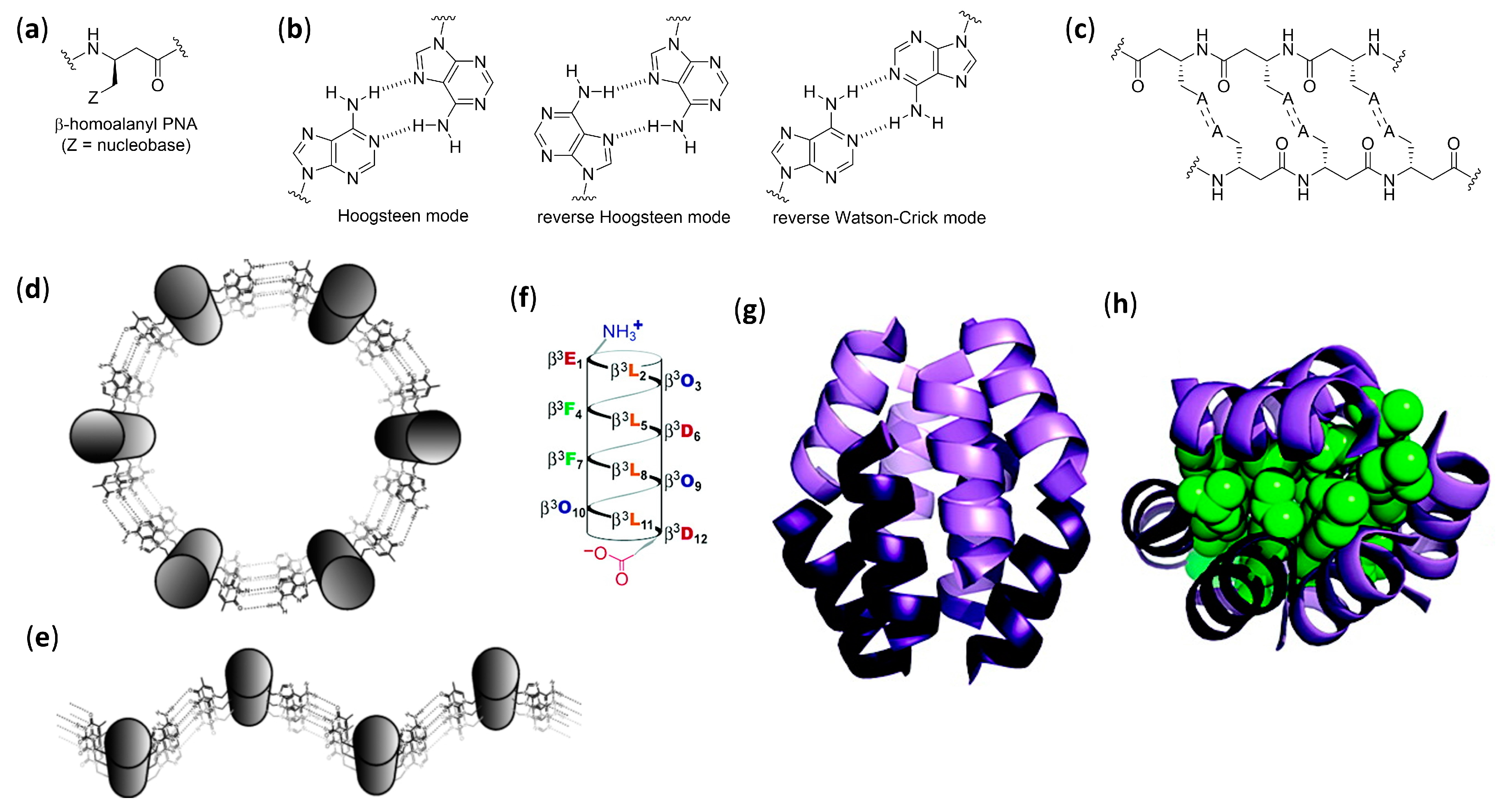
- Diederichsen, U.; Schmitt, H.W. β-Homoalanyl PNAs: Synthesis and indication of higher ordered structures. Angew. Chem. Int. Ed. 1998, 37, 302–305. [Google Scholar] [CrossRef]
- Diederichsen, U.; Schmitt, H.W. β-Homoalanyl-PNA: A special case of β-peptides with β-sheet-like backbone conformation; Organization in higher ordered structures. Eur. J. Org. Chem. 1998, 827–835. [Google Scholar] [CrossRef]
- Kritzer, J.A.; Tirado-Rives, J.; Hart, S.A.; Lear, J.D.; Jorgensen, W.L.; Schepartz, A. Relationship between side chain structure and 14-helix stability of β3-peptides in water. J. Am. Chem. Soc. 2005, 127, 167–178. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rathore, N.; Gellman, S.H.; De Pablo, J.J. Thermodynamic stability of β-peptide helices and the role of cyclic residues. Biophys. J. 2006, 91, 3425–3435. [Google Scholar] [CrossRef] [PubMed]
- Brückner, A.M.; Chakraborty, P.; Gellman, S.H.; Diederichsen, U. Molecular architecture with functionalized β-peptide helices. Angew. Chem. Int. Ed. 2003, 42, 4395–4399. [Google Scholar] [CrossRef]
- Chakraborty, P.; Diederichsen, U. Three-dimensional organization of helices: Design principles for nucleobase-functionalized β-peptides. Chem. Eur. J. 2005, 11, 3207–3216. [Google Scholar] [CrossRef] [PubMed]
- Weiß, A.; Diederichsen, U. Uniformly nucleobase-functionalized β-peptide helices: Watson-Crick pairing or nonspecific aggregation. Eur. J. Org. Chem. 2007, 5531–5539. [Google Scholar] [CrossRef]
- Srivastava, R.; Ray, A.K.; Diederichsen, U. Higher aggregation of β-peptide networks controlled by nucleobase pairing. Eur. J. Org. Chem. 2009, 4793–4800. [Google Scholar] [CrossRef]
- Raguse, T.L.; Lai, J.R.; LePlae, P.R.; Gellman, S.H. Toward β-peptide tertiary structure: Self-association of an amphiphilic 14-helix in aqueous solution. Org. Lett. 2001, 3, 3963–3966. [Google Scholar] [CrossRef]
- Curtis, R.; Pophale, R.; Deem, M. Monte Carlo simulations of the homopolypeptide pair potential of mean force. Fluid Phase Equilib. 2006, 241, 354–367. [Google Scholar] [CrossRef]
- Müller, M.M.; Windsor, M.A.; Pomerantz, W.C.; Gellman, S.H.; Hilvert, D. A rationally designed aldolase foldamer. Angew. Chem. Int. Ed. 2009, 48, 922–925. [Google Scholar] [CrossRef]
- Qiu, J.X.; Petersson, E.J.; Matthews, E.E.; Schepartz, A. Toward β-amino acid proteins: A cooperatively folded β-peptide quaternary structure. J. Am. Chem. Soc. 2006, 128, 11338–11339. [Google Scholar] [CrossRef]
- Daniels, D.S.; Petersson, E.J.; Qiu, J.X.; Schepartz, A. High-resolution structure of a β-peptide bundle. J. Am. Chem. Soc. 2007, 129, 1532–1533. [Google Scholar] [CrossRef]
- Goodman, J.L.; Petersson, E.J.; Daniels, D.S.; Qiu, J.X.; Schepartz, A. Biophysical and structural characterization of a robust octameric β-peptide bundle. J. Am. Chem. Soc. 2007, 129, 14746–14751. [Google Scholar] [CrossRef] [PubMed]
- Molski, M.A.; Goodman, J.L.; Craig, C.J.; Meng, H.; Kumar, K.; Schepartz, A. β-Peptide bundles with fluorous cores. J. Am. Chem. Soc. 2010, 132, 3658–3659. [Google Scholar] [CrossRef]
- Wang, P.S.P.; Nguyen, J.B.; Schepartz, A. Design and high-resolution structure of a β3-peptide bundle catalyst. J. Am. Chem. Soc. 2014, 136, 6810–6813. [Google Scholar] [CrossRef] [PubMed]
- Melicher, M.S.; Chu, J.; Walker, A.S.; Miller, S.J.; Baxter, R.H.G.; Schepartz, A. A β-boronopeptide bundle of known structure as a vehicle for polyol recognition. Org. Lett. 2013, 15, 5048–5051. [Google Scholar] [CrossRef] [PubMed]
- Melicher, M.S.; Walker, A.S.; Shen, J.; Miller, S.J.; Schepartz, A. Improved carbohydrate recognition in water with an electrostatically enhanced β-peptide bundle. Org. Lett. 2015, 17, 4718–4721. [Google Scholar] [CrossRef]
- Szolnoki, É.; Hetényi, A.; Mándity, I.M.; Fülöp, F.; Martinek, T.A. Foldameric β-H18/20P mixed helix stabilized by head-to-tail contacts: A way to higher-order structures. Eur. J. Org. Chem. 2013, 3555–3559. [Google Scholar] [CrossRef]
- Hetényi, A.; Mándity, I.M.; Martinek, T.A.; Tóth, G.K.; Fülöp, F. Chain-length-dependent helical motifs and self-association of β-peptides with constrained side chains. J. Am. Chem. Soc. 2005, 127, 547–553. [Google Scholar] [CrossRef]
- Clark, T.D.; Buehler, L.K.; Ghadiri, M.R. Self-assembling cyclic β3-peptide nanotubes as artificial transmembrane ion channels. J. Am. Chem. Soc. 1998, 120, 651–656. [Google Scholar] [CrossRef]
- Ghadiri, M.R.; Granja, J.R.; Milligan, R.A.; McRee, D.E.; Khazanovich, N. Self-assembling organic nanotubes based on a cyclic peptide architecture. Nature 1993, 366, 324–327. [Google Scholar] [CrossRef]
- Martinek, T.A.; Hetényi, A.; Fülöp, L.; Mándity, I.M.; Tóth, G.K.; Dékány, I.; Fülöp, F. Secondary structure dependent self-assembly of β-peptides into nanosized fibrils and membranes. Angew. Chem. Int. Ed. 2006, 45, 2396–2400. [Google Scholar] [CrossRef]
- Mándity, I.M.; Fülöp, L.; Vass, E.; Tóth, G.K.; Martinek, T.A.; Fülöp, F. Building β-peptide H10/12 foldamer helices with six-membered cyclic side-chains: Fine-tuning of folding and self-assembly. Org. Lett. 2010, 12, 5584–5587. [Google Scholar] [CrossRef]
- Mándity, I.M.; Monsignori, A.; Fülöp, L.; Forró, E.; Fülöp, F. Exploiting aromatic interactions for β-peptide foldamer helix stabilization: A significant design element. Chem. Eur. J. 2014, 20, 4591–4597. [Google Scholar] [CrossRef]
