
A Practical Perspective on the Roles of Solution NMR Spectroscopy in Drug Discovery


Abstract
1. Introduction
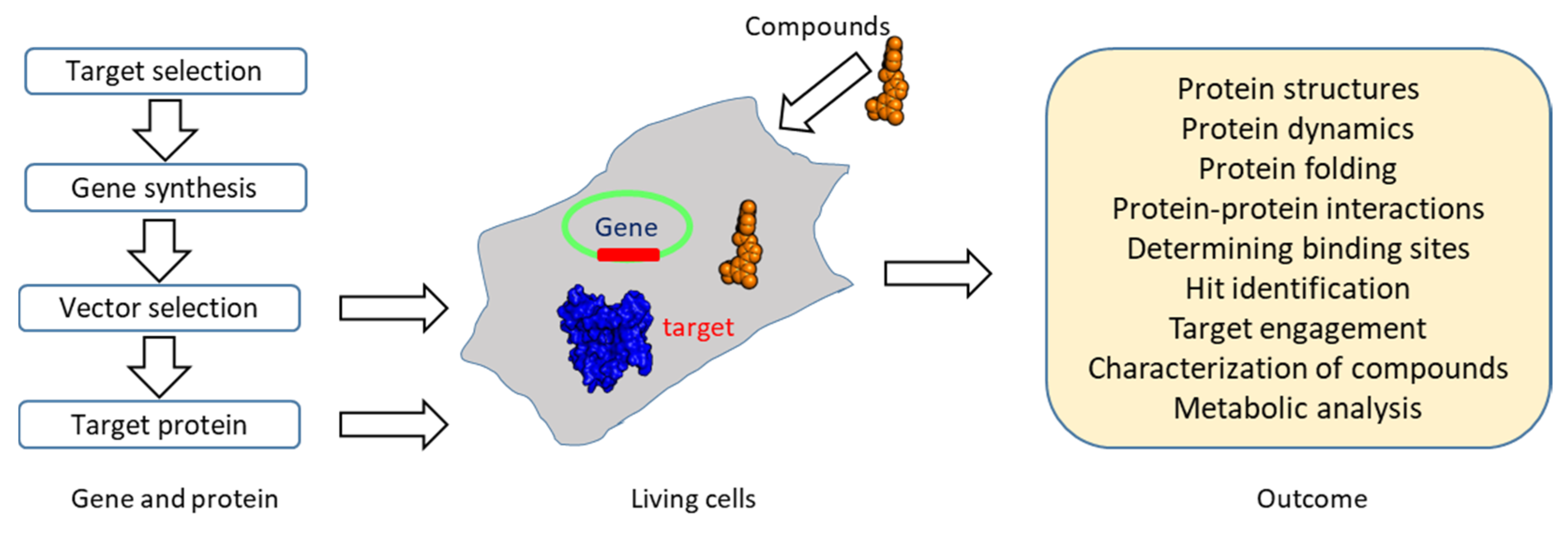
2. Advantages of NMR in Drug Discovery
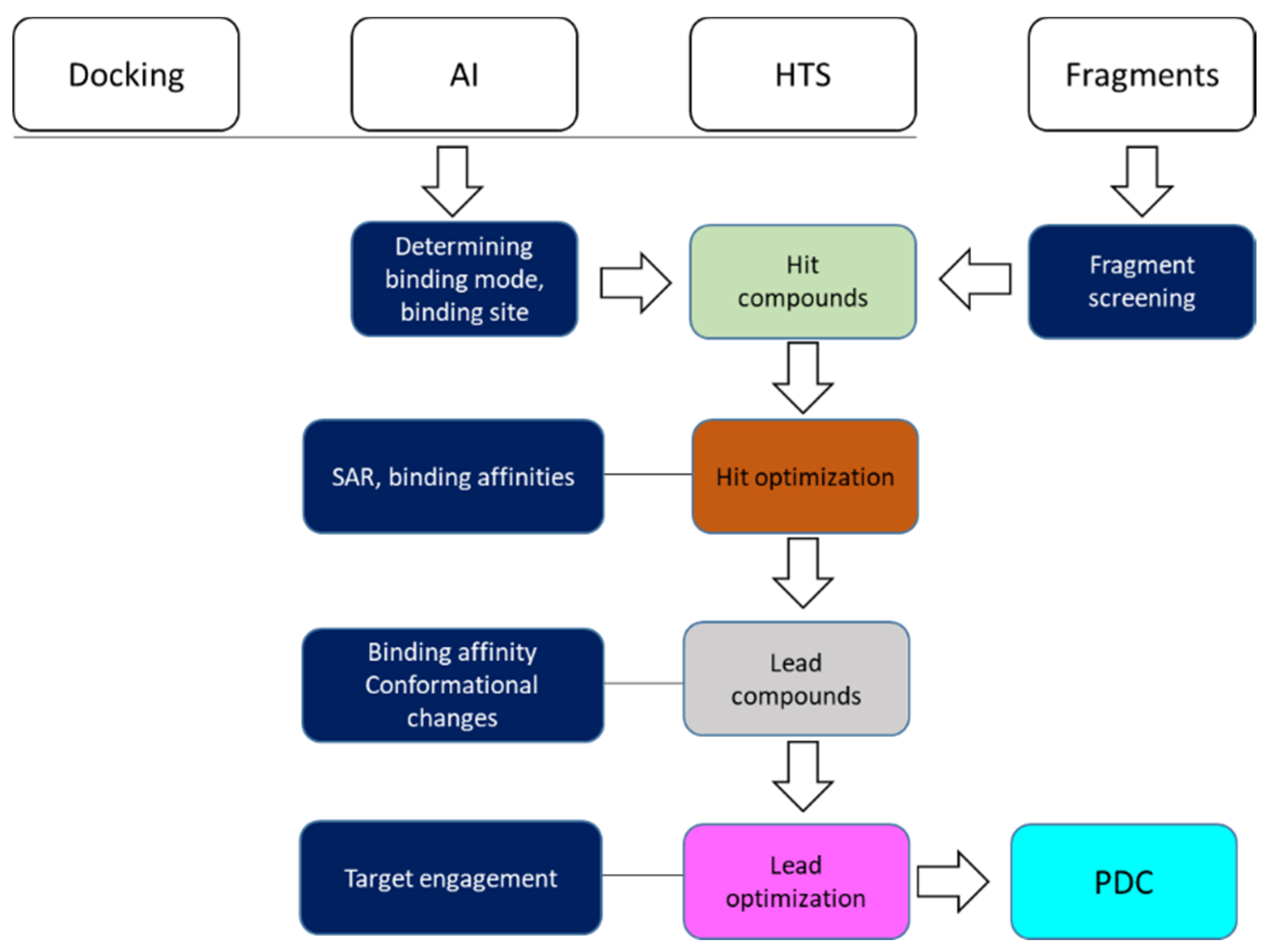
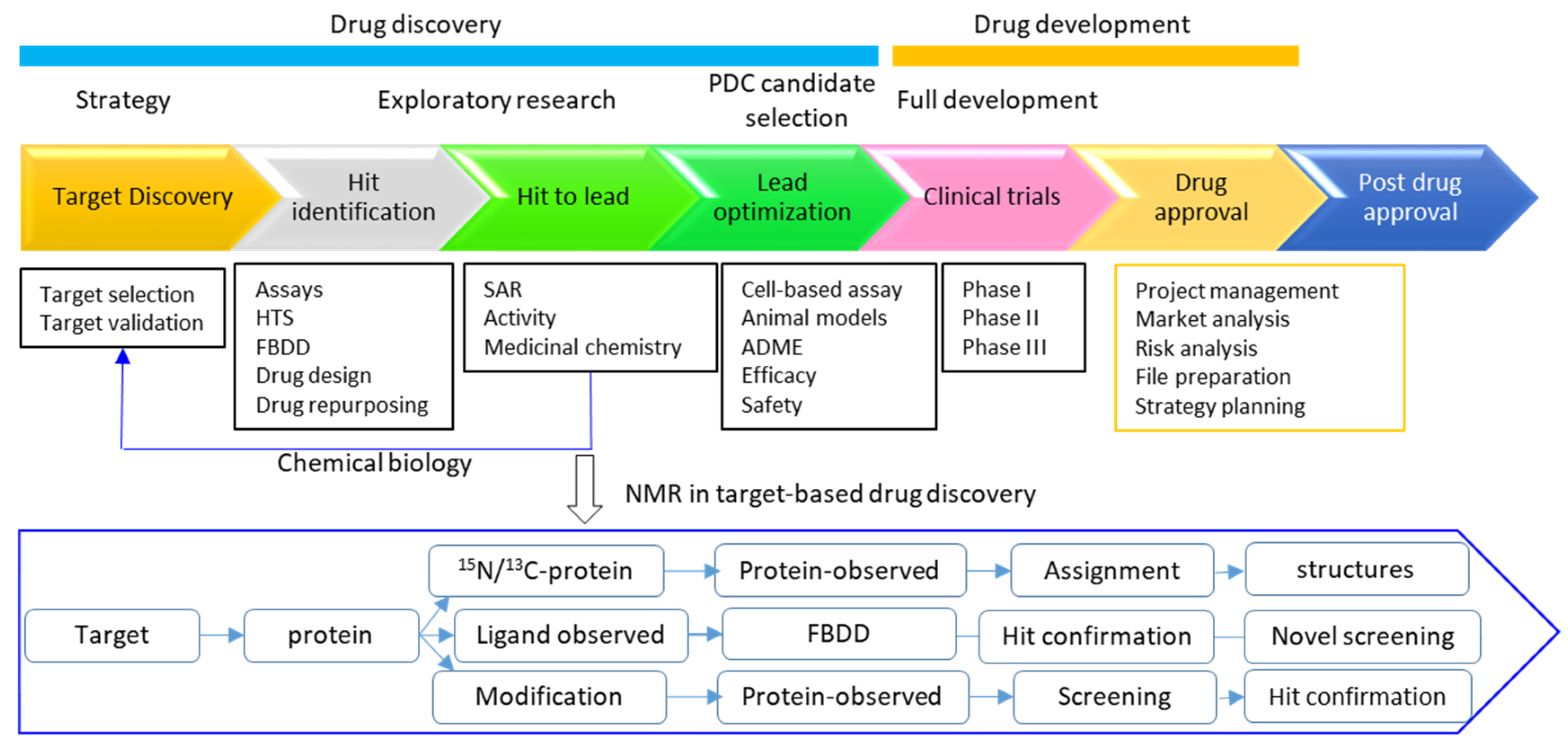
3. Roles of NMR in Drug Discovery
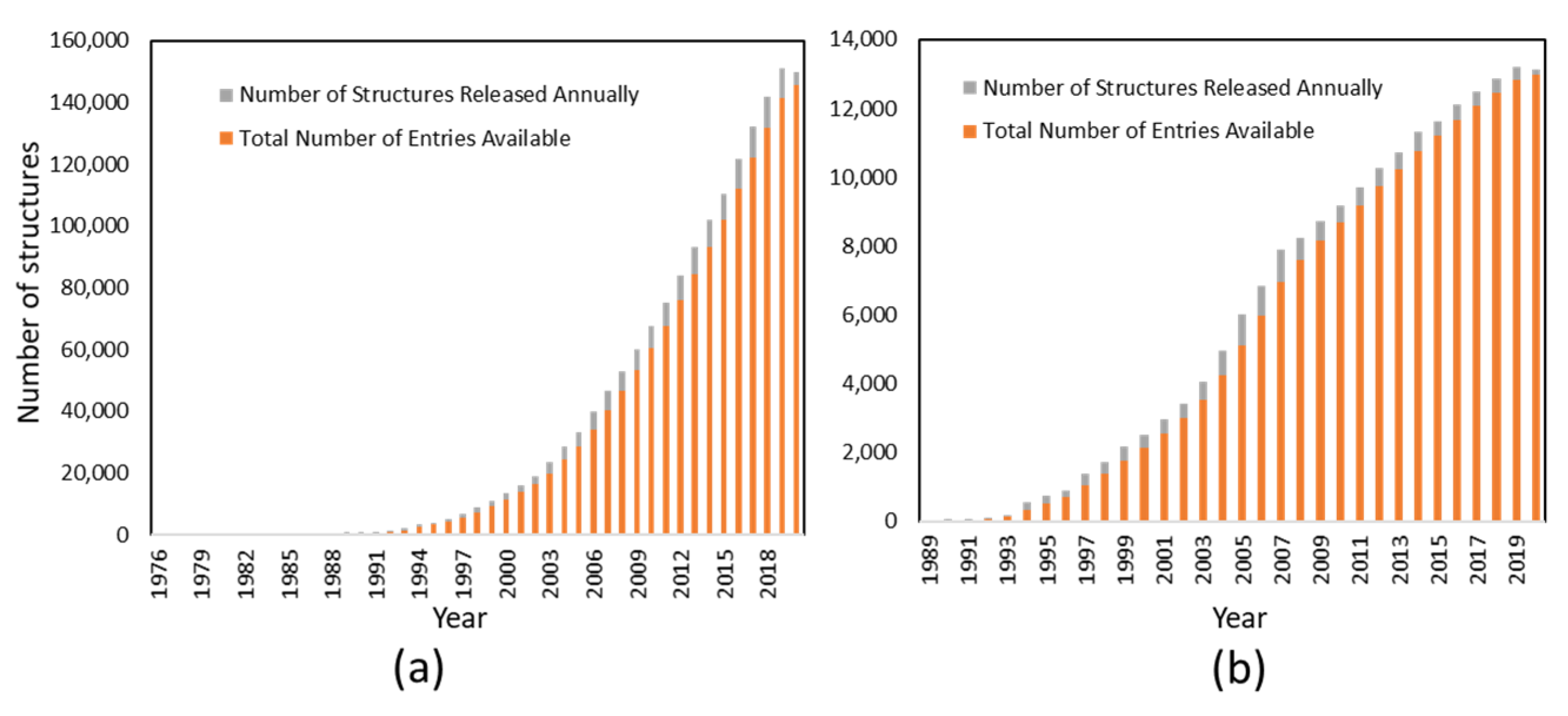
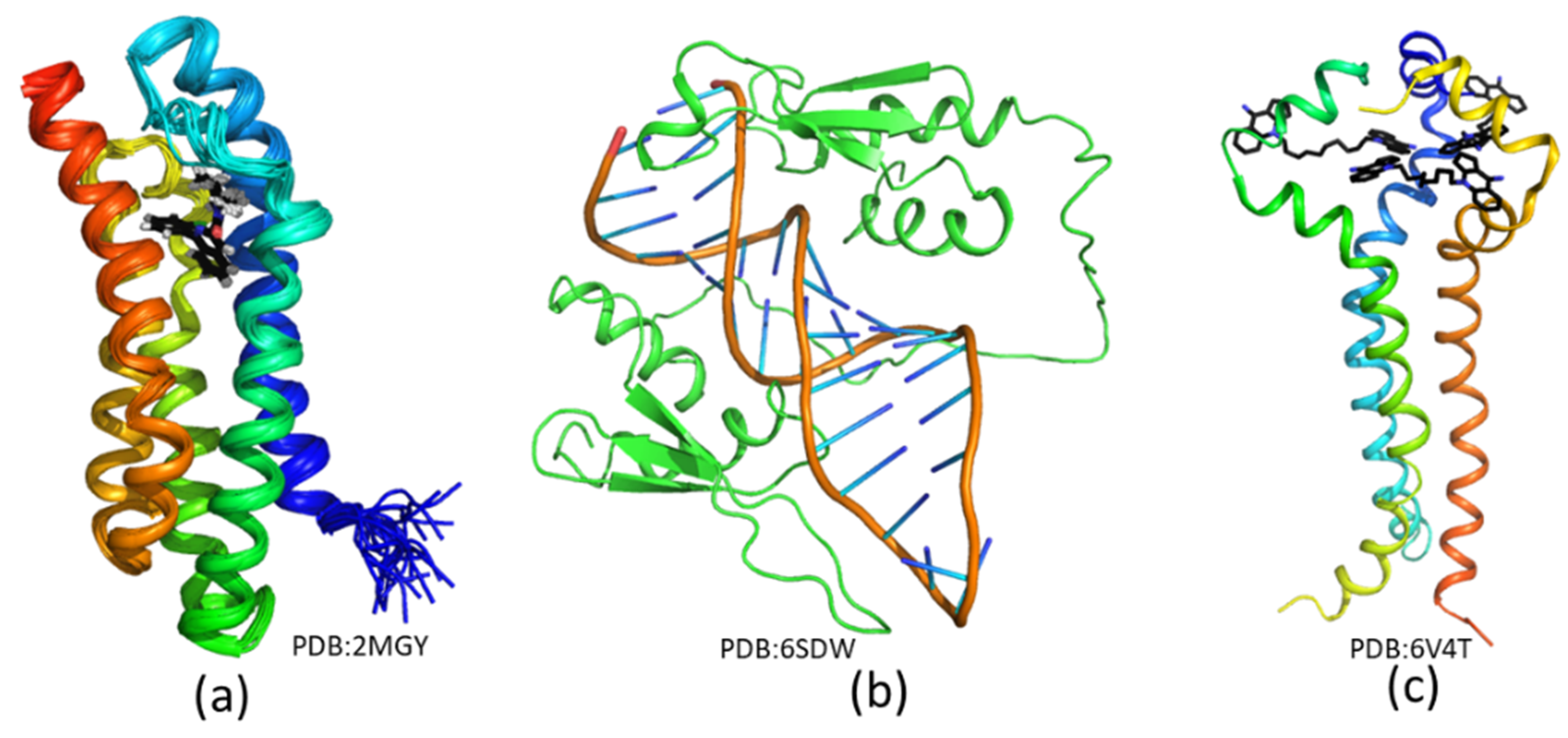
3.1. Structure Biology
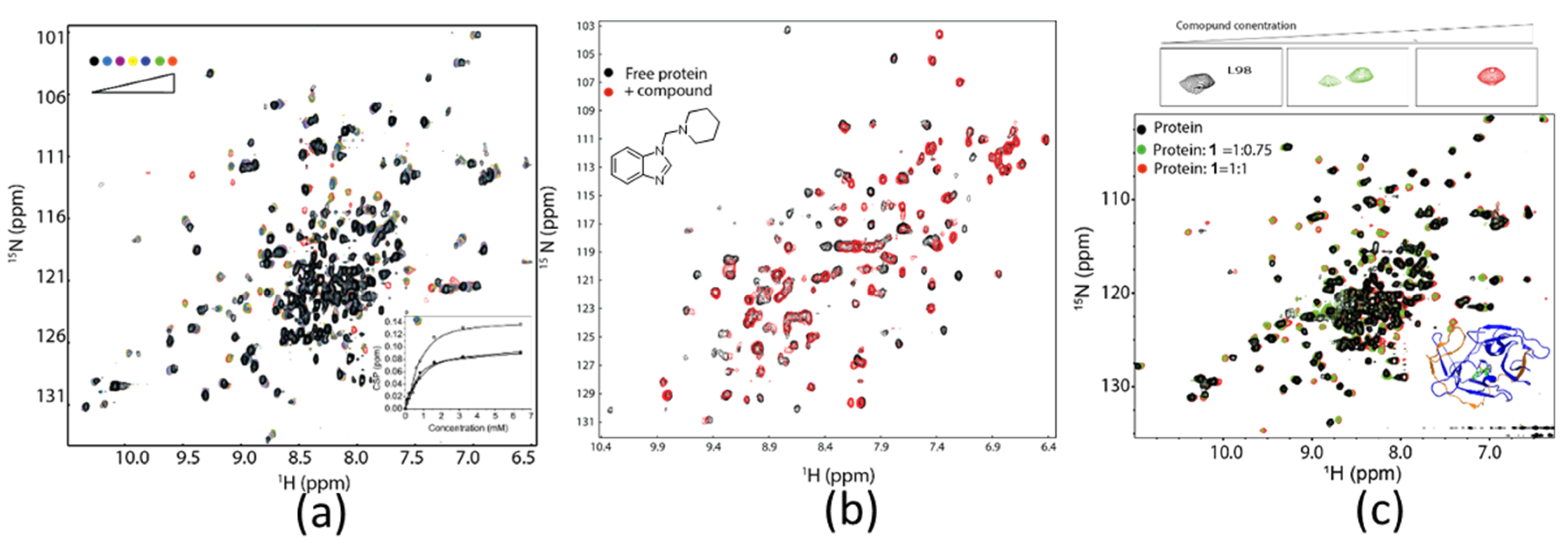
3.2. Hit Identification and Confirmation
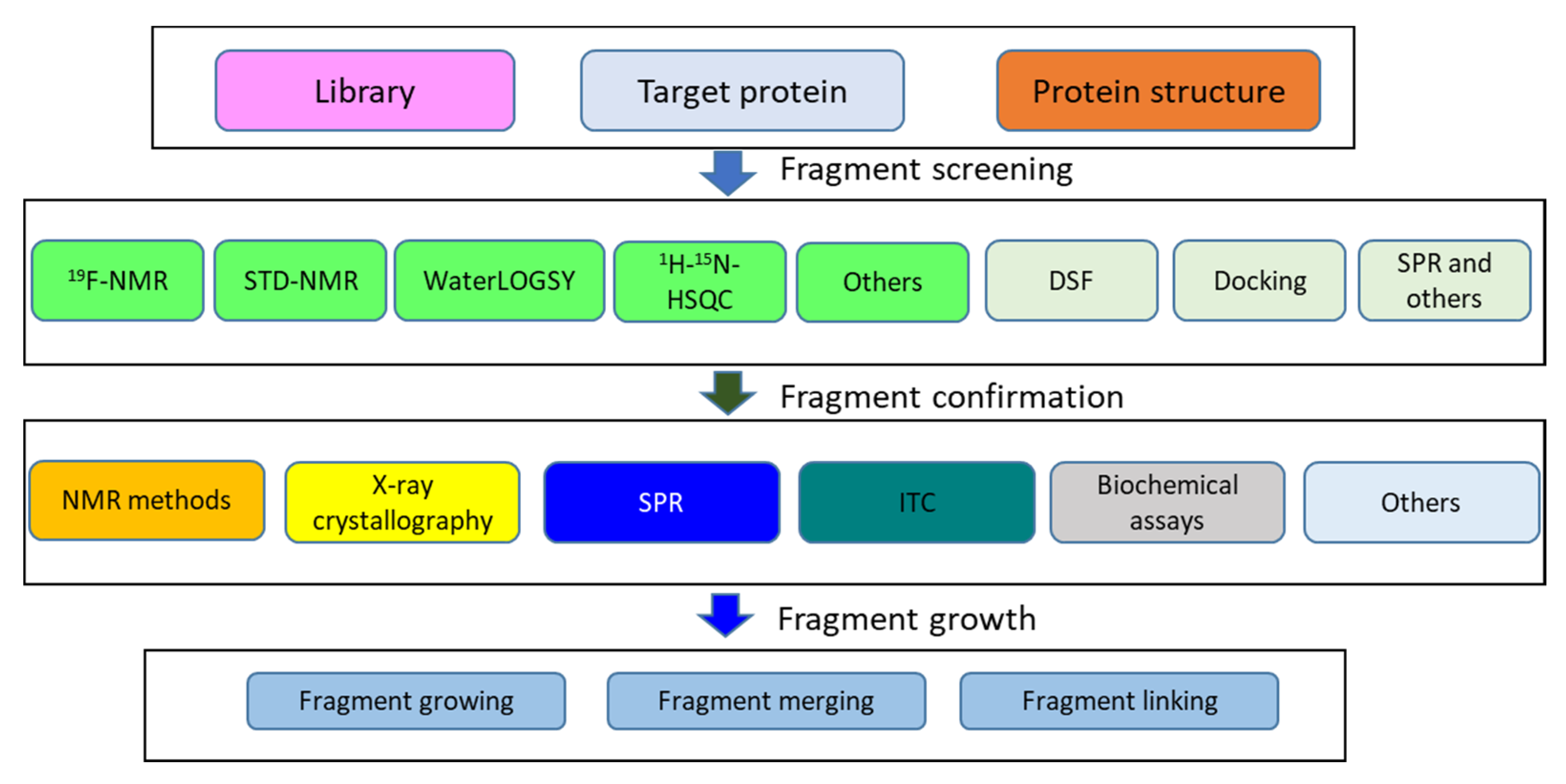
3.3. Fragment-Based Drug Discovery
3.4. Determining Ligand Binding Modes
3.4.1. Understand SAR in Drug Discovery
3.4.2. Solving Solution Structures of Protein-Ligand Complexes
3.4.3. Obtaining Structures through Docking
3.5. Target Engagement
3.6. Chemical Biology
4. Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Markwick, P.R.L.; Malliavin, T.; Nilges, M. Structural Biology by NMR: Structure, Dynamics, and Interactions. PLoS Comput. Biol. 2008, 4, e1000168. [Google Scholar] [CrossRef] [PubMed]
- Billeter, M.; Wagner, G.; Wuthrich, K. Solution NMR structure determination of proteins revisited. J. Biomol. NMR 2008, 42, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Alderson, T.R.; Kay, L.E. Unveiling invisible protein states with NMR spectroscopy. Curr. Opin. Struct. Biol. 2020, 60, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Sugiki, T.; Furuita, K.; Fujiwara, T.; Kojima, C. Current NMR Techniques for Structure-Based Drug Discovery. Molecules 2018, 23, 148. [Google Scholar] [CrossRef] [PubMed]
- Harner, M.J.; Mueller, L.; Robbins, K.J.; Reily, M.D. NMR in drug design. Arch. Biochem. Biophys. 2017, 628, 132–147. [Google Scholar] [CrossRef]
- Kay, L.E. New Views of Functionally Dynamic Proteins by Solution NMR Spectroscopy. J. Mol. Biol. 2016, 428, 323–331. [Google Scholar] [CrossRef]
- Jonas, J.; Ballard, L.; Nash, D. High-Resolution, High-Pressure NMR Studies of Proteins. Biophys. J. 1998, 75, 445–452. [Google Scholar] [CrossRef]
- Cho, M.-K.; Xiang, S.; Kim, H.-Y.; Becker, S.; Zweckstetter, M. Cold-Induced Changes in the Protein Ubiquitin. PLoS ONE 2012, 7, e37270. [Google Scholar] [CrossRef]
- Charlier, C.; Alderson, T.R.; Courtney, J.M.; Ying, J.; Anfinrud, P.; Bax, A. Study of protein folding under native conditions by rapidly switching the hydrostatic pressure inside an NMR sample cell. Proc. Natl. Acad. Sci. USA 2018, 115, E4169–E4178. [Google Scholar] [CrossRef]
- Jung, A.; Bamann, C.; Kremer, W.; Kalbitzer, H.R.; Brunner, E. High-temperature solution NMR structure of TmCsp. Protein Sci. 2004, 13, 342–350. [Google Scholar] [CrossRef][Green Version]
- Gayen, S.; Li, Q.; Kang, C. Solution NMR study of the transmembrane domain of single-span membrane proteins: Opportunities and strategies. Curr. Protein Pept. Sci. 2012, 13, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Li, Q. Solution NMR study of integral membrane proteins. Curr. Opin. Chem. Biol. 2011, 15, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Vanoye, C.G.; Welch, R.C.; Van Horn, W.D.; Sanders, C.R. Functional delivery of a membrane protein into oocyte membranes using bicelles. Biochemistry 2010, 49, 653–655. [Google Scholar] [CrossRef] [PubMed]
- Gayen, S.; Kang, C. Solution structure of a human minimembrane protein Ost4, a subunit of the oligosaccharyltransferase complex. Biochem. Biophys. Res. Commun. 2011, 409, 572–576. [Google Scholar] [CrossRef]
- Wallerstein, J.; Akke, M. Minute Additions of DMSO Affect Protein Dynamics Measurements by NMR Relaxation Experiments through Significant Changes in Solvent Viscosity. ChemPhysChem 2019, 20, 326–332. [Google Scholar] [CrossRef]
- Li, Q.; Wong, Y.L.; Kang, C. Solution structure of the transmembrane domain of the insulin receptor in detergent micelles. Biochim. Biophys. Acta 2014, 1838, 1313–1321. [Google Scholar] [CrossRef]
- Li, Q.; Wong, Y.L.; Huang, Q.; Kang, C. Structural insight into the transmembrane domain and the juxtamembrane region of the erythropoietin receptor in micelles. Biophys. J. 2014, 107, 2325–2336. [Google Scholar] [CrossRef]
- Sugiki, T.; Kobayashi, N.; Fujiwara, T. Modern Technologies of Solution Nuclear Magnetic Resonance Spectroscopy for Three-dimensional Structure Determination of Proteins Open Avenues for Life Scientists. Comput. Struct. Biotechnol. J. 2017, 15, 328–339. [Google Scholar] [CrossRef]
- Hyberts, S.G.; Arthanari, H.; Wagner, G. Applications of non-uniform sampling and processing. Top. Curr. Chem. 2012, 316, 125–148. [Google Scholar]
- Fernandez, C.; Wider, G. TROSY in NMR studies of the structure and function of large biological macromolecules. Curr. Opin. Struct. Biol. 2003, 13, 570–580. [Google Scholar] [CrossRef]
