
Weak Interactions and Conformational Changes in Core-Protonated A2- and Ax-Type Porphyrin Dications

 , , and
, , and 
Abstract
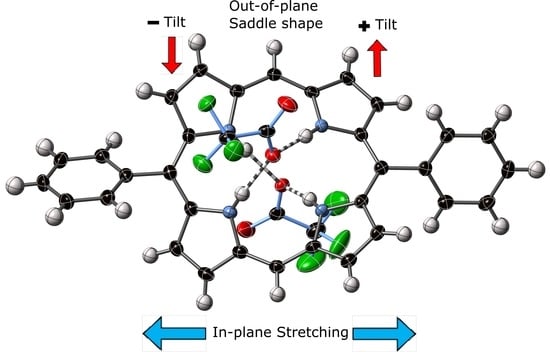
1. Introduction
2. Results and Discussion
2.1. Scope of Study
2.2. Database Analysis
2.3. Single Crystal X-ray Structure Determinations
2.3.1. 5,15-Diphenylporphyrin and its Dication Salt as Parent Compounds
2.3.2. Other 5,15-Disubstituted Porphyrins
2.3.3. A3-Type Porphyrins
2.4. Normal Structural Decomposition Analysis
2.5. Intermolecular Interactions
2.5.1. N–H⋯X Bonding from Cationic Porphyrins
2.5.2. C–H⋯O Bonding from Cationic Porphyrins
3. Materials and Methods
3.1. Synthesis
3.2. Crystallography
3.2.1. Instrumentation and Analysis
3.2.2. Refinement Details
3.3. Normal Structural Decomposition Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kadish, K.M.; Smith, K.M.; Guilard, R. (Eds.) The Porphyrin Handbook; Academic Press: New York, NY, USA, 2000; Volume 1–20, new edition 2013. [Google Scholar]
- Jurow, M.; Schuckman, A.E.; Batteas, J.D.; Drain, C.M. Porphyrins as Molecular Electronic Components of Functional Devices. Coord. Chem. Rev. 2010, 254, 2297–2310. [Google Scholar] [CrossRef] [PubMed]
- Senge, M.O.; Fazekas, M.; Notaras, E.G.A.; Blau, W.J.; Zawadzka, M.; Locos, O.B.; Mhuircheartaigh, E.M.N. Nonlinear optical properties of porphyrins. Adv. Mater. 2007, 19, 2737–2774. [Google Scholar] [CrossRef]
- Ethirajan, M.; Chen, Y.; Joshi, P.; Pandey, R.K. The role of porphyrin chemistry in tumor imaging and photodynamic therapy. Chem. Soc. Rev. 2011, 40, 340–362. [Google Scholar] [CrossRef]
- Scheidt, W.R.; Lee, Y.J. Recent Advances in the Stereochemistry of Metallotetrapyrroles. Struct. Bonding 1987, 64, 1–70. [Google Scholar]
- Kielmann, M.; Prior, C.; Senge, M.O. Porphyrins in troubled times: A spotlight on porphyrins and their metal complexes for explosives testing and CBRN defense. New J. Chem. 2018, 42, 7529–7550. [Google Scholar] [CrossRef]
- Senge, M.O.; MacGowan, S.A.; O’Brien, J.M. Conformational control of cofactors in nature-the influence of protein-induced macrocycle distortion on the biological function of tetrapyrroles. Chem. Commun. 2015, 51, 17031–17063. [Google Scholar] [CrossRef] [PubMed]
- Beletskaya, I.; Tyurin, V.S.; Tsivadze, A.Y.; Guilard, R.; Stern, C. Supramolecular chemistry of metalloporphyrins. Chem. Rev. 2009, 109, 1659–1713. [Google Scholar] [CrossRef]
- Kielmann, M.; Senge, M.O. Molecular Engineering of Free-Base Porphyrins as Ligands—The N-H ··· X Binding Motif in Tetrapyrroles. Angew. Chem. Int. Ed. 2019, 58, 418–441. [Google Scholar] [CrossRef]
- Barkigia, K.M.; Fajer, J.; Renner, M.W.; Dolores Berber, M.; Medforth, C.J.; Smith, K.M. Nonplanar Porphyrins. X-ray Structures of (2,3,7,8,12,13,17,18-Octaethyl- and -Octamethyl-5,10,15,20-tetraphenylporphinato)zinc(II). J. Am. Chem. Soc. 1990, 112, 8851–8857. [Google Scholar] [CrossRef]