- Martinek, T.A.; Tóth, G.K.; Vass, E.; Hollósi, M.; Fülöp, F. cis-2-Aminocyclopentanecarboxylic acid oligomers adopt a sheetlike structure: Switch from helix to nonpolar strand. Angew. Chem. Int. Ed. 2002, 41, 1718–1721. [Google Scholar] [CrossRef]
- Pomerantz, W.C.; Abbott, N.L.; Gellman, S.H. Lyotropic liquid crystals from designed helical β-peptides. J. Am. Chem. Soc. 2006, 128, 8730–8731. [Google Scholar] [CrossRef]
- Pomerantz, W.C.; Yuwono, V.M.; Pizzey, C.L.; Hartgerink, J.D.; Abbott, N.L.; Gellman, S.H. Nanofibers and lyotropic liquid crystals from a class of self-assembling β-peptides. Angew. Chem. Int. Ed. 2008, 47, 1241–1244. [Google Scholar] [CrossRef]
- Thiele, C.M.; Pomerantz, W.C.; Abbott, N.L.; Gellman, S.H. Lyotropic liquid crystalline phases from helical β-peptides as alignment media. Chem. Commun. 2011, 47, 502–504. [Google Scholar] [CrossRef]
- Pomerantz, W.C.; Yuwono, V.M.; Drake, R.; Hartgerink, J.D.; Abbott, N.L.; Gellman, S.H. Lyotropic liquid crystals formed from ACHC-rich β-Peptides. J. Am. Chem. Soc. 2011, 133, 13604–13613. [Google Scholar] [CrossRef]
- Pizzey, C.L.; Pomerantz, W.C.; Sung, B.J.; Yuwono, V.M.; Gellman, S.H.; Hartgerink, J.D.; Arun Yethiraj, A.L.; Abbott, N.L. Characterization of nanofibers formed by self-assembly of β-peptide oligomers using small angle x-ray scattering. J. Chem. Phys. 2008, 129, 09B603. [Google Scholar] [CrossRef]
- Mason, J.M.; Arndt, K.M. Coiled coil domains: Stability, specificity, and biological implications. ChemBioChem 2004, 5, 170–176. [Google Scholar] [CrossRef]
- Oshea, E.K.; Klemm, J.D.; Kim, P.S.; Alber, T. X-ray structure of the GCN4 leucine zipper, a two-stranded, parallel coiled coil. Science 1991, 254, 539–544. [Google Scholar] [CrossRef]
- Appella, D.H.; Christianson, L.A.; Karle, I.; Powell, D.R.; Gellman, S.H. β-Peptide foldamers: Robust helix formation in a new family of β-amino acid oligomers. J. Am. Chem. Soc. 1996, 118, 13071–13072. [Google Scholar] [CrossRef]
- Appella, D.H.; Christianson, L.A.; Karle, I.; Powell, D.R.; Gellman, S.H. Synthesis and characterization of trans-2-aminocyclohexanecarboxylic acid oligomers: An unnatural helical secondary structure and implications for β-peptide tertiary structure. J. Am. Chem. Soc. 1999, 121, 6206–6212. [Google Scholar] [CrossRef]
- Pomerantz, W.C.; Cadwell, K.D.; Hsu, Y.J.; Gellman, S.H.; Abbott, N.L. Sequence dependent behavior of amphiphilic β-peptides on gold surfaces. Chem. Mater. 2007, 19, 4436–4441. [Google Scholar] [CrossRef]
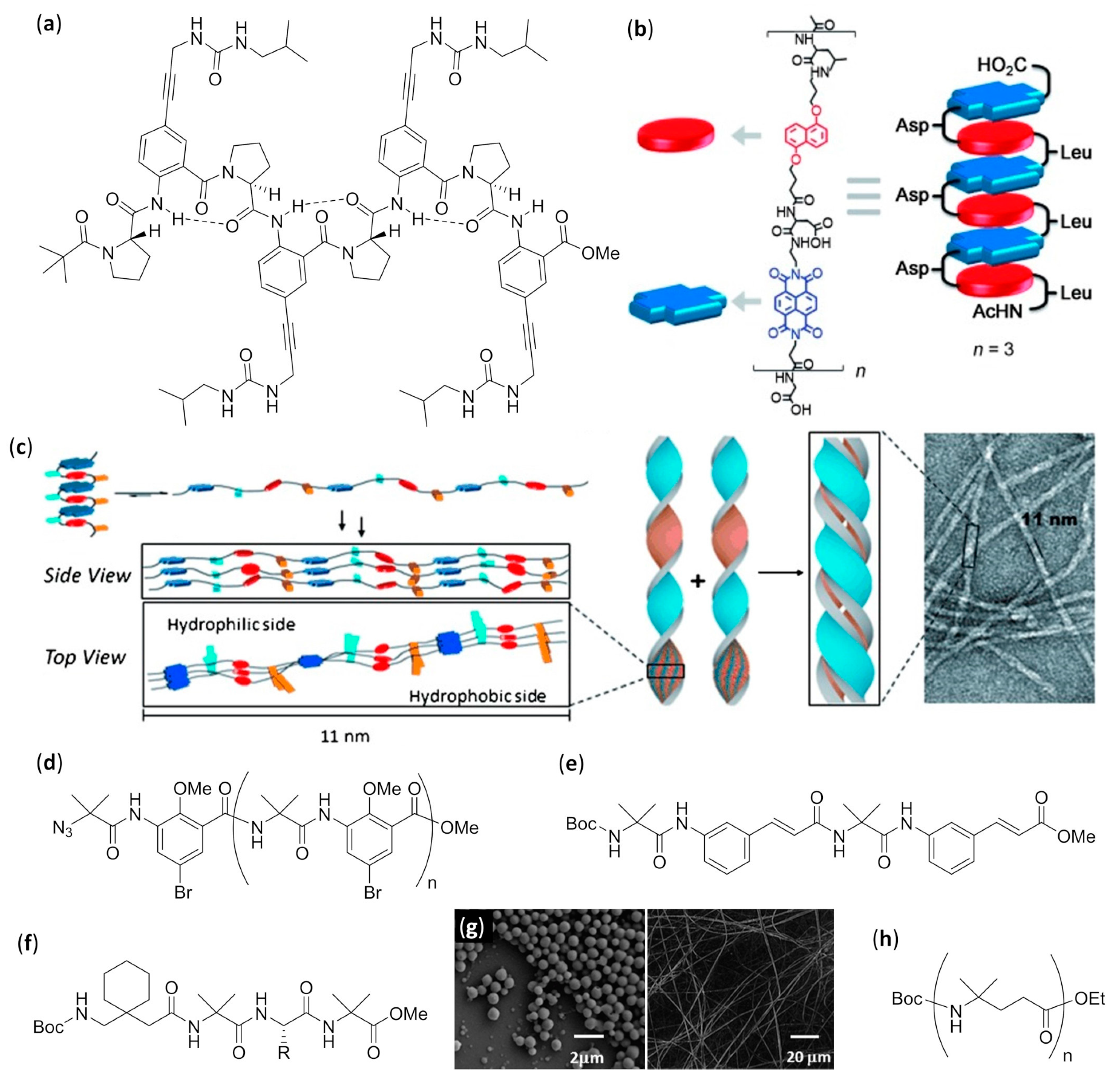
- Del Borgo, M.P.; Mechler, A.I.; Traore, D.; Forsyth, C.; Wilce, J.A.; Wilce, M.C.J.; Aguilar, M.-I.; Perlmutter, P. Supramolecular self-assembly of N-acetyl-capped β-peptides leads to nano- to macroscale fiber formation. Angew. Chem. Int. Ed. 2013, 52, 8266–8270. [Google Scholar] [CrossRef]
- Seoudi, R.S.; Del Borgo, M.P.; Kulkarni, K.; Perlmutter, P.; Aguilar, M.-I.; Mechler, A. Amino acid sequence controls the self-assembled superstructure morphology of N-acetylated tri-β3-peptides. Pure Appl. Chem. 2015, 87, 1021–1028. [Google Scholar] [CrossRef]
- Seoudi, R.S.; Dowd, A.; Del Borgo, M.; Kulkarni, K.; Perlmutter, P.; Aguilar, M.-I.; Mechler, A. Supramolecular self-assembly of 14-helical nanorods with tunable linear and dendritic hierarchical morphologies. New J. Chem. 2015, 39, 3280–3287. [Google Scholar] [CrossRef]
- Motamed, S.; Del Borgo, M.; Kulkarni, K.; Habila, N.; Zhou, K.; Perlmutter, P.; Forsythe, J.S.; Aguilar, M.-I. A self-assembling β-peptide hydrogel for neural tissue engineering. Soft Matter 2016, 12, 2243–2246. [Google Scholar] [CrossRef]
- Luder, K.; Kulkarni, K.; Lee, H.W.; Widdop, R.E.; Del Borgo, M.P.; Aguilar, M.-I. Decorated self-assembling β3-tripeptide foldamers form cell adhesive scaffolds. Chem. Commun. 2016, 52, 4549–4552. [Google Scholar] [CrossRef]
- Kulkarni, K.; Motamed, S.; Habila, N.; Perlmutter, P.; Forsythe, J.S.; Aguilar, M.-I.; Del Borgo, M. Orthogonal strategy for the synthesis of dual-functionalised β3-peptide based hydrogels. Chem. Commun. 2016, 52, 5844–5847. [Google Scholar] [CrossRef]
- Seoudi, R.S.; Hinds, M.G.; Wilson, D.J.D.; Adda, C.G.; Del Borgo, M.; Aguilar, M.-I.; Perlmutter, P.; Mechler, A. Self-assembled nanomaterials based on beta (β3) tetrapeptides. Nanotechnology 2016, 27, 135606. [Google Scholar] [CrossRef] [PubMed]
- Del Borgo, M.P.; Kulkarni, K.; Tonta, M.A.; Ratcliffe, J.L.; Seoudi, R.; Mechler, A.I.; Perlmutter, P.; Parkington, H.C.; Aguilar, M.-I. β3-Tripeptides act as sticky ends to self-assemble into a bioscaffold. APL Bioeng. 2018, 2, 026104. [Google Scholar] [CrossRef] [PubMed]
- Christofferson, A.J.; Al-Garawi, Z.S.; Todorova, N.; Turner, J.; Del Borgo, M.P.; Serpell, L.C.; Aguilar, M.-I.; Yarovsky, I. Identifying the coiled-coil triple helix structure of β-peptide nanofibers at atomic resolution. ACS Nano 2018, 12, 9101–9109. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, K.; Hung, J.; Fulcher, A.J.; Chan, A.H.P.; Hong, A.; Forsythe, J.S.; Aguilar, M.-I.; Wise, S.G.; Del Borgo, M.P. β3-Tripeptides co-assemble into fluorescent hydrogels for serial monitoring in vivo. ACS Biomater. Sci. Eng. 2018, 4, 3843–3847. [Google Scholar] [CrossRef]
- Motamed, S.; Del Borgo, M.P.; Zhou, K.; Kulkarni, K.; Crack, P.J.; Merson, T.D.; Aguilar, M.-I.; Finkelstein, D.I.; Forsythe, J.S. Migration and differentiation of neural stem cells diverted from the subventricular zone by an injectable self-assembling β-peptide hydrogel. Front. Bioeng. Biotechnol. 2019, 7, 315. [Google Scholar] [CrossRef] [PubMed]
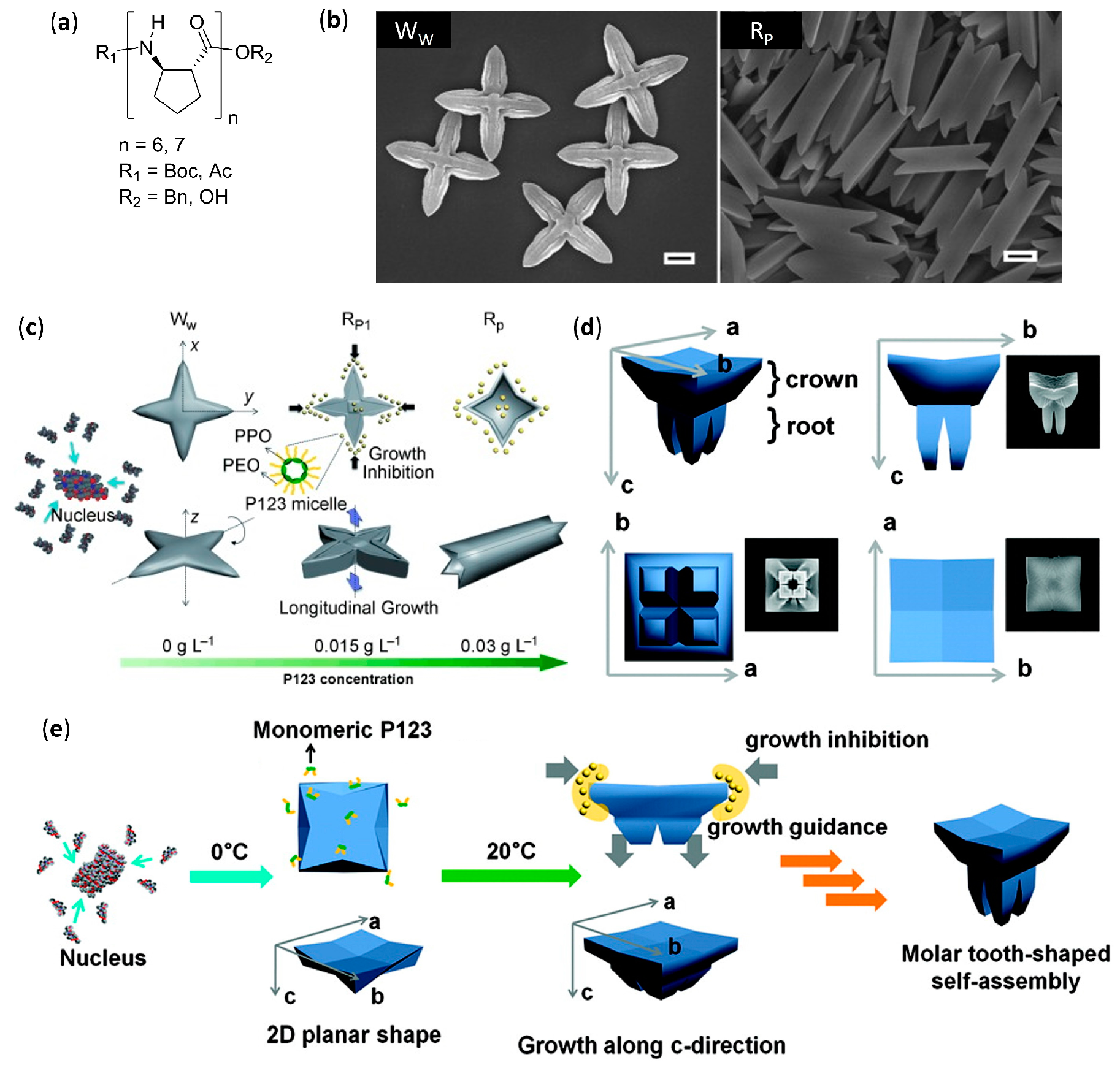