- Hiller, S.; Wagner, G. The role of solution NMR in the structure determinations of VDAC-1 and other membrane proteins. Curr. Opin. Struct. Biol. 2009, 19, 396–401. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Takeuchi, K.; Baskaran, K.; Arthanari, H. Structure determination using solution NMR: Is it worth the effort? J. Magn. Reson. 2019, 306, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Opella, S.J.; Marassi, F.M. Applications of NMR to membrane proteins. Arch. Biochem. Biophys. 2017, 628, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Opella, S.J.; Nevzorov, A.; Mesleb, M.F.; Marassi, F.M. Structure determination of membrane proteins by NMR spectroscopy. Biochem. Cell Biol. 2002, 80, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Yushmanov, V.E.; Tang, P. NMR studies of drug interaction with membranes and membrane-associated proteins. Biosci. Rep. 2002, 22, 175–196. [Google Scholar] [CrossRef]
- Gawrisch, K.; Eldho, N.V.; Polozov, I.V. Novel NMR tools to study structure and dynamics of biomembranes. Chem. Phys. Lipids 2002, 116, 135–151. [Google Scholar] [CrossRef]
- Chill, J.H.; Naider, F. A solution NMR view of protein dynamics in the biological membrane. Curr. Opin. Struct. Biol. 2011, 21, 627–633. [Google Scholar] [CrossRef]
- Barrett, P.J.; Chen, J.; Cho, M.-K.; Kim, J.-H.; Lu, Z.; Mathew, S.; Peng, D.; Song, Y.; Van Horn, W.D.; Zhuang, T.; et al. The Quiet Renaissance of Protein Nuclear Magnetic Resonance. Biochemistry 2013, 52, 1303–1320. [Google Scholar] [CrossRef]
- Mak, K.-K.; Pichika, M.R. Artificial intelligence in drug development: Present status and future prospects. Drug Discov. Today 2019, 24, 773–780. [Google Scholar] [CrossRef]
- Ke, Y.-Y.; Peng, T.-T.; Yeh, T.-K.; Huang, W.-Z.; Chang, S.-E.; Wu, S.-H.; Hung, H.-C.; Hsu, T.-A.; Lee, S.-J.; Song, J.-S.; et al. Artificial intelligence approach fighting COVID-19 with repurposing drugs. Biomed. J. 2020. [Google Scholar] [CrossRef]
- Nabuurs, S.B.; Spronk, C.A.; Krieger, E.; Maassen, H.; Vriend, G.; Vuister, G.W. Quantitative evaluation of experimental NMR restraints. J. Am. Chem. Soc. 2003, 125, 12026–12034. [Google Scholar] [CrossRef] [PubMed]
- Lipsitz, R.S.; Tjandra, N. Residual dipolar couplings in NMR structure analysis. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 387–413. [Google Scholar] [CrossRef]
- Varani, G.; Chen, Y.; Leeper, T.C. NMR studies of protein-nucleic acid interactions. Methods Mol. Biol. 2004, 278, 289–312. [Google Scholar] [PubMed]
- Berjanskii, M.V.; Wishart, D.S. A simple method to predict protein flexibility using secondary chemical shifts. J. Am. Chem. Soc. 2005, 127, 14970–14971. [Google Scholar] [CrossRef] [PubMed]
- Carlomagno, T. Ligand-target interactions: What can we learn from NMR? Annu. Rev. Biophys. Biomol. Struct. 2005, 34, 245–266. [Google Scholar] [CrossRef]
- Xu, Y.; Zheng, Y.; Fan, J.S.; Yang, D. A new strategy for structure determination of large proteins in solution without deuteration. Nat. Methods 2006, 3, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Kateb, F.; Pelupessy, P.; Bodenhausen, G. Measuring fast hydrogen exchange rates by NMR spectroscopy. J. Magn. Reson. 2007, 184, 108–113. [Google Scholar] [CrossRef][Green Version]
- Hansen, D.F.; Vallurupalli, P.; Kay, L.E. Using relaxation dispersion NMR spectroscopy to determine structures of excited, invisible protein states. J. Biomol. NMR 2008, 41, 113–120. [Google Scholar] [CrossRef]
- Kitevski-LeBlanc, J.L.; Evanics, F.; Prosser, R.S. Approaches for the measurement of solvent exposure in proteins by 19F NMR. J. Biomol. NMR 2009, 45, 255–264. [Google Scholar] [CrossRef]
- Ma, J.H.; Gruschus, J.M.; Tjandra, N. N-15-H-1 Scalar Coupling Perturbation: An Additional Probe for Measuring Structural Changes Due to Ligand Binding. J. Am. Chem. Soc. 2009, 131, 9884–9885. [Google Scholar] [CrossRef][Green Version]
- O’Connell, M.R.; Gamsjaeger, R.; Mackay, J.P. The structural analysis of protein-protein interactions by NMR spectroscopy. Proteomics 2009, 9, 5224–5232. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Bax, A. Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR 2013, 56, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Williamson, M.P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 73, 1–16. [Google Scholar] [CrossRef]
- Lepre, C.A.; Moore, J.M.; Peng, J.W. Theory and applications of NMR-based screening in pharmaceutical research. Chem. Rev. 2004, 104, 3641–3676. [Google Scholar] [CrossRef] [PubMed]
- Gossert, A.D.; Jahnke, W. NMR in drug discovery: A practical guide to identification and validation of ligands interacting with biological macromolecules. Prog. Nucl. Magn. Reson. Spectrosc. 2016, 97, 82–125. [Google Scholar] [CrossRef]
- Niesen, F.H.; Berglund, H.; Vedadi, M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2007, 2, 2212. [Google Scholar] [CrossRef]