- Medforth, C.J.; Senge, M.O.; Smith, K.M.; Sparks, L.D.; Shelnutt, J.A. Nonplanar Distortion Modes for Highly Substituted Porphyrins. J. Am. Chem. Soc. 1992, 114, 9859–9869. [Google Scholar] [CrossRef]
- Senge, M.O.; Kalisch, W.W. Synthesis and Structural Characterization of Nonplanar Tetraphenylporphyrins and Their Metal Complexes with Graded Degrees of β-Ethyl Substitution. Inorg. Chem. 1997, 36, 6103–6116. [Google Scholar] [CrossRef] [PubMed]
- Senge, M.O. Exercises in molecular gymnastics - Bending, stretching and twisting porphyrins. Chem. Commun. 2006, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Sessler, J.L.; Camiolo, S.; Gale, P.A. Pyrrolic and polypyrrolic anion binding agents. Coord. Chem. Rev. 2003, 240, 17–55. [Google Scholar] [CrossRef]
- Norvaiša, K.; Kielmann, M.; Senge, M.O. Porphyrins as Colorimetric and Photometric Biosensors in Modern Bioanalytical Systems. ChemBioChem 2020, in press. [Google Scholar] [CrossRef]
- Roucan, M.; Kielmann, M.; Connon, S.J.; Bernhard, S.S.R.; Senge, M.O. Conformational control of nonplanar free base porphyrins: Towards bifunctional catalysts of tunable basicity. Chem. Commun. 2017, 54, 26–29. [Google Scholar] [CrossRef]
- Rybicka-Jasińska, K.; Shan, W.; Zawada, K.; Kadish, K.M.; Gryko, D. Porphyrins as Photoredox Catalysts: Experimental and Theoretical Studies. J. Am. Chem. Soc. 2016, 138, 15451–15458. [Google Scholar] [CrossRef]
- Costa e Silva, R.; da Silva, L.O.; de Andrade Bartolomeu, A.; Brocksom, T.J.; de Oliveira, K.T. Recent applications of porphyrins as photocatalysts in organic synthesis: Batch and continuous flow approaches. Beilstein J. Org. Chem. 2020, 16, 917–955. [Google Scholar] [CrossRef]
- Fischer, H.; Orth, H. Die Chemie des Pyrrols; Akademische Verlagsgesellschaft: Leipzig, Germany, 1937. [Google Scholar]
- Stone, A.; Fleischer, E.B. The molecular and crystal structure of porphyrin diacids. J. Am. Chem. Soc. 1968, 90, 2735–2748. [Google Scholar] [CrossRef]
- Senge, M.O.; Forsyth, T.P.; Nguyen, L.T.; Smith, K.M. Sterically Strained Porphyrins—Influence of Core Protonation and Peripheral Substitution on the Conformation of Tetra-meso-, Octa-β-, and Dodeca-Substituted Porphyrin Dications. Angew. Chem. Int. Ed. Engl. 1995, 33, 2485–2487. [Google Scholar] [CrossRef]
- Cheng, B.; Munro, O.Q.; Marques, H.M.; Scheidt, W.R. An analysis of porphyrin molecular flexibility - Use of porphyrin diacids. J. Am. Chem. Soc. 1997, 119, 10732–10742. [Google Scholar] [CrossRef]
- Senge, M.O.; Kalisch, W.W. Structure and Conformation of Tetra-meso-, Octa-β-, and Dodecasubstituted 22,24-Dihydroporphyrins (Porphyrin Dications). Z. Naturforsch. 1999, 54b, 943–959. [Google Scholar] [CrossRef]
- Senge, M.O. A Conformational Study of 5,10,15,20-Tetraalkyl-22+,24+-porphyrindiium Salts (Dication Salts). Z. Naturforsch. 2000, 55b, 336–344. [Google Scholar] [CrossRef]