- Kwon, S.; Jeon, A.; Yoo, S.H.; Chung, I.S.; Lee, H.S. Unprecedented molecular architectures by the controlled self-assembly of a β-peptide foldamer. Angew. Chem. Int. Ed. 2010, 49, 8232–8236. [Google Scholar] [CrossRef]
- Kwon, S.; Shin, H.S.; Gong, J.; Eom, J.-H.; Jeon, A.; Yoo, S.H.; Chung, I.S.; Cho, S.J.; Lee, H.-S. Self-assembled peptide architecture with a tooth shape: Folding into shape. J. Am. Chem. Soc. 2011, 133, 17618–17621. [Google Scholar] [CrossRef]
- Kwon, S.; Kang, K.; Jeon, A.; Park, J.H.; Choi, I.S.; Lee, H.S. Evaporation-induced self-assembly of trans-2-aminocyclopentanecarboxylic acid hexamers. Tetrahedron 2012, 68, 4368–4373. [Google Scholar] [CrossRef]
- Kim, J.; Kwon, S.; Kim, S.H.; Lee, C.-K.; Lee, J.-H.; Cho, S.J.; Lee, H.-S.; Ihee, H. Microtubes with rectangular cross-section by self-assembly of a short β-peptide foldamer. J. Am. Chem. Soc. 2012, 134, 20573–20576. [Google Scholar] [CrossRef]
- Yoo, S.H.; Eom, T.; Kwon, S.; Gong, J.; Kim, J.; Cho, S.J.; Driver, R.W.; Lee, Y.; Kim, H.; Lee, H.-S. Foldecture as a core material with anisotropic surface characteristics. J. Am. Chem. Soc. 2015, 137, 2159–2162. [Google Scholar] [CrossRef]
- Kwon, S.; Kim, B.J.; Lim, H.-K.; Kang, K.; Yoo, S.H.; Gong, J.; Yoon, E.; Lee, J.; Choi, I.S.; Kim, H.; et al. Magnetotactic molecular architectures from self-assembly of β-peptide foldamers. Nat. Commun. 2015, 6, 8747. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Eom, T.; Lee, W.; Roy, A.; Kwon, S.; Kim, H.; Lee, H.-S. Self-assembly of a β-peptide foldamer: The role of the surfactant in three-dimensional shape selection. ChemPlusChem 2019, 84, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Appella, D.H.; Christianson, L.A.; Klein, D.A.; Richards, M.R.; Powell, D.R.; Gellman, S.H. Synthesis and structural characterization of helix-forming β-peptides: trans-2-Aminocyclopentanecarboxylic acid oligomers. J. Am. Chem. Soc. 1999, 121, 7574–7581. [Google Scholar] [CrossRef]
- Eddleston, M.D.; Jones, W. Formation of tubular crystals of pharmaceutical compounds. Cryst. Growth Des. 2010, 10, 365–370. [Google Scholar] [CrossRef]
- De, S.; Chi, B.; Granier, T.; Qi, T.; Maurizot, V.; Huc, I. Designing cooperatively folded abiotic uni- and multimolecular helix bundles. Nat. Chem. 2018, 10, 51–57. [Google Scholar] [CrossRef]
- Zhao, Y.; Connor, A.L.; Sobiech, T.A.; Gong, B. Effects of oligomer length, solvents, and temperature on the self-association of aromatic oligoamide foldamers. Org. Lett. 2018, 20, 5486–5489. [Google Scholar] [CrossRef]
- Berl, V.; Huc, I.; Khoury, R.G.; Lehn, J.-M. Helical molecular programming: Folding of oligopyridine-dicarboxamides into molecular single helices. Chem. Eur. J. 2001, 7, 2798–2809. [Google Scholar] [CrossRef]
- Buffeteau, T.; Ducasse, L.; Poniman, L.; Delsuc, N.; Huc, I. Vibrational circular dichroism and ab initio structure elucidation of an aromatic foldamer. Chem. Commun. 2006, 2714–2716. [Google Scholar] [CrossRef]
- Berl, V.; Huc, I.; Khoury, R.G.; Krische, M.J.; Lehn, J.-M. Interconversion of single and double helices formed from synthetic molecular strands. Nature 2000, 407, 720–723. [Google Scholar] [CrossRef]
- Berl, V.; Huc, I.; Khoury, R.G.; Lehn, J.-M. Helical molecular programming: Supramolecular double helices by dimerization of helical oligopyridine-dicarboxamide strands. Chem. Eur. J. 2001, 7, 2810–2820. [Google Scholar] [CrossRef]
- Acocella, A.; Venturini, A.; Zerbetto, F. Pyridinedicarboxamide strands form double helices via an activated slippage mechanism. J. Am. Chem. Soc. 2004, 126, 2362–2367. [Google Scholar] [CrossRef]
- Baptiste, B.; Zhu, J.; Haldar, D.; Kauffmann, B.; Léger, J.-M.; Huc, I. Hybridization of long pyridine-carboxamide oligomers into multi-turn double helices: Slow strand association and dissociation, solvent dependence, and solid state structures. Chem. Asian J. 2010, 5, 1364–1375. [Google Scholar]
- Dolain, C.; Zhan, C.; Léger, J.M.; Daniels, L.; Huc, I. Folding directed N-oxidation of oligopyridine-dicarboxamide strands and hybridization of oxidized oligomers. J. Am. Chem. Soc. 2005, 127, 2400–2401. [Google Scholar] [CrossRef]
- Zhan, C.; Léger, J.-M.; Huc, I. Cross-hybridization of pyridinedicarboxamide helical strands and their N-oxides. Chem. Eur. J. 2006, 45, 4625–4628. [Google Scholar]
- Berni, E.; Kauffmann, B.; Bao, C.; Lefeuvre, J.; Bassani, D.M.; Huc, I. Assessing the mechanical properties of a molecular spring. Chem. Eur. J. 2007, 13, 8463–8469. [Google Scholar] [CrossRef]
- Singleton, M.L.; Pirotte, G.; Kauffmann, B.; Ferrand, Y.; Huc, I. Increasing the size of an aromatic helical foldamer cavity by strand intercalation. Angew. Chem. Int. Ed. 2014, 53, 13140–13144. [Google Scholar] [CrossRef]
- Berni, E.; Garric, J.; Lamit, C.; Kauffmann, B.; Léger, J.-M.; Huc, I. Interpenetrating single helical capsules. Chem. Commun. 2008, 1968–1970. [Google Scholar] [CrossRef]
- Bao, C.; Gan, Q.; Kauffmann, B.; Jiang, H.; Huc, I. A self-assembled foldamer capsule: Combining single and double helical segments in one aromatic amide sequence. Chem. Eur. J. 2009, 15, 11530–11536. [Google Scholar] [CrossRef]
- Gan, Q.; Bao, C.; Kauffmann, B.; Grélard, A.; Xiang, J.; Liu, S.; Huc, I.; Jiang, H. Quadruple and double helices of 8-fluoroquinoline oligoamides. Angew. Chem. Int. Ed. 2008, 47, 1715–1718. [Google Scholar] [CrossRef]
- Gan, Q.; Li, F.; Li, G.; Kauffmann, B.; Xiang, J.; Huc, I.; Jiang, H. Heteromeric double helix formation by cross-hybridization of chloro and fluoro-substituted quinoline oligoamides. Chem. Commun. 2010, 46, 297–299. [Google Scholar] [CrossRef]