- Du, X.; Li, Y.; Xia, Y.-L.; Ai, S.-M.; Liang, J.; Sang, P.; Ji, X.-L.; Liu, S.-Q. Insights into Protein–Ligand Interactions: Mechanisms, Models, and Methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef]
- McFedries, A.; Schwaid, A.; Saghatelian, A. Methods for the Elucidation of Protein-Small Molecule Interactions. Chem. Biol. 2013, 20, 667–673. [Google Scholar] [CrossRef]
- Maity, S.; Gundampati, R.K.; Suresh Kumar, T.K. NMR Methods to Characterize Protein-Ligand Interactions. Nat. Prod. Commun. 2019, 14. [Google Scholar] [CrossRef]
- Ludwig, C.; Guenther, U.L. Ligand based NMR methods for drug discovery. Front. Biosci. 2009, 14, 4565–4574. [Google Scholar] [CrossRef]
- Bista, M.; Kowalska, K.; Janczyk, W.; Domling, A.; Holak, T.A. Robust NMR screening for lead compounds using tryptophan-containing proteins. J. Am. Chem. Soc. 2009, 131, 7500–7501. [Google Scholar] [CrossRef] [PubMed]
- Powers, R.; Mercier, K.A.; Copeland, J.C. The application of FAST-NMR for the identification of novel drug discovery targets. Drug Discov. Today 2008, 13, 172–179. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pellecchia, M.; Bertini, I.; Cowburn, D.; Dalvit, C.; Giralt, E.; Jahnke, W.; James, T.L.; Homans, S.W.; Kessler, H.; Luchinat, C.; et al. Perspectives on NMR in drug discovery: A technique comes of age. Nat. Rev. Drug Discov. 2008, 7, 738–745. [Google Scholar] [CrossRef] [PubMed]
- Reibarkh, M.; Malia, T.J.; Hopkins, B.T.; Wagner, G. Identification of individual protein-ligand NOEs in the limit of intermediate exchange. J. Biomol. NMR 2006, 36, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Reibarkh, M.; Malia, T.J.; Wagner, G. NMR distinction of single- and multiple-mode binding of small-molecule protein ligands. J. Am. Chem. Soc. 2006, 128, 2160–2161. [Google Scholar] [CrossRef]
- Dalvit, C.; Vulpetti, A. Ligand-Based Fluorine NMR Screening: Principles and Applications in Drug Discovery Projects. J. Med. Chem. 2018, 62, 2218–2244. [Google Scholar] [CrossRef]
- Huth, J.R.; Sun, C.; Sauer, D.R.; Hajduk, P.J. Utilization of NMR-derived fragment leads in drug design. Methods Enzym. 2005, 394, 549–571. [Google Scholar]
- Shangary, S.; Wang, S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: A novel approach for cancer therapy. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 223–241. [Google Scholar] [CrossRef]
- Leone, M.; Rodriguez-Mias, R.A.; Pellecchia, M. Selective incorporation of 19F-labeled Trp side chains for NMR-spectroscopy-based ligand-protein interaction studies. Chembiochem 2003, 4, 649–650. [Google Scholar] [CrossRef]
- Murray, C.W.; Rees, D.C. The rise of fragment-based drug discovery. Nat. Chem. 2009, 1, 187–192. [Google Scholar] [CrossRef]
- Dalvit, C.; Mongelli, N.; Papeo, G.; Giordano, P.; Veronesi, M.; Moskau, D.; Kummerle, R. Sensitivity improvement in 19F NMR-based screening experiments: Theoretical considerations and experimental applications. J. Am. Chem. Soc. 2005, 127, 13380–13385. [Google Scholar] [CrossRef] [PubMed]
- Jahnke, W.; Floersheim, P.; Ostermeier, C.; Zhang, X.; Hemmig, R.; Hurth, K.; Uzunov, D.P. NMR Reporter Screening for the Detection of High-Affinity Ligands. Angew. Chem. Int. Ed. 2002, 41, 3420–3423. [Google Scholar] [CrossRef]
- Singh, A. Atomic-level in-cell protein NMR. Nat. Methods 2019, 16, 676. [Google Scholar] [CrossRef]
- Stadmiller, S.S.; Pielak, G.J. The Expanding Zoo of In-Cell Protein NMR. Biophys. J. 2018, 115, 1628–1629. [Google Scholar] [CrossRef] [PubMed]
- Luchinat, E.; Banci, L. In-Cell NMR in Human Cells: Direct Protein Expression Allows Structural Studies of Protein Folding and Maturation. Acc. Chem. Res. 2018, 51, 1550–1557. [Google Scholar] [CrossRef]
- Dotsch, V. In-cell NMR Spectroscopy for the Investigation of the Conformation of Macromolecules. Biophys. J. 2018, 114, 400a. [Google Scholar] [CrossRef]
- Luchinat, E.; Banci, L. A Unique Tool for Cellular Structural Biology: In-cell NMR. J. Biol. Chem. 2016, 291, 3776–3784. [Google Scholar] [CrossRef]
- Delius, J.; Frank, O.; Hofmann, T. Label-free quantitative 1H NMR spectroscopy to study low-affinity ligand–protein interactions in solution: A contribution to the mechanism of polyphenol-mediated astringency. PLoS ONE 2017, 12, e0184487. [Google Scholar] [CrossRef]
- Brancaccio, D.; Di Maro, S.; Cerofolini, L.; Giuntini, S.; Fragai, M.; Luchinat, C.; Tomassi, S.; Limatola, A.; Russomanno, P.; Merlino, F.; et al. HOPPI-NMR: Hot-Peptide-Based Screening Assay for Inhibitors of Protein–Protein Interactions by NMR. Acs Med. Chem. Lett. 2020, 11, 1047–1053. [Google Scholar] [CrossRef]
- Vanwetswinkel, S.; Heetebrij, R.J.; van Duynhoven, J.; Hollander, J.G.; Filippov, D.V.; Hajduk, P.J.; Siegal, G. TINS, Target Immobilized NMR Screening: An Efficient and Sensitive Method for Ligand Discovery. Chem. Biol. 2005, 12, 207–216. [Google Scholar] [CrossRef]