- Juillard, S.; Ferrand, Y.; Simonneaux, G.; Toupet, L. Molecular structure of simple mono- and diphenyl meso-substituted porphyrin diacids: Influence of protonation and substitution on the distorsion. Tetrahedron 2005, 61, 3489–3495. [Google Scholar] [CrossRef]
- Manka, J.S.; Lawrence, D.S. High yield synthesis of 5,15-diarylporphyrins. Tetrahedron Lett. 1989, 30, 6989–6992. [Google Scholar] [CrossRef]
- Brückner, C.; Posakony, J.J.; Johnson, C.K.; Boyle, R.W.; James, B.R.; Dolphin, D. Novel and improved syntheses of 5,15-diphenylporphyrin and its dipyrrolic precursors. J. Porphyr. Phthalocyanines 1998, 2, 455–465. [Google Scholar] [CrossRef]
- Senge, M.O. Stirring the porphyrin alphabet soup—Functionalization reactions for porphyrins. Chem. Commun. 2011, 47, 1943–1960. [Google Scholar] [CrossRef]
- Yorimitsu, H.; Osuka, A. Organometallic approaches for direct modification of peripheral C-H bonds in porphyrin cores. Asian J. Org. Chem. 2013, 2, 356–373. [Google Scholar] [CrossRef]
- Shanmugathasan, S.; Johnson, C.K.; Edwards, C.; Matthews, E.K.; Dolphin, D.; Boyle, R.W. Regioselective halogenation and palladium-catalysed couplings on 5,15-diphenylporphyrin. J. Porphyr. Phthalocyanines 2000, 4, 228–232. [Google Scholar] [CrossRef]
- Senge, M.O.; Fazekas, M.; Pintea, M.; Zawadzka, M.; Blau, W.J. 5,15-A2B2- and 5,15-A2BC-type porphyrins with donor and acceptor groups for use in nonlinear optics and photodynamic therapy. Eur. J. Org. Chem. 2011, 5797–5816. [Google Scholar] [CrossRef]
- Gehrold, A.C.; Bruhn, T.; Schneider, H.; Radius, U.; Bringmann, G. Monomeric Chiral and Achiral Basket-Handle Porphyrins: Synthesis, Structural Features, and Arrested Tautomerism. J. Org. Chem. 2015, 80, 12359–12378. [Google Scholar] [CrossRef]
- Senge, M.O.; Feng, X. Regioselective reaction of 5,15-disubstituted porphyrins with organolithium reagents-Synthetic access to 5,10,15-trisubstituted porphyrins and directly meso-meso-linked bisporphyrins. J. Chem. Soc. Perkin Trans. 1 2000, 3615–3621. [Google Scholar] [CrossRef]
- Brown, A.; Langton, M.J.; Kilah, N.L.; Thompson, A.L.; Beer, P.D. Chloride-Anion-Templated Synthesis of a Strapped-Porphyrin-Containing Catenane Host System. Chem. Eur. J. 2015, 21, 17664–17675. [Google Scholar] [CrossRef] [PubMed]
- Roy, I.; Bobbala, S.; Young, R.M.; Beldjoudi, Y.; Nguyen, M.T.; Cetin, M.M.; Cooper, J.A.; Allen, S.; Anamimoghadam, O.; Scott, E.A.; et al. A Supramolecular Approach for Modulated Photoprotection, Lysosomal Delivery, and Photodynamic Activity of a Photosensitizer. J. Am. Chem. Soc. 2019, 141, 12296–12304. [Google Scholar] [CrossRef] [PubMed]
- Bond, A.D.; Feeder, N.; Redman, J.E.; Teat, S.J.; Sanders, J.K.M. Molecular Conformation and Intermolecular Interactions in the Crystal Structures of Free-Base 5,15-Diarylporphyrins. Cryst. Growth Des. 2002, 2, 27–39. [Google Scholar] [CrossRef]