- Ferrand, Y.; Kendhale, A.M.; Garric, J.; Kauffmann, B.; Huc, I. Parallel and antiparallel triple helices of naphthyridine oligoamides. Angew. Chem. Int. Ed. 2010, 49, 1778–1781. [Google Scholar] [CrossRef] [PubMed]
- Chandramouli, N.; Ferrand, Y.; Kaufmann, B.; Huc, I. Citric acid encapsulation by a double-helical foldamer in competitive solvents. Chem. Commun. 2016, 52, 3939–3942. [Google Scholar] [CrossRef] [PubMed]
- Denisov, S.A.; Gan, Q.; Wang, X.; Scarpantonio, L.; Ferrand, Y.; Kauffmann, B.; Jonusauskas, G.; Huc, I.; McClenaghan, N.D. Electronic energy transfer modulation in a dynamic foldaxane: Proof-of-principle of a lifetime-based conformation probe. Angew. Chem. Int. Ed. 2016, 55, 1328–1333. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wicher, B.; Ferrand, Y.; Huc, I. Orchestrating directional molecular motions: Kinetically controlled supramolecular pathways of a helical host on rodlike guests. J. Am. Chem. Soc. 2017, 139, 9350–9358. [Google Scholar] [CrossRef] [PubMed]
- Ferrand, Y.; Gan, Q.; Kauffmann, B.; Jiang, H.; Huc, I. Template-induced screw motions within an aromatic amide foldamer double helix. Angew. Chem. Int. Ed. 2011, 50, 7572–7575. [Google Scholar] [CrossRef]
- Gan, Q.; Ferrand, Y.; Chandramouli, N.; Kauffmann, B.; Aube, C.; Dubreuil, D.; Huc, I. Identification of a foldaxane kinetic byproduct during guest-induced single to double helix conversion. J. Am. Chem. Soc. 2012, 134, 15656–15659. [Google Scholar] [CrossRef]
- Tanaka, Y.; Katagiri, H.; Furusho, Y.; Yashima, E. A modular strategy to artificial double helices. Angew. Chem. Int. Ed. 2005, 44, 3867–3870. [Google Scholar] [CrossRef]
- Ito, H.; Furusho, Y.; Hasegawa, T.; Yashima, E. Sequence-and chain-length-specific complementary double-helix formation. J. Am. Chem. Soc. 2008, 130, 14008–14015. [Google Scholar] [CrossRef]
- Yamada, H.; Furusho, Y.; Ito, H.; Yashima, E. Complementary double helix formation through template synthesis. Chem. Commun. 2010, 46, 3487–3489. [Google Scholar] [CrossRef][Green Version]
- Shang, J.; Gan, Q.; Dawson, S.J.; Rosu, F.; Jiang, H.; Ferrand, Y.; Huc, I. Self-association of aromatic oligoamide foldamers into double helices in water. Org. Lett. 2014, 16, 4992–4995. [Google Scholar] [CrossRef]
- Méndez-Ardoy, A.; Markandeya, N.; Li, X.; Tsai, Y.-T.; Pecastaings, G.; Buffeteau, T.; Maurizot, V.; Muccioli, L.; Castet, F.; Huc, I.; et al. Multi-dimensional charge transport in supramolecular helical foldamer assemblies. Chem. Sci. 2017, 8, 7251–7257. [Google Scholar] [CrossRef] [PubMed]
- Adam, C.; Faour, L.; Bonnin, V.; Breton, T.; Levillain, E.; Sallé, M.; Gautier, C.; Canevet, D. Supramolecular chemistry of helical foldamers at the solid-liquid interface: Self-assembled monolayers and anion recognition. Chem. Commun. 2019, 55, 8426–8429. [Google Scholar] [CrossRef] [PubMed]
- Faour, L.; Adam, C.; Gautier, C.; Goeb, S.; Allain, M.; Levillain, E.; Canevet, D.; Sallé, M. Redox-controlled hybridization of helical foldamers. Chem. Commun. 2019, 55, 5743–5746. [Google Scholar] [CrossRef] [PubMed]
- Hein, R.; Borissov, A.; Smith, M.D.; Beer, P.D.; Davis, J.J. A halogen-bonding foldamer molecular film for selective reagentless anion sensing in water. Chem. Commun. 2019, 55, 4849–4852. [Google Scholar] [CrossRef]
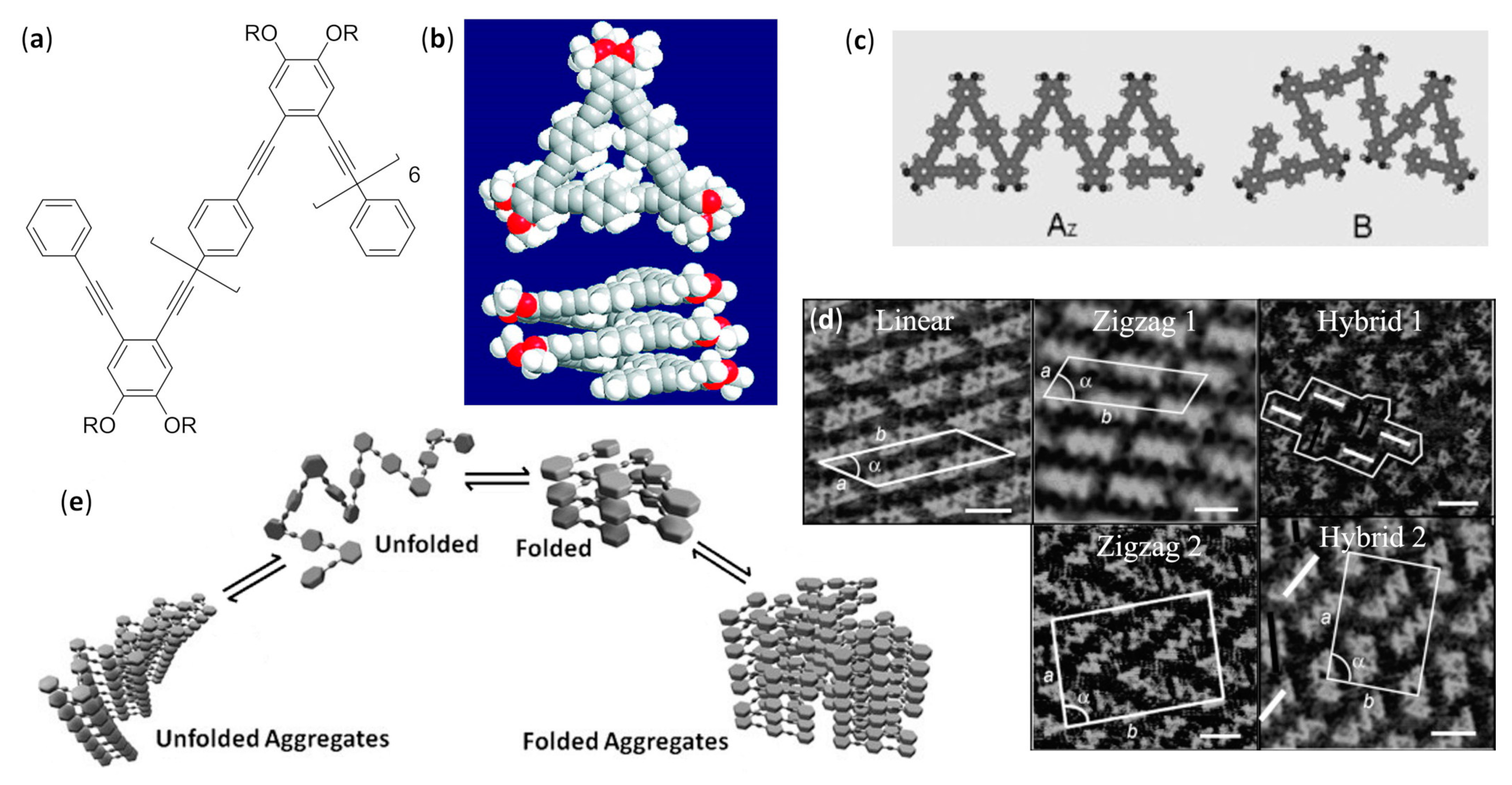
- Zhu, N.; Hu, W.; Han, S.; Wang, Q.; Zhao, D. Folding a conjugated chain: Oligo(o-phenyleneethynylene-alt-p-phenyleneethynylene). Org. Lett. 2008, 10, 4283–4286. [Google Scholar] [CrossRef]
- Shen, Y.T.; Zhu, N.; Zhang, X.M.; Deng, K.; Feng, W.; Yan, Q.; Lei, S.; Zhao, D.; Zeng, Q.D.; Wang, C. A foldamer at the liquid/graphite interface: The effect of interfacial interactions, solvent, concentration, and temperature. Chem. Eur. J. 2011, 17, 7061–7068. [Google Scholar] [CrossRef]
- Shen, Y.T.; Zhu, N.B.; Zhang, X.M.; Lei, S.; Wei, Z.; Li, M.; Zhao, D.; Zeng, Q.D.; Wang, C. Assemblies at the liquid-solid interface: Chirality expression from molecular conformers. ChemPhysChem 2013, 14, 92–95. [Google Scholar] [CrossRef]
- Luo, Z.; Zhu, N.; Zhao, D. Helical folding competing with unfolded aggregation in phenylene ethynylene foldamers. Chem. Eur. J. 2016, 22, 11028–11034. [Google Scholar] [CrossRef]
- Cai, W.; Wang, G.T.; Xu, Y.X.; Jiang, X.K.; Li, Z.T. Vesicles and organogels from foldamers: A solvent-modulated self-assembling process. J. Am. Chem. Soc. 2008, 130, 6936–6937. [Google Scholar] [CrossRef]
- You, L.Y.; Wang, G.T.; Jiang, X.K.; Li, Z.T. Hydrogen bonded aromatic hydrazide foldamers for the self-assembly of vesicles and gels. Tetrahedron 2009, 65, 9494–9504. [Google Scholar] [CrossRef]
- Li, C.; Ren, S.F.; Hou, J.L.; Yi, H.P.; Zhu, S.Z.; Jiang, X.K.; Li, Z.T. F···H-N hydrogen bonding driven foldamers: Efficient receptors for dialkylammonium ions. Angew. Chem. Int. Ed. 2005, 44, 5725–5729. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xiao, Z.Y.; Hou, J.L.; Wang, G.T.; Jiang, X.K.; Li, Z.T. Self-assembly of vesicles from the stacking of a dipodal F/H–N hydrogen bonded arylamide foldamer. Tetrahedron 2009, 65, 10544–10551. [Google Scholar] [CrossRef]
- Yan, T.; Li, F.; Tian, J.; Wang, L.; Luo, Q.; Hou, C.; Dong, Z.; Xu, J.; Liu, J. Biomimetic pulsating vesicles with both pH-tunable membrane permeability and light-triggered disassembly-re-assembly behaviors prepared by supra-amphiphilic helices. ACS Appl. Mater. Interfaces 2019, 11, 30566–30574. [Google Scholar] [CrossRef]