- Shuker, S.B.; Hajduk, P.J.; Meadows, R.P.; Fesik, S.W. Discovering high-affinity ligands for proteins: SAR by NMR. Science 1996, 274, 1531–1534. [Google Scholar] [CrossRef] [PubMed]
- Medek, A.; Hajduk, P.J.; Mack, J.; Fesik, S.W. The use of differential chemical shifts for determining the binding site location and orientation of protein-bound ligands. J. Am. Chem. Soc. 2000, 122, 1241–1242. [Google Scholar] [CrossRef]
- Jaremko, L.; Jaremko, M.; Giller, K.; Becker, S.; Zweckstetter, M. Structure of the mitochondrial translocator protein in complex with a diagnostic ligand. Science 2014, 343, 1363–1366. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wong, Y.L.; Ng, F.M.; Liu, B.; Wong, Y.X.; Poh, Z.Y.; Liu, S.; Then, S.W.; Lee, M.Y.; Ng, H.Q.; et al. Escherichia coli topoisomerase IV E subunit and an inhibitor binding mode revealed by NMR spectroscopy. J. Biol. Chem. 2016, 291, 17743–17753. [Google Scholar] [CrossRef] [PubMed]
- Su, X.C.; Ozawa, K.; Yagi, H.; Lim, S.P.; Wen, D.; Ekonomiuk, D.; Huang, D.; Keller, T.H.; Sonntag, S.; Caflisch, A.; et al. NMR study of complexes between low molecular mass inhibitors and the West Nile virus NS2B-NS3 protease. Febs. J. 2009, 276, 4244–4255. [Google Scholar] [CrossRef] [PubMed]
- Viegas, A.; Manso, J.; Nobrega, F.L.; Cabrita, E.J. Saturation-Transfer Difference (STD) NMR: A Simple and Fast Method for Ligand Screening and Characterization of Protein Binding. J. Chem. Educ. 2011, 88, 990–994. [Google Scholar] [CrossRef]
- Dalvit, C.; Fogliatto, G.; Stewart, A.; Veronesi, M.; Stockman, B. WaterLOGSY as a method for primary NMR screening: Practical aspects and range of applicability. J. Biomol. NMR 2001, 21, 349–359. [Google Scholar] [CrossRef]
- Dalvit, C.; Pevarello, P.; Tatò, M.; Veronesi, M.; Vulpetti, A.; Sundström, M. Identification of compounds with binding affinity to proteins via magnetization transfer from bulk water*. J. Biomol. NMR 2000, 18, 65–68. [Google Scholar] [CrossRef]
- Ni, F. Recent developments in transferred NOE methods. Prog. Nucl. Magn. Reson. Spectrosc. 1994, 26, 517–606. [Google Scholar] [CrossRef]
- Fejzo, J.; Lepre, C.A.; Peng, J.W.; Bemis, G.W.; Ajay; Murcko, M.A.; Moore, J.M. The SHAPES strategy: An NMR-based approach for lead generation in drug discovery. Chem. Biol. 1999, 6, 755–769. [Google Scholar] [CrossRef]
- Becattini, B.; Pellecchia, M. SAR by ILOEs: An NMR-Based Approach to Reverse Chemical Genetics. Chem. A Eur. J. 2006, 12, 2658–2662. [Google Scholar] [CrossRef] [PubMed]
- Derrick, T.S.; McCord, E.F.; Larive, C.K. Analysis of Protein/Ligand Interactions with NMR Diffusion Measurements: The Importance of Eliminating the Protein Background. J. Magn. Reson. 2002, 155, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Skora, L.; Jahnke, W. 19F-NMR-Based Dual-Site Reporter Assay for the Discovery and Distinction of Catalytic and Allosteric Kinase Inhibitors. Acs. Med. Chem. Lett. 2017, 8, 632–635. [Google Scholar] [CrossRef] [PubMed]
- Dalvit, C.; Flocco, M.; Veronesi, M.; Stockman, B.J. Fluorine-NMR competition binding experiments for high-throughput screening of large compound mixtures. Comb. Chem. High Throughput Screen 2002, 5, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Dalvit, C.; Flocco, M.; Knapp, S.; Mostardini, M.; Perego, R.; Stockman, B.J.; Veronesi, M.; Varasi, M. High-throughput NMR-based screening with competition binding experiments. J. Am. Chem. Soc. 2002, 124, 7702–7709. [Google Scholar] [CrossRef]
- Hajduk, P.J.; Olejniczak, E.T.; Fesik, S.W. One-Dimensional Relaxation- and Diffusion-Edited NMR Methods for Screening Compounds That Bind to Macromolecules. J. Am. Chem. Soc. 1997, 119, 12257–12261. [Google Scholar] [CrossRef]
- Fraenkel, Y.; Navon, G.; Aronheim, A.; Gershoni, J.M. Direct measurement of agonist binding to genetically engineered peptides of the acetylcholine receptor by selective T1 NMR relaxation. Biochemistry 1990, 29, 2617–2622. [Google Scholar] [CrossRef]
- Lee, M.D.; Loh, C.T.; Shin, J.; Chhabra, S.; Dennis, M.L.; Otting, G.; Swarbrick, J.D.; Graham, B. Compact, hydrophilic, lanthanide-binding tags for paramagnetic NMR spectroscopy. Chem. Sci. 2015, 6, 2614–2624. [Google Scholar] [CrossRef]
- Liang, B.; Bushweller, J.H.; Tamm, L.K. Site-directed parallel spin-labeling and paramagnetic relaxation enhancement in structure determination of membrane proteins by solution NMR spectroscopy. J. Am. Chem. Soc. 2006, 128, 4389–4397. [Google Scholar] [CrossRef]
- Jahnke, W.; Rüdisser, S.; Zurini, M. Spin Label Enhanced NMR Screening. J. Am. Chem. Soc. 2001, 123, 3149–3150. [Google Scholar] [CrossRef]
- Prestegard, J.H.; al-Hashimi, H.M.; Tolman, J.R. NMR structures of biomolecules using field oriented media and residual dipolar couplings. Q. Rev. Biophys. 2000, 33, 371–424. [Google Scholar] [CrossRef] [PubMed]