- Senge, M.O. 5,15-Bis(4-pentyloxyphenyl)porphyrin. Acta Cryst. 2013, E69, o1048. [Google Scholar] [CrossRef]
- Marin, D.M.; Castaneda, J.; Kaushal, M.; Kaouk, G.; Jones, D.S.; Walter, M.G. Spatially resolved micro-photoluminescence imaging of porphyrin single crystals. Chem. Phys. Lett. 2016, 659, 137–141. [Google Scholar] [CrossRef]
- Zhou, L.; Xu, Z.X.; Zhou, Y.; Feng, Y.; Zhou, X.G.; Xiang, H.F.; Roy, V.A.L. Structure-charge transport relationship of 5,15-dialkylated porphyrins. Chem. Commun. 2012, 48, 5139–5141. [Google Scholar] [CrossRef]
- Paul-Roth, C.O.; Letessier, J.; Juillard, S.; Simonneaux, G.; Roisnel, T.; Rault-Berthelot, J. Synthesis, solid-state molecular structure and polymerization of a trans-substituted meso-porphyrin with thienyl pendant arms. J. Mol. Struct. 2008, 872, 105–112. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge structural database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef]
- Senge, M.O. Database of Tetrapyrrole Crystal Structure Determinations. In The Porphyrin Handbook; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; Academic Press: San Diego, CA, USA, 2000; Volume 10, pp. 1–218. [Google Scholar]
- Senge, M.O.; Medforth, C.J.; Forsyth, T.P.; Lee, D.A.; Olmstead, M.M.; Jentzen, W.; Pandey, R.K.; Shelnutt, J.A.; Smith, K.M. Comparative Analysis of the Conformations of Symmetrically and Asymmetrically Decaand Undecasubstituted Porphyrins Bearing Meso-Alkyl or -Aryl Groups. Inorg. Chem. 1997, 36, 1149–1163. [Google Scholar] [CrossRef]
- Dragelj, J.L.; Janjić, G.V.; Veljković, D.Ž.; Zarić, S.D. Crystallographic and ab initio study of pyridine CH–O interactions: Linearity of the interactions and influence of pyridine classical hydrogen bonds. CrystEngComm 2013, 15, 10481–10489. [Google Scholar] [CrossRef]
- Medforth, C.J.; Senge, M.O.; Forsyth, T.P.; Hobbs, J.D.; Shelnutt, J.A.; Smith, K.M. Conformational Study of 2,3,5,7,8,12,13,15,17,18-Decaalkylporphyrins. Inorg. Chem. 1994, 33, 3865–3872. [Google Scholar] [CrossRef]
- Ogoshi, H.; Setsune, J.; Omura, T.; Yoshida, Z. Novel rhodium(I)-porphyrin complexes and organorhodium(III)-porphyrin complexes. IV. J. Am. Chem. Soc. 1975, 97, 6461–6466. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, A.; Furuta, H.; Osuka, A. The first bis-Rh(I) metal complex of N-confused porphyrin. Chem. Commun. 2001, 1666–1667. [Google Scholar] [CrossRef]
- Kato, K.; Cha, W.; Oh, J.; Furukawa, K.; Yorimitsu, H.; Kim, D.; Osuka, A. Spontaneous Formation of an Air-Stable Radical upon the Direct Fusion of Diphenylmethane to a Triarylporphyrin. Angew. Chem. Int. Ed. 2016, 55, 8711–8714. [Google Scholar] [CrossRef]
- Lang, P.; Pfrunder, M.; Quach, G.; Braun-Cula, B.; Moore, E.G.; Schwalbe, M. Sensitized Photochemical CO2 Reduction by Hetero-Pacman Compounds Linking a Re I Tricarbonyl with a Porphyrin Unit. Chem. Eur. J. 2019, 25, 4509–4519. [Google Scholar] [CrossRef]