- Yan, T.; Li, F.; Qi, S.; Tian, J.; Tian, R.; Hou, J.; Luo, Q.; Dong, Z.; Xu, J.; Liu, J. Light-responsive vesicles for enantioselective release of chiral drugs prepared from a supra-amphiphilic M-helix. Chem. Commun. 2020, 56, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, C.; Qi, S.; Deng, X.; Yang, B.; Liu, J.; Dong, Z. A switchable helical capsule for encapsulation and release of potassium ion. J. Org. Chem. 2018, 83, 1898–1902. [Google Scholar] [CrossRef] [PubMed]
- Pfukwa, R.; Kouwer, P.H.J.; Rowan, A.E.; Klumperman, B. Templated hierarchical self-assembly of poly(p-aryltriazole) foldamers. Angew. Chem. Int. Ed. 2013, 52, 11040–11044. [Google Scholar] [CrossRef]
- Gan, Q.; Ying, W.; Hua, J. Twisted helical microfibers by hierarchical self-assembly of an aromatic oligoamide foldamer. Chin. J. Chem. 2013, 31, 651–656. [Google Scholar] [CrossRef]
- Cai, W.; Wang, G.T.; Du, P.; Wang, R.X.; Jiang, X.K.; Li, Z.T. Foldamer organogels: A circular dichroism study of glucose-mediated dynamic helicity induction and amplification. J. Am. Chem. Soc. 2008, 130, 13450–13459. [Google Scholar] [CrossRef]
- Green, M.M.; Reidy, M.P.; Johnson, R.J.; Darling, G.; O’Leary, D.J.; Willson, G. Macromolecular stereochemistry: The out-of-proportion influence of optically active comonomers on the conformational characteristics of polyisocyanates. The sergeants and soldiers experiment. J. Am. Chem. Soc. 1989, 111, 6452–6454. [Google Scholar] [CrossRef]
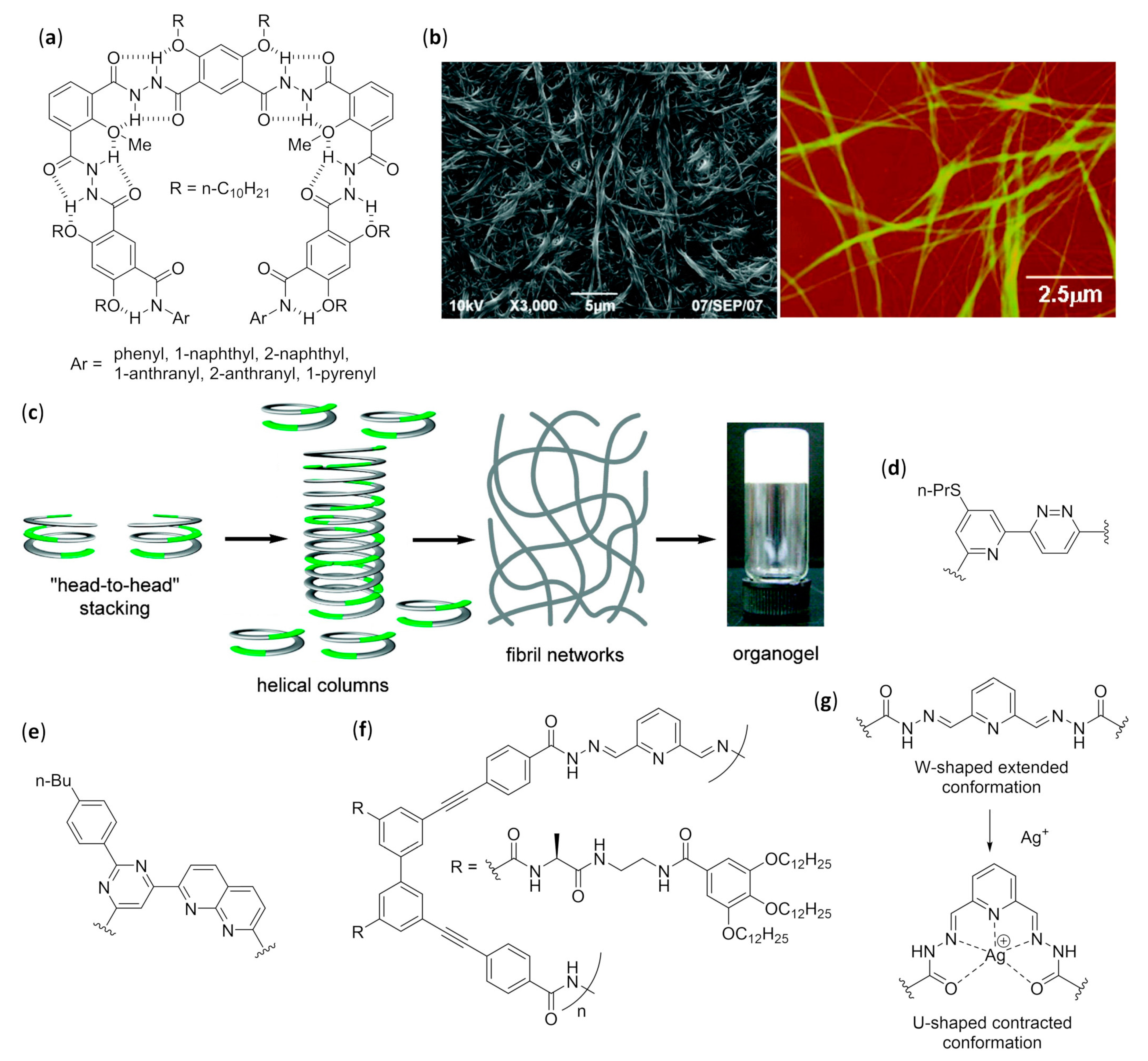
- Cuccia, L.A.; Ruiz, E.; Lehn, J.-M.; Homo, J.-C.; Schmutz, M. Helical self-organization and hierarchical self-assembly of an oligoheterocyclic pyridine-pyridazine strand into extended supramolecular fibers. Chem. Eur. J. 2002, 8, 3448–3457. [Google Scholar] [CrossRef]
- Cuccia, L.A.; Lehn, J.-M.; Homo, J.-C.; Schmutz, M. Encoded helical self-organization and self-assembly into helical fibers of an oligoheterocyclic pyridine-pyridazine molecular strand. Angew. Chem. Int. Ed. 2000, 39, 233–237. [Google Scholar] [CrossRef]
- Azeroual, S.; Surprenant, J.; Lazzara, T.D.; Kocun, M.; Tao, Y.; Cuccia, L.A.; Lehn, J.-M. Mirror symmetry breaking and chiral amplification in foldamer-based supramolecular helical aggregates. Chem. Commun. 2012, 48, 2292–2294. [Google Scholar] [CrossRef]
- Petitjean, A.; Puntoriero, F.; Campagna, S.; Juris, A.; Lehn, J.-M. Multicomponent supramolecular devices: Synthesis, optical, and electronic properties of bridged bis-dirhodium and-diruthenium complexes. Eur. J. Inorg. Chem. 2006, 3878–3892. [Google Scholar] [CrossRef]
- Petitjean, A.; Cuccia, L.A.; Schmutz, M.; Lehn, J.-M. Naphthyridine-based helical foldamers and macrocycles: Synthesis, cation binding, and supramolecular assemblies. J. Org. Chem. 2008, 73, 2481–2495. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, S.; Ousaka, N.; Yashima, E. Allosteric regulation of metal-binding sites inside an optically-active helical foldamer and its tubular assemblies. Chem. Commun. 2018, 54, 2417–2420. [Google Scholar] [CrossRef] [PubMed]
- Ratjen, L.; Lehn, J.-M. Reversible photo-, metallo-and thermo-induced morphological dynamics of bis-acylhydrazones. RSC Adv. 2014, 4, 50554–50557. [Google Scholar] [CrossRef]
- Pike, S.J.; Diemer, V.; Raftery, J.; Webb, S.J.; Clayden, J. Designing foldamer–foldamer interactions in solution: The roles of helix length and terminus functionality in promoting the self-association of aminoisobutyric acid oligomers. Chem. Eur. J. 2014, 20, 15981–15990. [Google Scholar] [CrossRef] [PubMed]
- Venkatraman, J.; Shankaramma, S.C.; Balaram, P. Design of folded peptides. Chem. Rev. 2001, 101, 3131–3152. [Google Scholar] [CrossRef]
- Zych, A.; Iverson, B.L. Synthesis and conformational characterization of tethered, self-complexing 1,5-dialkoxynaphthalene/1,4,5,8-naphthalenetetracarboxylic diimide systems. J. Am. Chem. Soc. 2000, 122, 8898–8909. [Google Scholar] [CrossRef]
- Gabriel, G.J.; Iverson, B.L. Aromatic oligomers that form hetero duplexes in aqueous solution. J. Am. Chem. Soc. 2002, 124, 15174–15175. [Google Scholar] [CrossRef]
- Gong, B.; Yan, Y.; Zeng, H.; Skrzypczak-Jankun, E.; Kim, Y.W.; Zhu, J.; Ickes, H. A new approach for the design of supramolecular recognition units: Hydrogen-bonded molecular duplexes. J. Am. Chem. Soc. 1999, 121, 5607–5608. [Google Scholar] [CrossRef]
- Zeng, H.; Miller, R.S.; Flowers, R.A.; Gong, B. A highly stable, six-hydrogen-bonded molecular duplex. J. Am. Chem. Soc. 2000, 122, 2635–2644. [Google Scholar] [CrossRef]
- Zeng, H.; Ickes, H.; Flowers, R.A.; Gong, B. Sequence specificity of hydrogen-bonded molecular duplexes. J. Org. Chem. 2001, 66, 3574–3583. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cao, R.; Shen, J.; Detchou, C.S.F.; Zhong, Y.; Wang, H.; Zou, S.; Huang, Q.; Lian, C.; Wang, Q.; et al. Hydrogen-bonded duplexes with lengthened linkers. Org. Lett. 2018, 20, 1555–1558. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Yang, X.; Flowers, R.A.; Gong, B. A noncovalent approach to antiparallel β-sheet formation. J. Am. Chem. Soc. 2002, 124, 2903–2910. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Martinovic, S.; Smith, R.D.; Gong, B. Duplex foldamers from assembly induced folding. J. Am. Chem. Soc. 2003, 125, 9932–9933. [Google Scholar] [CrossRef]
- Nowick, J.S.; Tsai, J.H.; Bui, Q.-C.D.; Maitra, S. A chemical model of a protein β-sheet dimer. J. Am. Chem. Soc. 1999, 121, 8409–8410. [Google Scholar] [CrossRef]