- Carulla, N.; Caddy, G.L.; Hall, D.R.; Zurdo, J.; Gairí, M.; Feliz, M.; Giralt, E.; Robinson, C.V.; Dobson, C.M. Molecular recycling within amyloid fibrils. Nature 2005, 436, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Nakanishi, T.; Kami, K.; Arata, Y.; Shimada, I. A novel NMR method for determining the interfaces of large protein–protein complexes. Nat. Struct. Biol. 2000, 7, 220–223. [Google Scholar] [PubMed]
- Tanaka, T.; Ikeya, T.; Kamoshida, H.; Suemoto, Y.; Mishima, M.; Shirakawa, M.; Güntert, P.; Ito, Y. High-Resolution Protein 3D Structure Determination in Living Eukaryotic Cells. Angew. Chem. Int. Ed. 2019, 58, 7284–7288. [Google Scholar] [CrossRef] [PubMed]
- Bleicher, K.H.; Böhm, H.-J.; Müller, K.; Alanine, A.I. Hit and lead generation: Beyond high-throughput screening. Nat. Rev. Drug Discov. 2003, 2, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Shimada, I.; Ueda, T.; Kofuku, Y.; Eddy, M.T.; Wuthrich, K. GPCR drug discovery: Integrating solution NMR data with crystal and cryo-EM structures. Nat. Rev. Drug Discov. 2019, 18, 59–82. [Google Scholar] [CrossRef] [PubMed]
- Jahnke, W. Perspectives of biomolecular NMR in drug discovery: The blessing and curse of versatility. J. Biomol. NMR 2007, 39, 87–90. [Google Scholar] [CrossRef]
- Li, Y.; Kang, C. Solution NMR Spectroscopy in Target-Based Drug Discovery. Molecules 2017, 22, 1399. [Google Scholar]
- Göbl, C.; Madl, T.; Simon, B.; Sattler, M. NMR approaches for structural analysis of multidomain proteins and complexes in solution. Prog. Nucl. Magn. Reson. Spectrosc. 2014, 80, 26–63. [Google Scholar] [CrossRef]
- Evangelidis, T.; Nerli, S.; Nováček, J.; Brereton, A.E.; Karplus, P.A.; Dotas, R.R.; Venditti, V.; Sgourakis, N.G.; Tripsianes, K. Automated NMR resonance assignments and structure determination using a minimal set of 4D spectra. Nat. Commun. 2018, 9, 384. [Google Scholar] [CrossRef]
- Sanders, C.R.; Sonnichsen, F. Solution NMR of membrane proteins: Practice and challenges. Magn. Reson. Chem. 2006, 44, S24–S40. [Google Scholar] [CrossRef]
- Konrat, R. NMR contributions to structural dynamics studies of intrinsically disordered proteins. J. Magn. Reson. 2014, 241, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, E.B.; Cook, E.C.; Showalter, S.A. Application of NMR to studies of intrinsically disordered proteins. Arch. Biochem. Biophys. 2017, 628, 57–70. [Google Scholar] [CrossRef]
- Hilty, C.; Wider, G.; Fernandez, C.; Wuthrich, K. Stereospecific assignments of the isopropyl methyl groups of the membrane protein OmpX in DHPC micelles. J. Biomol. NMR 2003, 27, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Pellecchia, M.; Sem, D.S.; Wuthrich, K. NMR in drug discovery. Nat. Rev. Drug Discov. 2002, 1, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Pervushin, K.; Riek, R.; Wider, G.; Wuthrich, K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. USA 1997, 94, 12366–12371. [Google Scholar] [CrossRef]
- Kaptein, R.; Wagner, G. NMR studies of membrane proteins. J. Biomol. NMR 2015, 61, 181–184. [Google Scholar] [CrossRef][Green Version]
- Danmaliki, G.I.; Hwang, P.M. Solution NMR spectroscopy of membrane proteins. Biochim. Et Biophys. Acta (Bba)-Biomembr. 2020, 1862, 183356. [Google Scholar] [CrossRef]
- Nitsche, C.; Otting, G. NMR studies of ligand binding. Curr. Opin. Struct. Biol. 2018, 48, 16–22. [Google Scholar] [CrossRef]
- Lange, O.F.; Rossi, P.; Sgourakis, N.G.; Song, Y.; Lee, H.W.; Aramini, J.M.; Ertekin, A.; Xiao, R.; Acton, T.B.; Montelione, G.T.; et al. Determination of solution structures of proteins up to 40 kDa using CS-Rosetta with sparse NMR data from deuterated samples. Proc. Nat. Acad. Sci. USA 2012, 109, 10873–10878. [Google Scholar] [CrossRef]
- Vernon, R.; Shen, Y.; Baker, D.; Lange, O.F. Improved chemical shift based fragment selection for CS-Rosetta using Rosetta3 fragment picker. J. Biomol. NMR 2013, 57, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Roy, A. Early Probe and Drug Discovery in Academia: A Minireview. High-Throughput 2018, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Dahlin, J.L.; Nissink, J.W.M.; Strasser, J.M.; Francis, S.; Higgins, L.; Zhou, H.; Zhang, Z.; Walters, M.A. PAINS in the Assay: Chemical Mechanisms of Assay Interference and Promiscuous Enzymatic Inhibition Observed during a Sulfhydryl-Scavenging HTS. J. Med. Chem. 2015, 58, 2091–2113. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.; Walters, M.A. Chemistry: Chemical con artists foil drug discovery. Nature 2014, 513, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Erlanson, D.A.; Fesik, S.W.; Hubbard, R.E.; Jahnke, W.; Jhoti, H. Twenty years on: The impact of fragments on drug discovery. Nat. Rev. Drug Discov. 2016, 15, 605–619. [Google Scholar] [CrossRef]