- Ryan, A.; Gehrold, A.; Perusitti, R.; Pintea, M.; Fazekas, M.; Locos, O.B.; Blaikie, F.; Senge, M.O. Porphyrin dimers and arrays. Eur. J. Org. Chem. 2011, 5817–5844. [Google Scholar] [CrossRef]
- Wojaczyński, J.; Stȩpień, M.; Latos-Grazyński, L. Monomeric and dimeric iron(III) complexes of 5-hydroxy-10,15,20-triphenylporphyrin: Formation of cyano and pyridine complexes of (5-oxo-10,15,20-triphenylphlorin)iron. Eur. J. Inorg. Chem. 2002, 1806–1815. [Google Scholar] [CrossRef]
- Jentzen, W.; Ma, J.G.; Shelnutt, J.A. Conservation of the conformation of the porphyrin macrocycle in hemoproteins. Biophys. J. 1998, 74, 753–763. [Google Scholar] [CrossRef]
- Shelnutt, J.A. Normal-coordinate structural decomposition and the vibronic spectra of porphyrins. J. Porphyr. Phthalocyanines 2001, 5, 300–311. [Google Scholar] [CrossRef]
- Schindler, J.; Kupfer, S.; Ryan, A.A.; Flanagan, K.J.; Senge, M.O.; Dietzek, B. Sterically induced distortions of nickel (II) porphyrins–Comprehensive investigation by DFT calculations and resonance Raman spectroscopy. Coord. Chem. Rev. 2018, 360, 1–16. [Google Scholar] [CrossRef]
- Gouterman, M. Spectra of Porphyrins. J. Mol. Spectrosc. 1961, 6, 138–163. [Google Scholar] [CrossRef]
- Senge, M.O. N-H Hydrogen Bonding in Porphyrins-from Conformational Design to Supramolecular Chemistry. ECS Trans. 2015, 66, 1–10. [Google Scholar] [CrossRef]
- Belcher, W.J.; Fabre, M.; Farhan, T.; Steed, J.W. Pyridinium CH⋯anion and π-stacking interactions in modular tripodal anion binding hosts: ATP binding and solid-state chiral induction. Org. Biomol. Chem. 2006, 4, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Steiner, T. The Hydrogen Bond in the Solid State. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Steiner, T. C-H⋯O hydrogen bonding in crystals. Crystallogr. Rev. 2003, 9, 177–228. [Google Scholar] [CrossRef]
- Itoh, Y.; Nakashima, Y.; Tsukamoto, S.; Kurohara, T.; Suzuki, M.; Sakae, Y.; Oda, M.; Okamoto, Y.; Suzuki, T. N+-C-H···O Hydrogen bonds in protein-ligand complexes. Sci. Rep. 2019, 9, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Ryppa, C.; Senge, M.O.; Hatscher, S.S.; Kleinpeter, E.; Wacker, P.; Schilde, U.; Wiehe, A. Synthesis of mono- And disubstituted porphyrins: A- and 5,10-A2-type systems. Chem. Eur. J. 2005, 11, 3427–3442. [Google Scholar] [CrossRef]
- Wiehe, A.; Shaker, Y.M.; Brandt, J.C.; Mebs, S.; Senge, M.O. Lead structures for applications in photodynamic therapy. Part 1: Synthesis and variation of m-THPC (Temoporfin) related amphiphilic A2BC-type porphyrins. Tetrahedron 2005, 61, 5535–5564. [Google Scholar] [CrossRef]
- Sugita, N.; Tsuchiya, I.; Takanami, T. Palladium-catalyzed cross-coupling reactions of brominated porphyrins with functionalized organomagnesium reagents: Direct preparation of functional-group-bearing free base porphyrins. Heterocycles 2016, 93, 483–511. [Google Scholar] [CrossRef]