- Nowick, J.S.; Chung, D.M.; Maitra, K.; Maitra, S.; Stigers, K.D.; Sun, Y. An unnatural amino acid that mimics a tripeptide β-strand and forms β-sheetlike hydrogen-bonded dimers. J. Am. Chem. Soc. 2000, 122, 7654–7661. [Google Scholar] [CrossRef]
- Nowick, J.S.; Lam, K.S.; Khasanova, T.V.; Kemnitzer, W.E.; Maitra, S.; Mee, H.T.; Liu, R. An unnatural amino acid that induces β-sheet folding and interaction in peptides. J. Am. Chem. Soc. 2002, 124, 4972–4973. [Google Scholar] [CrossRef]
- Nowick, J.S.; Chung, D.M. Sequence-selective molecular recognition between β sheets. Angew. Chem. Int. Ed. 2003, 42, 1765–1768. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, Z.Y.; Yi, Y.P.; Xiang, J.F.; Chen, C.F.; Wan, L.J.; Shuai, Z.G. Helical molecular duplex strands: Multiple hydrogen-bond-mediated assembly of self-complementary oligomeric hydrazide derivatives. J. Org. Chem. 2007, 72, 4936–4946. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhong, Y.; Connor, A.L.; Miller, D.P.; Cao, R.; Shen, J.; Song, B.; Baker, E.S.; Tang, Q.; Pulavarti, S.V.S.R.K.; et al. Folding and assembly of short α, β, γ-hybrid peptides: Minor variations in sequence and drastic differences in higher-level structures. J. Am. Chem. Soc. 2019, 141, 14239–14248. [Google Scholar] [CrossRef]
- Misra, R.; Dey, S.; Reja, R.M.; Gopi, H.N. Artificial β-double helices from achiral γ-peptides. Angew. Chem. Int. Ed. 2018, 57, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, R.; Debnath, M.; Maji, K.; Haldar, D. Solvent assisted structural diversity: Supramolecular sheet and double helix of a short aromatic γ-peptide. RSC Adv. 2015, 5, 76257–76262. [Google Scholar] [CrossRef]
- Amorín, M.; Castedo, L.; Granja, J.R. Folding control in cyclic peptides through N-methylation pattern selection: Formation of antiparallel β-sheet dimers, double reverse turns and supramolecular helices by 3α,γ cyclic peptides. Chem. Eur. J. 2008, 14, 2100–2111. [Google Scholar] [CrossRef] [PubMed]
- Horne, W.S.; Price, J.L.; Keck, J.L.; Gellman, S.H. Helix bundle quaternary structure from α/β-peptide foldamers. J. Am. Chem. Soc. 2007, 129, 4178–4180. [Google Scholar] [CrossRef]
- Price, J.L.; Horne, W.S.; Gellman, S.H. Discrete heterogeneous quaternary structure formed by α/β-peptide foldamers and α-peptides. J. Am. Chem. Soc. 2007, 129, 6376–6377. [Google Scholar] [CrossRef]
- Giuliano, M.W.; Horne, W.S.; Gellman, S.H. An α/β-peptide helix bundle with a pure β3-amino acid core and a distinctive quaternary structure. J. Am. Chem. Soc. 2009, 131, 9860–9861. [Google Scholar] [CrossRef]
- Price, J.L.; Horne, W.S.; Gellman, S.H. Structural consequences of β-amino acid preorganization in a self-assembling α/β-peptide: Fundamental studies of foldameric helix bundles. J. Am. Chem. Soc. 2010, 132, 12378–12387. [Google Scholar] [CrossRef]
- Horne, W.S.; Price, J.L.; Gellman, S.H. Interplay among side chain sequence, backbone composition, and residue rigidification in polypeptide folding and assembly. Proc. Natl. Acad. Sci. USA 2008, 105, 9151–9156. [Google Scholar] [CrossRef]
- Harbury, P.B.; Zhang, T.; Kim, P.S.; Alber, T. A switch between two-, three-, and four-stranded coiled coils in GCN4 leucine zipper mutants. Science 1993, 262, 1401–1407. [Google Scholar] [CrossRef] [PubMed]
- Collie, G.W.; Pulka-Ziach, K.; Lombardo, C.M.; Fremaux, J.; Rosu, F.; Decossas, M.; Mauran, L.; Lambert, O.; Gabelica, V.; Mackereth, C.D.; et al. Shaping quaternary assemblies of water-soluble non-peptide helical foldamers by sequence manipulation. Nat. Chem. 2015, 7, 871–878. [Google Scholar] [CrossRef]
- Lombardo, C.M.; Collie, G.W.; Pulka-Ziach, K.; Rosu, F.; Gabelica, V.; Mackereth, C.D.; Guichard, G. Anatomy of an oligourea six-helix bundle. J. Am. Chem. Soc. 2016, 138, 10522–10530. [Google Scholar] [CrossRef] [PubMed]
- Collie, G.W.; Pulka-Ziach, K.; Guichard, G. In situ iodination and X-ray crystal structure of a foldamer helical bundle. Chem. Commun. 2016, 52, 1202–1205. [Google Scholar] [CrossRef]
- Collie, G.W.; Bailly, R.; Pulka-Ziach, K.; Lombardo, C.M.; Mauran, L.; Taib-Maamar, N.; Dessolin, J.; Mackereth, C.D.; Guichard, G. Molecular recognition within the cavity of a foldamer helix bundle: Encapsulation of primary alcohols in aqueous conditions. J. Am. Chem. Soc. 2017, 139, 6128–6137. [Google Scholar] [CrossRef]
- Segman, S.; Lee, M.-R.; Vaiser, V.; Gellman, S.H.; Rapaport, H. Highly stable pleated-sheet secondary structure in assemblies of amphiphilic α/β-peptides at the air–water interface. Angew. Chem. Int. Ed. 2010, 49, 716–719. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.A.; Choi, S.H.; Guzei, I.A.; Gellman, S.H. Residue requirements for helical folding in short α/β-peptides: Crystallographic characterization of the 11-helix in an optimized sequence. J. Am. Chem. Soc. 2005, 127, 13130–13131. [Google Scholar] [CrossRef]
- Rapaport, H. Ordered peptide assemblies at interfaces. Supramol. Chem. 2006, 18, 445–454. [Google Scholar] [CrossRef]
- Segman-Magidovich, S.; Lee, M.-r.; Vaiser, V.; Struth, B.; Gellman, S.H.; Rapaport, H. Sheet-like assemblies of charged amphiphilic α/β-peptides at the air–water interface. Chem. Eur. J. 2011, 17, 14857–14866. [Google Scholar] [CrossRef]
- Schuurmans, N.; Hiroshi Uji-i, H.; Mamdouh, W.; De Schryver, F.C.; Feringa, B.L.; Van Esch, J.; De Feyter, S. Design and STM investigation of intramolecular folding in self-assembled monolayers on the surface. J. Am. Chem. Soc. 2004, 126, 13884–13885. [Google Scholar] [CrossRef][Green Version]