- Sabbah, M.; Mendes, V.; Vistal, R.G.; Dias, D.M.G.; Záhorszká, M.; Mikušová, K.; Korduláková, J.; Coyne, A.G.; Blundell, T.L.; Abell, C. Fragment-Based Design of Mycobacterium tuberculosis InhA Inhibitors. J. Med. Chem. 2020, 63, 4749–4761. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.-Y.; Ng, F.M.; Tan, Y.W.; Poulsen, A.; Seetoh, W.; Lin, G.; Kang, C.; Then, S.W.; Ahmad, N.H.; Wong, Y.L.; et al. Application of Fragment-Based Drug Discovery against DNA Gyrase B. ChemPlusChem 2015, 80, 1250–1254. [Google Scholar] [CrossRef]
- Robson-Tull, J. Biophysical screening in fragment-based drug design: A brief overview. Biosci. Horiz. Int. J. Stud. Res. 2019, 11, hzy015. [Google Scholar] [CrossRef]
- Jacquemard, C.; Kellenberger, E. A bright future for fragment-based drug discovery: What does it hold? Expert Opin. Drug Discov. 2019, 14, 413–416. [Google Scholar] [CrossRef]
- Harner, M.J.; Frank, A.O.; Fesik, S.W. Fragment-based drug discovery using NMR spectroscopy. J. Biomol. NMR 2013, 56, 65–75. [Google Scholar] [CrossRef]
- Hajduk, P.J.; Meadows, R.P.; Fesik, S.W. NMR-based screening in drug discovery. Q. Rev. Biophys. 1999, 32, 211–240. [Google Scholar] [CrossRef]
- Singh, M.; Tam, B.; Akabayov, B. NMR-Fragment Based Virtual Screening: A Brief Overview. Molecules 2018, 23, 233. [Google Scholar] [CrossRef] [PubMed]
- Poppler, A.C. Filling Blank Spots on the Map: Identification of Ligand Binding Modes and Interacting Water Molecules for Brd4-BD1 by WaterLOGSY Titrations. J. Med. Chem. 2017, 60, 8706–8707. [Google Scholar] [CrossRef] [PubMed]
- Lingel, A.; Vulpetti, A.; Reinsperger, T.; Proudfoot, A.; Denay, R.; Frommlet, A.; Henry, C.; Hommel, U.; Gossert, A.; Luy, B.; et al. Comprehensive and High-Throughput Exploration of Chemical Space Using Broadband 19F NMR-Based Screening. Angew. Chem. Int. Ed. 2020. [Google Scholar] [CrossRef]
- Norton, R.S.; Leung, E.W.W.; Chandrashekaran, I.R.; MacRaild, C.A. Applications of 19F-NMR in Fragment-Based Drug Discovery. Molecules 2016, 21, 860. [Google Scholar] [CrossRef]
- Stadmiller, S.S.; Aguilar, J.S.; Waudby, C.A.; Pielak, G.J. Rapid Quantification of Protein-Ligand Binding via 19F NMR Lineshape Analysis. Biophys. J. 2020, 118, 2537–2548. [Google Scholar] [CrossRef]
- Gee, C.T.; Arntson, K.E.; Urick, A.K.; Mishra, N.K.; Hawk, L.M.L.; Wisniewski, A.J.; Pomerantz, W.C.K. Protein-observed 19F-NMR for fragment screening, affinity quantification and druggability assessment. Nat. Protoc. 2016, 11, 1414. [Google Scholar] [CrossRef]
- Becker, W.; Bhattiprolu, K.C.; Gubensäk, N.; Zangger, K. Investigating Protein–Ligand Interactions by Solution Nuclear Magnetic Resonance Spectroscopy. ChemPhysChem 2018, 19, 895–906. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, Y.; Loh, Y.R.; Phoo, W.W.; Hung, A.W.; Kang, C.; Luo, D. Crystal structure of unlinked NS2B-NS3 protease from Zika virus. Science 2016, 354, 1597–1600. [Google Scholar] [CrossRef]
- Li, Y.; Liu, S.; Ng, E.Y.; Li, R.; Poulsen, A.; Hill, J.; Pobbati, A.V.; Hung, A.W.; Hong, W.; Keller, T.H.; et al. Structural and ligand-binding analysis of the YAP-binding domain of transcription factor TEAD4. Biochem. J. 2018, 475, 2043–2055. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Z.; Phoo, W.W.; Loh, Y.R.; Li, R.; Yang, H.Y.; Jansson, A.E.; Hill, J.; Keller, T.H.; Nacro, K.; et al. Structural Insights into the Inhibition of Zika Virus NS2B-NS3 Protease by a Small-Molecule Inhibitor. Structure 2018, 26, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Fesik, S.W.; Zuiderweg, E.R.; Olejniczak, E.T.; Gampe, R.T. Jr. NMR methods for determining the structures of enzyme/inhibitor complexes as an aid in drug design. Biochem. Pharm. 1990, 40, 161–167. [Google Scholar] [CrossRef]
- Sirockin, F.; Sich, C.; Improta, S.; Schaefer, M.; Saudek, V.; Froloff, N.; Karplus, M.; Dejaegere, A. Structure activity relationship by NMR and by computer: A comparative study. J. Am. Chem. Soc. 2002, 124, 11073–11084. [Google Scholar] [CrossRef] [PubMed]
- Tikhonova, I.G.; Costanzi, S. Unraveling the structure and function of G protein-coupled receptors through NMR spectroscopy. Curr. Pharm. Des. 2009, 15, 4003–4016. [Google Scholar] [CrossRef]
- Proudfoot, A.; Bussiere, D.E.; Lingel, A. High-Confidence Protein–Ligand Complex Modeling by NMR-Guided Docking Enables Early Hit Optimization. J. Am. Chem. Soc. 2017, 139, 17824–17833. [Google Scholar] [CrossRef]
- Bai, F.; Morcos, F.; Cheng, R.R.; Jiang, H.; Onuchic, J.N. Elucidating the druggable interface of protein−protein interactions using fragment docking and coevolutionary analysis. Proc. Natl. Acad. Sci. USA 2016, 113, E8051–E8058. [Google Scholar] [CrossRef]
- Wang, T.; Wu, M.B.; Chen, Z.J.; Chen, H.; Lin, J.P.; Yang, L.R. Fragment-Based Drug Discovery and Molecular Docking in Drug Design. Curr. Pharm. Biotechnol. 2015, 16, 11–25. [Google Scholar] [CrossRef]