- Nowak-Król, A.; Plamont, R.; Canard, G.; Edzang, J.A.; Gryko, D.T.; Balaban, T. An efficient synthesis of porphyrins with different meso substituents that avoids scrambling in aqueous media. Chem. Eur. J. 2015, 21, 1488–1498. [Google Scholar] [CrossRef] [PubMed]
- Takanami, T.; Hayashi, M.; Chijimatsu, H.; Inoue, W.; Suda, K. Palladium-catalyzed cyanation of porphyrins utilizing cyanoethylzinc bromide as an efficient cyanide ion source. Org. Lett. 2005, 7, 3937–3940. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.; Qu, W.; Liang, W.; Tai, F.; Guo, K.; Leung, C.W.; Lo, Y.H.; Wong, W.Y. Porphyrin-based metallopolymers: Synthesis, characterization and pyrolytic study for the generation of magnetic metal nanoparticles. J. Mater. Chem. C 2016, 4, 5010–5018. [Google Scholar] [CrossRef]
- Senge, M.O.; Pintea, M.; Ryan, A.A. Synthesis and crystal structure of a meso-meso directly linked bisporphyrin. Z. Naturforsch. 2011, 66b, 553–558. [Google Scholar] [CrossRef][Green Version]
- Ryan, A.A.; Senge, M.O. Synthesis and functionalization of triply fused porphyrin dimers. Eur. J. Org. Chem. 2013, 3700–3711. [Google Scholar] [CrossRef]
- Hope, H. X-ray crystallography—A fast, first-resort analytical tool. Progr. Inorg. Chem. 1994, 41, 1–19. [Google Scholar] [CrossRef]
- Rigaku. CrystalClear; Rigaku Corporation: Tokyo, Japan, 2011. [Google Scholar]
- Bruker. SAINT; Bruker AXS Inc.: Madison, WI, USA, 2012. [Google Scholar]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt graphical user interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef]
- Bruno, I.J.; Cole, J.C.; Edgington, P.R.; Kessler, M.; Macrae, C.F.; McCabe, P.; Pearson, J.; Taylor, R. New software for searching the Cambridge Structural Database and visualizing crystal structures. Acta Cryst. 2002, B58, 389–397. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | H211 | H211·CH2Cl2 | [H411][CF3CO2]2 | [H411][CF3CO2]2 ·2CF3CO2H | H213·⅓(CH2Cl2) | [H413][CF3CO2]2 | [H414][CF3CO2]2 | H215 |
|---|---|---|---|---|---|---|---|---|
| CCCDC # | 2012807 | 2012815 | 2012814 | 2012823 | 2012810 | 2012809 | 2012811 | 2012821 |
| Empirical Formula | C32H22N4 | C33H24Cl2N4 | C36H24F6N4O4 | C40H26F12N4O8 | C30.33H34.67Cl0.67N4 | C34H36F6N4O4 | C42H28F6N4O4 | C40H38N4O2 |
| Formula Weight g·mol−1 | 462.53 | 547.46 | 690.59 | 918.65 | 478.92 | 678.67 | 766.68 | 606.74 |
| Crystal system | triclinic | monoclinic | monoclinic | monoclinic | trigonal | monoclinic | triclinic | monoclinic |
| Space Group | P | P21/c | P21/c | P21/c | R | P21/c | P | P21/c |
| Z | 1 | 4 | 4 | 8 | 9 | 2 | 2 | 2 |
| λ (Å) | 0.71073 | 0.71075 | 0.71073 | 0.71073 | 0.71075 | 0.71075 | 1.54178 | 0.71073 |
| T (K) | 90(2) | 108(2) | 90(2) | 100(2) | 108(2) | 108(2) | 100(2) | 90(2) |
| a (Å) | 6.6737(6) | 18.190(7) | 10.9500(6) | 18.0414(17) | 19.1340(7) | 12.149(4) | 10.3071(4) | 9.5377(2) |