- Klymchenko, A.S.; Schuurmans, N.; Van Der Auweraer, M.; Feringa, B.L.; Van Esch, J.; De Feyter, S. Scanning tunnelling microscopy of a foldamer prototype at the liquid/solid interface: Water/Au(111) versus 1-octanol/graphite. New J. Chem. 2006, 30, 1420–1428. [Google Scholar] [CrossRef][Green Version]
- Li, M.; Gobbo, C.; De Cat, I.; Eelkema, R.; Vanaverbeke, B.; Lazzaroni, R.; De Feyter, S.; Van Esch, J. Molecular patterning at a liquid/solid interface: The foldamer approach. Langmuir 2011, 27, 13598–13605. [Google Scholar] [CrossRef]
- Gobbo, C.; Li, M.; Mali, K.S.; Van Esch, J.H.; De Feyter, S. Preprogrammed 2D folding of conformationally flexible oligoamides: Foldamers with multiple turn elements. ACS Nano 2012, 6, 10684–10698. [Google Scholar] [CrossRef] [PubMed]
- Pulka-Ziach, K.S.; Sęk, S. α-Helicomimetic foldamers as electron transfer mediators. Nanoscale 2017, 9, 14913–14920. [Google Scholar] [CrossRef]
- Sepunaru, L.; Refaely-Abramson, S.; Lovrinčić, R.; Gavrilov, Y.; Agrawal, P.; Levy, Y.; Kronik, L.; Pecht, I.; Sheves, M.; Cahen, D. Electronic transport via homopeptides: The role of side chains and secondary structure. J. Am. Chem. Soc. 2015, 137, 9617–9626. [Google Scholar] [CrossRef] [PubMed]
- Pulka-Ziach, K.; Puszko, A.K.; Juhaniewicz-Debinska, J.; Sek, S. Electron transport and a rectifying effect of oligourea foldamer films entrapped within nanoscale junctions. J. Phys. Chem. C 2019, 123, 1136–1141. [Google Scholar] [CrossRef]
- Nam, K.T.; Shelby, S.A.; Choi, P.H.; Marciel, A.B.; Chen, R.; Tan, L.; Chu, T.K.; Mesch, R.A.; Lee, B.-C.; Connolly, M.D.; et al. Free-floating ultrathin two-dimensional crystals from sequence-specific peptoid polymers. Nat. Mater. 2010, 9, 454–460. [Google Scholar] [CrossRef]
- Olivier, G.K.; Cho, A.; Sanii, B.; Connolly, M.D.; Tran, H.; Zuckermann, R.N. Antibody-mimetic peptoid nanosheets for molecular recognition. ACS Nano 2013, 7, 9276–9286. [Google Scholar] [CrossRef]
- Robertson, E.J.; Proulx, C.; Su, J.K.; Garcia, R.L.; Yoo, S.; Nehls, E.M.; Connolly, M.D.; Taravati, L.; Zuckermann, R.N. Molecular engineering of the peptoid nanosheet hydrophobic core. Langmuir 2016, 32, 11946–11957. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, S.C.; Kline, M.A.; Grzincic, E.M.; Tresca, B.W.; Cardiel, J.; Karbaschi, M.; Dehigaspitiya, D.C.; Chen, Y.; Udumula, V.; et al. Discovery of stable and selective antibody mimetics from combinatorial libraries of polyvalent, loop-functionalized peptoid nanosheets. ACS Nano 2020, 14, 185–195. [Google Scholar] [CrossRef]
- Sanii, B.; Kudirka, R.; Cho, A.; Venkateswaran, N.; Olivier, G.K.; Olson, A.M.; Tran, H.; Harada, R.M.; Tan, L.; Zuckermann, R.N. Shaken, not stirred: Collapsing a peptoid monolayer to produce free-floating, stable nanosheets. J. Am. Chem. Soc. 2011, 133, 20808–20815. [Google Scholar] [CrossRef] [PubMed]
- Mannige, R.V.; Haxton, T.K.; Proulx, C.; Robertson, E.J.; Battigelli, A.; Butterfoss, G.L.; Zuckermann, R.N.; Whitelam, S. Peptoid nanosheets exhibit a new secondary-structure motif. Nature 2015, 526, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.C.; Battigelli, A.; Connolly, M.D.; Edison, J.; Spencer, R.K.; Whitelam, S.; Zuckermann, R.N.; Paravastu, A.K. Evidence for cis amide bonds in peptoid nanosheets. J. Phys. Chem. Lett. 2018, 9, 2574–2578. [Google Scholar] [CrossRef] [PubMed]
- Edison, J.R.; Spencer, R.K.; Butterfoss, G.L.; Hudson, B.C.; Hochbaum, A.I.; Paravastu, A.K.; Zuckermann, R.N.; Whitelam, S. Conformations of peptoids in nanosheets result from the interplay of backbone energetics and intermolecular interactions. Proc. Natl. Acad. Sci. USA 2018, 115, 5647–5651. [Google Scholar] [CrossRef]
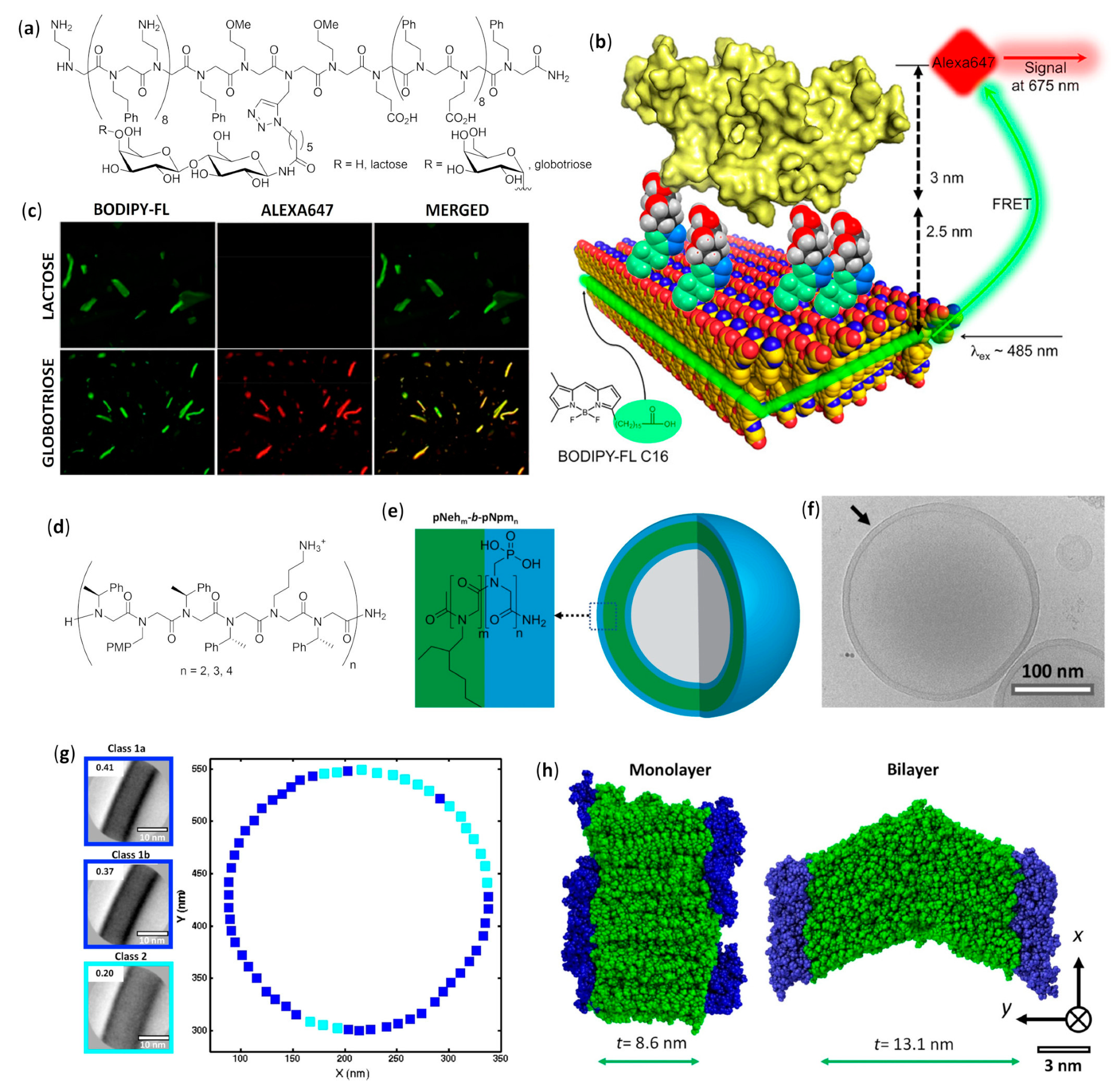
- Battigelli, A.; Kim, J.H.; Dehigaspitiya, D.C.; Proulx, C.; Robertson, E.J.; Murray, D.J.; Rad, B.; Kirshenbaum, K.; Zuckermann, R.N. Glycosylated peptoid nanosheets as a multivalent scaffold for protein recognition. ACS Nano 2018, 12, 2455–2465. [Google Scholar] [CrossRef]
- Hebert, M.L.; Shah, D.S.; Blake, P.; Turner, J.P.; Servoss, S.L. Tunable peptoid microspheres: Effects of side chain chemistry and sequence. Org. Biomol. Chem. 2013, 11, 4459–4464. [Google Scholar] [CrossRef]
- Hebert, M.L.; Shah, D.S.; Blake, P.; Servoss, S.L. Uniform and robust peptoid microsphere coatings. Coatings 2013, 3, 98–107. [Google Scholar] [CrossRef]
- Perez Bakovic, G.R.; Roberts, J.L.; Colford, B.; Joyce, M.; Servoss, S.L. Peptoid microsphere coatings: The effects of helicity, temperature, pH, and ionic strength. Biopolymers 2019, 110, e23283. [Google Scholar] [CrossRef]
- Jiang, X.; Spencer, R.K.; Sun, J.; Ophus, C.; Zuckermann, R.N.; Downing, K.H.; Balsara, N.P. Resolving the morphology of peptoid vesicles at the 1 nm length scale using cryogenic electron microscopy. J. Phys. Chem. B 2019, 123, 1195–1205. [Google Scholar] [CrossRef]
- Bermúdez, H.; Hammer, D.A.; Discher, D.E. Effect of bilayer thickness on membrane bending rigidity. Langmuir 2004, 20, 540–543. [Google Scholar] [CrossRef]
- Ingole, T.S.; Kale, S.S.; Santhosh Babu, S.; Sanjayan, G.J. Self-assembled vesicles of urea-tethered foldamer as a hydrophobic drug carrier. Chem. Commun. 2016, 52, 10771–10774. [Google Scholar] [CrossRef]
- Kale, S.S.; Kotmale, A.S.; Kumar Dutta, A.; Pal, S.; Rajamohanan, P.R.; Sanjayan, G.J. Conformational modulation of Ant–Pro oligomers using chirality alteration of proline residues. Org. Biomol. Chem. 2012, 10, 8426–8433. [Google Scholar] [CrossRef] [PubMed]