- Kim, Y.M.; Gayen, S.; Kang, C.; Joy, J.; Huang, Q.; Chen, A.S.; Wee, J.L.; Ang, M.J.; Lim, H.A.; Hung, A.W.; et al. NMR analysis of a novel enzymatically active unlinked dengue NS2B-NS3 protease complex. J. Biol. Chem. 2013, 288, 12891–12900. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Z.; Phoo, W.W.; Loh, Y.R.; Wang, W.; Liu, S.; Chen, M.W.; Hung, A.W.; Keller, T.H.; Luo, D.; et al. Structural Dynamics of Zika Virus NS2B-NS3 Protease Binding to Dipeptide Inhibitors. Structure 2017, 25, 1242–1250. [Google Scholar] [CrossRef]
- Erlanson, D.A.; Davis, B.J.; Jahnke, W. Fragment-Based Drug Discovery: Advancing Fragments in the Absence of Crystal Structures. Cell Chem. Biol. 2019, 26, 9–15. [Google Scholar] [CrossRef]
- de la Cruz, L.; Nguyen, T.H.; Ozawa, K.; Shin, J.; Graham, B.; Huber, T.; Otting, G. Binding of low molecular weight inhibitors promotes large conformational changes in the dengue virus NS2B-NS3 protease: Fold analysis by pseudocontact shifts. J. Am. Chem. Soc. 2011, 133, 19205–19215. [Google Scholar] [CrossRef] [PubMed]
- Quek, J.P.; Liu, S.; Zhang, Z.; Li, Y.; Ng, E.Y.; Loh, Y.R.; Hung, A.W.; Luo, D.; Kang, C. Identification and structural characterization of small molecule fragments targeting Zika virus NS2B-NS3 protease. Antivir. Res. 2020, 175, 104707. [Google Scholar] [CrossRef] [PubMed]
- Yadav, D.K.; Zigáčková, D.; Zlobina, M.; Klumpler, T.; Beaumont, C.; Kubíčková, M.; Vaňáčová, Š.; Lukavsky, P.J. Staufen1 reads out structure and sequence features in ARF1 dsRNA for target recognition. Nucleic Acids Res. 2019, 48, 2091–2106. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Frey, G.; Fu, Q.; Lavine, C.L.; Scott, D.A.; Seaman, M.S.; Chou, J.J.; Chen, B. HIV-1 fusion inhibitors targeting the membrane-proximal external region of Env spikes. Nat. Chem. Biol. 2020, 16, 529–537. [Google Scholar] [CrossRef]
- Orts, J.; Gossert, A.D. Structure determination of protein-ligand complexes by NMR in solution. Methods 2018, 138, 3–25. [Google Scholar] [CrossRef]
- Schmitz, C.; Vernon, R.; Otting, G.; Baker, D.; Huber, T. Protein structure determination from pseudocontact shifts using ROSETTA. J. Mol. Biol. 2012, 416, 668–677. [Google Scholar] [CrossRef]
- de Vries, S.J.; van Dijk, M.; Bonvin, A.M. The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 2010, 5, 883–897. [Google Scholar] [CrossRef]
- Chen, W.-N.; Otting, G. Using tert-Butyl Groups in a Ligand To Identify Its Binding Site on a Protein. Acs Med. Chem. Lett. 2018, 9, 109–113. [Google Scholar] [CrossRef]
- Vlach, J.; Eastep, G.N.; Ghanam, R.H.; Watanabe, S.M.; Carter, C.A.; Saad, J.S. Structural basis for targeting avian sarcoma virus Gag polyprotein to the plasma membrane for virus assembly. J. Biol. Chem. 2018, 293, 18828–18840. [Google Scholar] [CrossRef]
- Durham, T.B.; Blanco, M.-J. Target Engagement in Lead Generation. Bioorganic Med. Chem. Lett. 2015, 25, 998–1008. [Google Scholar] [CrossRef]
- Simon, G.M.; Niphakis, M.J.; Cravatt, B.F. Determining target engagement in living systems. Nat. Chem. Biol. 2013, 9, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Schürmann, M.; Janning, P.; Ziegler, S.; Waldmann, H. Small-Molecule Target Engagement in Cells. Cell Chem. Biol. 2016, 23, 435–441. [Google Scholar] [CrossRef]
- Martinez Molina, D.; Jafari, R.; Ignatushchenko, M.; Seki, T.; Larsson, E.A.; Dan, C.; Sreekumar, L.; Cao, Y.; Nordlund, P. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 2013, 341, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Jafari, R.; Almqvist, H.; Axelsson, H.; Ignatushchenko, M.; Lundback, T.; Nordlund, P.; Molina, D.M. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc. 2014, 9, 2100–2122. [Google Scholar] [CrossRef]
- Dubach, J.M.; Kim, E.; Yang, K.; Cuccarese, M.; Giedt, R.J.; Meimetis, L.G.; Vinegoni, C.; Weissleder, R. Quantitating drug-target engagement in single cells in vitro and in vivo. Nat. Chem. Biol. 2017, 13, 168–173. [Google Scholar] [CrossRef]
- Kang, C. Applications of In-Cell NMR in Structural Biology and Drug Discovery. Int. J. Mol. Sci. 2019, 20, 139. [Google Scholar] [CrossRef] [PubMed]
- Reckel, S.; Lohr, F.; Dotsch, V. In-cell NMR spectroscopy. Chembiochem 2005, 6, 1601–1606. [Google Scholar] [CrossRef]
- Luchinat, E.; Banci, L. In-cell NMR: A topical review. IUCrJ 2017, 4, 108–118. [Google Scholar] [CrossRef]
- Veronesi, M.; Giacomina, F.; Romeo, E.; Castellani, B.; Ottonello, G.; Lambruschini, C.; Garau, G.; Scarpelli, R.; Bandiera, T.; Piomelli, D.; et al. Fluorine nuclear magnetic resonance-based assay in living mammalian cells. Anal. Biochem. 2016, 495, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Serber, Z.; Corsini, L.; Durst, F.; Dotsch, V. In-cell NMR spectroscopy. Methods Enzym. 2005, 394, 17–41. [Google Scholar]