| b (Å) | 9.7034(9) | 13.682(5) | 8.3278(5) | 37.627(4) | 19.1340(7) | 10.177(3) | 10.4727(4) | 18.7751(5) |
| c (Å) | 10.1696(10) | 10.688(4) | 34.2043(18) | 12.3290(12) | 18.5994(9) | 14.348(4) | 16.9482(7) | 9.8937(4) |
| α (°) | 61.894(2) | 90 | 90 | 90 | 90 | 90 | 98.049(2) | 90 |
| β (°) | 87.318(2) | 99.159(6) | 94.1210(10) | 108.688(2) | 90 | 115.88(2) | 96.564(2) | 118.8140(10) |
| γ (°) | 76.631(2) | 90 | 90 | 90 | 120 | 90 | 99.110(2) | 90 |
| V (Å3) | 563.67(9) | 2626.1(17) | 3111.0(3) | 7928.3(13) | 5897.1(5) | 1596.1(9) | 1771.06(12) | 1552.32(8) |
| ρcalc (g·cm−3) | 1.363 | 1.385 | 1.474 | 1.539 | 1.214 | 1.412 | 1.438 | 1.298 |
| μ(Mo Kα), mm−1 | 0.082 | 0.082 | 0.279 | 0.122 | 0.145 | 0.137 | 0.117 | 0.981 |
| Independent reflections | 2934 | 2934 | 4623 | 8371 | 21398 | 3847 | 4335 | 6004 |
| Data/restraints/parameters | 2934/166/0 | 2934/0/166 | 4623/2/358 | 8371/1/463 | 21398/232/1247 | 3847/14/185 | 4335/133/295 | 6004/0/517 |
| R1 (I > 2σ(I)) | 0.0379 | 0.0954 | 0.0588 | 0.0798 | 0.0804 | 0.0621 | 0.0487 | 0.042 |
| wR2 (all) | 0.1082 | 0.1818 | 0.1505 | 0.2282 | 0.235 | 0.2237 | 0.1275 | 0.1243 |
| GoF | 1.029 | 1.224 | 0.998 | 1.298 | 1.073 | 0.994 | 1.009 | 1.005 |
| # | [H417][CF3CO2]2 ·2CF3CO2H | [H418][ClO4]2 | [H418][ClO4]2 | [H418][CF3CO2]2 ·2CF3CO2H | [H419][CF3CO2]2 ·2CF3CO2H | H219 | [H420][ClO4]2 |
|---|---|---|---|---|---|---|---|
| CCCDC # | 2012817 | 2012813 | 2012812 | 2012822 | 2012819 | 2012808 | 2012816 |
| Empirical Formula | C42H30F12N4O10 | C32H22Br2Cl2N4O8 | C32H22Br2Cl2N4O8 | C40H24Br2F12N4O8 | C42H30F12N4O8S2 | C36H24N4S | C32H22.81Br1.19Cl2N4O8 |
| Formula Weight g·mol−1 | 978.70 | 821.25 | 821.25 | 1076.45 | 1010.82 | 544.65 | 757.46 |
| Crystal system | triclinic | orthorhombic | orthorhombic | triclinic | triclinic | monoclinic | monoclinic |
| Space Group | P | Pbca | Pbca | P | P | P21/c | P21/c |
| Z | 2 | 8 | 8 | 2 | 2 | 4 | 4 |
| λ (Å) | 0.71073 | 1.54178 | 1.54178 | 0.71073 | 0.71073 | 1.54178 | 1.54178 |
| T (K) | 108(2) | 100(2) | 100(2) | 100(2) | 296(2) | 293(2) | 100(2) |
| a (Å) | 12.609(4) | 14.6001(5) | 14.5948(6) | 12.691(3) | 12.844(3) | 12.7897(7) | 9.5151(7) |
| b (Å) | 13.290(4) | 20.2708(7) | 20.2652(8) | 13.110(3) | 13.378(3) | 18.5630(11) | 21.4363(15) |
| c (Å) | 13.901(4) | 21.2555(8) | 21.2737(9) | 13.926(3) | 14.088(3) | 12.9972(8) | 15.0162(10) |
| α (°) | 107.622(3) | 90 | 90 | 107.04(3) | 107.629(4) | 90 | 90 |
| β (°) | 106.525(2) | 90 | 90 | 107.06(3) | 107.249(4) | 119.381(4) | 104.174(2) |
| γ (°) | 98.348(3) | 90 | 90 | 99.59(3) | 99.277(5) | 90 | 90 |
| V (Å3) | 2059.1(11) | 6290.7(4) | 6292.0(4) | 2035.3(9) | 2117.8(7) | 2688.8(3) | 2969.6(4) |
| ρcalc (g·cm−3) | 1.579 | 1.734 | 1.734 | 1.756 | 1.585 | 1.345 | 1.694 |
| μ(Mo Kα), mm−1 | 0.148 | 5.353 | 5.352 | 2.106 | 0.238 | 1.326 | 4.385 |