- Marafon, G.; Crisma, M.; Moretto, A. Intrinsically photoswitchable α/β peptides toward two-state foldamers. Angew. Chem. Int. Ed. 2018, 57, 10217–10220. [Google Scholar] [CrossRef] [PubMed]
- Marafon, G.; Crisma, M.; Moretto, A. Tunable E−Z photoisomerization in α,β-peptide foldamers featuring multiple (E/Z)-3-aminoprop-2-enoic acid units. Org. Lett. 2019, 21, 4182–4186. [Google Scholar] [CrossRef] [PubMed]
- Bradford, V.J.; Iverson, B.L. Amyloid-like behavior in abiotic, amphiphilic foldamers. J. Am. Chem. Soc. 2008, 130, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- Peebles, C.; Piland, R.; Iverson, B.L. More than meets the eyes: Conformational switching of a stacked dialkoxynaphthalene-naphthalenetetra-carboxylic diimide (DAN-NDI) foldamer to an NDIcx-NDI fibril aggregate. Chem. Eur. J. 2013, 19, 11598–11602. [Google Scholar] [CrossRef]
- Baruah, K.P.; Sreedevi, N.K.; Majumdar, B.; Pasricha, R.; Poddar, P.; Gonnade, R.; Ravindranathan, S.; Sanjayan, G.J. Sheet-forming abiotic hetero foldamers. Chem. Commun. 2008, 712–714. [Google Scholar] [CrossRef]
- Solà, J.; Bolte, M.; Alfonso, I. Metal driven assembly of peptidic foldamers: Formation of molecular tapes. CrystEngComm 2016, 18, 3793–3798. [Google Scholar] [CrossRef]
- Debnath, M.; Das, T.; Podder, D.; Haldar, D. α,ε-Hybrid peptide foldamers: Self-assembly of peptide with trans carbon−carbon double bonds in the backbone and its saturated analogue. ACS Omega 2018, 3, 8760–8768. [Google Scholar] [CrossRef]
- Marafon, G.; Menegazzo, I.; De Zotti, M.; Crisma, M.; Toniolo, C.; Moretto, A. Tuning morphological architectures generated through living supramolecular assembly of a helical foldamer end-capped with two complementary nucleobases. Soft Matter 2017, 13, 4231–4240. [Google Scholar] [CrossRef]
- Konda, M.; Kauffmann, B.; Rasale, D.B.; Das, A.K. Structural and morphological diversity of self-assembled synthetic γ-amino acid containing peptides. Org. Biomol. Chem. 2016, 14, 4089–4102. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, S.V.; Gopi, H.N. Remarkable thermoresponsive nanofibers from γ-peptides. Chem. Commun. 2013, 49, 9179–9181. [Google Scholar] [CrossRef]
- Murnen, H.K.; Rosales, A.M.; Jaworski, J.N.; Segalman, R.A.; Zuckermann, R.N. Hierarchical self-assembly of a biomimetic diblock copolypeptoid into homochiral superhelices. J. Am. Chem. Soc. 2010, 132, 16112–16119. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Jian, T.; Ding, Y.-H.; Chen, Y.; Mu, P.; Wang, L.; Chen, C.-L. Solid-phase synthesis of three-armed star-shaped peptoids and their hierarchical self-assembly. Biopolymers 2019, 110, e23258. [Google Scholar] [CrossRef]
- Jin, H.; Ding, Y.H.; Wang, M.; Song, Y.; Liao, Z.; Newcomb, C.J.; Wu, X.; Tang, X.Q.; Li, Z.; Lin, Y.; et al. Designable and dynamic single-walled stiff nanotubes assembled from sequence-defined peptoids. Nat. Commun. 2018, 9, 270. [Google Scholar] [CrossRef] [PubMed]
- Eom, J.H.; Gong, J.; Jeong, R.; Driver, R.W.; Lee, H.S. Parallelogram plate shaped foldecture from the controlled self-assembly of α/β-peptide foldamer. Solid State Sci. 2015, 48, 39–43. [Google Scholar] [CrossRef]
- Eom, J.H.; Gong, J.; Kwon, S.; Jeon, A.; Jeong, R.; Driver, R.W.; Lee, H.S. A hollow foldecture with truncated trigonal bipyramid shape from the self-assembly of an 11-helical foldamer. Angew. Chem. Int. Ed. 2015, 54, 13204–13207. [Google Scholar] [CrossRef]
- Eom, J.H.; Jeong, R.; Gong, J.; Driver, R.W.; Lee, H.S. Foldectures from the self-assembly of racemic foldamers. Bull. Korean Chem. Soc. 2015, 36, 2583–2584. [Google Scholar] [CrossRef]
- Gong, J.; Eom, J.H.; Jeong, R.; Driver, R.W.; Lee, H.S. Structural analysis of the foldecture derived from racemic peptide foldamers. Solid State Sci. 2017, 70, 1–5. [Google Scholar] [CrossRef]
- Lim, D.; Kim, H.; Gong, J.; Eom, J.H.; Yoon, E.; Driver, R.W.; Baik, M.H.; Lee, H.S. Directing foldamer self-assembly with a cyclopropanoyl cap. Chem. Eur. J. 2019, 25, 2226–2233. [Google Scholar] [CrossRef]
- Yoon, E.; Gong, J.; Jung, Y.; Lee, W.; Driver, R.W.; Lee, H.-S. Unambiguous characterization of anisotropic foldamer packing in a foldecture with an elongated hexagonal plate shape. Chem. Commun. 2016, 52, 5250–5253. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, S.V.; Misra, R.; Gopi, H.N. Foldamers to nanotubes: Influence of amino acid side chains in the hierarchical assembly of α,γ4-hybrid peptide helices. Chem. Eur. J. 2014, 20, 16523–16528. [Google Scholar] [CrossRef] [PubMed]
- Misra, R.; Reja, R.M.; Narendra, L.V.; George, G.; Raghothama, S.; Gopi, H.N. Exploring structural features of folded peptide architectures in the construction of nanomaterials. Chem. Commun. 2016, 52, 9597–9600. [Google Scholar] [CrossRef] [PubMed]
























© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rinaldi, S. The Diverse World of Foldamers: Endless Possibilities of Self-Assembly. Molecules 2020, 25, 3276. https://doi.org/10.3390/molecules25143276
Rinaldi S. The Diverse World of Foldamers: Endless Possibilities of Self-Assembly. Molecules. 2020; 25(14):3276. https://doi.org/10.3390/molecules25143276
Chicago/Turabian StyleRinaldi, Samuele. 2020. "The Diverse World of Foldamers: Endless Possibilities of Self-Assembly" Molecules 25, no. 14: 3276. https://doi.org/10.3390/molecules25143276
APA StyleRinaldi, S. (2020). The Diverse World of Foldamers: Endless Possibilities of Self-Assembly. Molecules, 25(14), 3276. https://doi.org/10.3390/molecules25143276