| Independent reflections | 11910 | 5464 | 2759 | 10952 | 13232 | 4136 | 5092 |
| Data/restraints/parameters | 11910/4/633 | 5464/152/485 | 2759/18/449 | 10952/2/610 | 13232/1/633 | 4136/230/413 | 5092/1/447 |
| R1 (I > 2σ(I)) | 0.0664 | 0.0375 | 0.0333 | 0.0844 | 0.0504 | 0.0474 | 0.0505 |
| wR2 (all) | 0.2236 | 0.1018 | 0.0771 | 0.1767 | 0.1436 | 0.1341 | 0.125 |
| GoF | 1.028 | 1.021 | 1.055 | 1.188 | 1.029 | 0.932 | 1.253 |
| # | [H421][CF3CO2]2·2CF3CO2H | [H422][MeSO4]2·¼H2O | [H823][ClO4]4·2H2O | [H824][CF3CO2]4·14H2O |
|---|---|---|---|---|
| CCCDC # | 2012820 | 2012818 | 2012824 | 2012825 |
| Empirical Formula | C42H28Br2F12N4O8 | C50H38.50N4O8.25S2 | C80H82Cl4N8O22 | C92H86F24N8O30 |
| Formula Weight g·mol−1 | 1104.5 | 891.47 | 1649.33 | 2239.68 |
| Crystal system | monoclinic | triclinic | monoclinic | monoclinic |
| Space Group | P21/n | P | C2/c | C2/c |
| Z | 4 | 2 | 4 | 4 |
| λ (Å) | 1.54178 | 1.54178 | 0.71073 | 0.71073 |
| T (K) | 100(2) | 100(2) | 100(2) | 100(2) |
| a (Å) | 12.5000(9) | 8.0315(4) | 28.7894(11) | 36.144(11) |
| b (Å) | 16.3340(12) | 13.8957(7) | 17.0458(6) | 13.956(4) |
| c (Å) | 20.9760(14) | 20.1428(9) | 16.0437(6) | 24.094(7) |
| α (°) | 90 | 99.575(3) | 90 | 90 |
| β (°) | 91.671(3) | 99.419(3) | 94.7000(10) | 126.501(5) |
| γ (°) | 90 | 104.745(3) | 90 | 90 |
| V (Å3) | 4281.0(5) | 2093.07(18) | 7846.8(5) | 9770(5) |
| ρcalc (g·cm−3) | 1.714 | 1.414 | 1.396 | 1.523 |
| μ(Mo Kα), mm−1 | 3.41 | 1.687 | 0.232 | 0.142 |
| Independent reflections | 7322 | 6701 | 9042 | 8925 |
| Data/restraints/parameters | 7322/1/636 | 6701/741/673 | 9042/297/612 | 8925/196/696 |
| R1 (I > 2σ(I)) | 0.0313 | 0.0460 | 0.0448 | 0.1181 |
| wR2 (all) | 0.082 | 0.1259 | 0.129 | 0.3597 |
| GoF | 1.037 | 1.037 | 1.070 | 1.687 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kingsbury, C.J.; Flanagan, K.J.; Eckhardt, H.-G.; Kielmann, M.; Senge, M.O. Weak Interactions and Conformational Changes in Core-Protonated A2- and Ax-Type Porphyrin Dications. Molecules 2020, 25, 3195. https://doi.org/10.3390/molecules25143195
Kingsbury CJ, Flanagan KJ, Eckhardt H-G, Kielmann M, Senge MO. Weak Interactions and Conformational Changes in Core-Protonated A2- and Ax-Type Porphyrin Dications. Molecules. 2020; 25(14):3195. https://doi.org/10.3390/molecules25143195
Chicago/Turabian StyleKingsbury, Christopher J., Keith J. Flanagan, Hans-Georg Eckhardt, Marc Kielmann, and Mathias O. Senge. 2020. "Weak Interactions and Conformational Changes in Core-Protonated A2- and Ax-Type Porphyrin Dications" Molecules 25, no. 14: 3195. https://doi.org/10.3390/molecules25143195
APA StyleKingsbury, C. J., Flanagan, K. J., Eckhardt, H.-G., Kielmann, M., & Senge, M. O. (2020). Weak Interactions and Conformational Changes in Core-Protonated A2- and Ax-Type Porphyrin Dications. Molecules, 25(14), 3195. https://doi.org/10.3390/molecules25143195