- Selenko, P.; Wagner, G. Looking into live cells with in-cell NMR spectroscopy. J. Struct. Biol. 2007, 158, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Selenko, P.; Wagner, G. NMR mapping of protein interactions in living cells. Nat. Methods 2006, 3, 80–81. [Google Scholar] [CrossRef] [PubMed]
- Freedberg, D.I.; Selenko, P. Live cell NMR. Annu. Rev. Biophys. 2014, 43, 171–192. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; McLeod, S.; MacCormack, K.; Sriram, S.; Gao, N.; Breeze, A.L.; Hu, J. Real-Time Monitoring of New Delhi Metallo-β-Lactamase Activity in Living Bacterial Cells by 1H NMR Spectroscopy. Angew. Chem. Int. Ed. 2014, 53, 2130–2133. [Google Scholar] [CrossRef] [PubMed]
- Majumder, S.; Xue, J.; DeMott, C.M.; Reverdatto, S.; Burz, D.S.; Shekhtman, A. Probing Protein Quinary Interactions by In-Cell Nuclear Magnetic Resonance Spectroscopy. Biochemistry 2015, 54, 2727–2738. [Google Scholar] [CrossRef] [PubMed]
- Borcherds, W.; Theillet, F.X.; Katzer, A.; Finzel, A.; Mishall, K.M.; Powell, A.T.; Wu, H.; Manieri, W.; Dieterich, C.; Selenko, P.; et al. Disorder and residual helicity alter p53-Mdm2 binding affinity and signaling in cells. Nat. Chem. Biol. 2014, 10, 1000–1002. [Google Scholar] [CrossRef]
- Bouvier, G.; Simenel, C.; Jang, J.; Kalia, N.P.; Choi, I.; Nilges, M.; Pethe, K.; Izadi-Pruneyre, N. Target engagement and binding mode of an anti-tuberculosis drug to its bacterial target deciphered in whole living cells by NMR. Biochemistry 2018, 58, 526–533. [Google Scholar] [CrossRef]
- Viennet, T.; Viegas, A.; Kuepper, A.; Arens, S.; Gelev, V.; Petrov, O.; Grossmann, T.N.; Heise, H.; Etzkorn, M. Selective Protein Hyperpolarization in Cell Lysates Using Targeted Dynamic Nuclear Polarization. Angew. Chem. Int. Ed. 2016, 55, 10746–10750. [Google Scholar] [CrossRef]
- Kumar, A.; Kuhn, L.T.; Balbach, J. In-Cell NMR: Analysis of Protein-Small Molecule Interactions, Metabolic Processes, and Protein Phosphorylation. Int. J. Mol. Sci. 2019, 20, 378. [Google Scholar] [CrossRef]
- Schreiber, S.L. A Chemical Biology View of Bioactive Small Molecules and a Binder-Based Approach to Connect Biology to Precision Medicines. Isr. J. Chem. 2019, 59, 52–59. [Google Scholar] [CrossRef]
- Kang, C.; Keller, T.H. Probing biological mechanisms with chemical tools. Pharm. Res. 2020, 153, 104656. [Google Scholar] [CrossRef] [PubMed]
- Cobb, S.L.; Murphy, C.D. 19F NMR applications in chemical biology. J. Fluor. Chem. 2009, 130, 132–143. [Google Scholar] [CrossRef]
- Horst, R.; Liu, J.J.; Stevens, R.C.; Wuthrich, K. beta(2)-adrenergic receptor activation by agonists studied with (1)(9)F NMR spectroscopy. Angew Chem. Int. Ed. Engl. 2013, 52, 10762–10765. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Van Eps, N.; Zimmer, M.; Ernst, O.P.; Scott Prosser, R. Activation of the A2A adenosine G-protein-coupled receptor by conformational selection. Nature 2016, 533, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Horst, R.; Katritch, V.; Stevens, R.C.; Wuthrich, K. Biased signaling pathways in beta2-adrenergic receptor characterized by 19F-NMR. Science 2012, 335, 1106–1110. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Yang, J.; Li, H.; Sun, H.; Liu, J.; Wang, J. Conformational change study of dengue virus NS2B-NS3 protease using 19F NMR spectroscopy. Biochem. Biophys. Res. Commun. 2015, 461, 677–680. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiments | Applications | References |
|---|---|---|
| 1H-NMR | Screening, protein-ligand interactions | [68,69] |
| 1H-NMR, TINS 1 | Screening | [70] |
| 1H-NMR, qNMR | Protein-ligand interactions | [68] |
| 2D HSQC 1 | Screening, hit confirmation, map ligand binding sites | [71,72] |
| NOESY 1 | Hit confirmation, structure determination | [73] |
| Filtered-NOESY | Hit confirmation, structure determination | [74,75] |
| STD-NMR 1 | Screening, hit confirmation | [76] |
| WaterLOGSY | Screening, hit confirmation | [77,78] |
| Transferred-NOE | Hit confirmation, structure determination | [79,80] |
| ILOEs 1 | Characterizing ligand bindings. | [81] |
| DOSY 1 | Hit confirmation | [82] |
| 19F-NMR 1 | Screening, hit confirmation | [56,83,84,85] |
| FAXS | Screening, hit confirmation | [62,83] |
| T2 CMPG | Screening, hit confirmation | [86] |
| T1 relaxation | Screening, hit confirmation | [86,87] |
| PRE 1 | Hit confirmation, structure determination | [88,89] |
| SLAPSTIC 1 | Screening | [90] |
| RDC 1 | Structure determination | [91] |
| H/D exchange | Binding characterization | [92] |
| Cross-saturation | Protein–protein interactions | [93] |
| In-cell NMR 1 | Protein structure and ligand binding in living cells. | [94] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Q.; Kang, C. A Practical Perspective on the Roles of Solution NMR Spectroscopy in Drug Discovery. Molecules 2020, 25, 2974. https://doi.org/10.3390/molecules25132974
Li Q, Kang C. A Practical Perspective on the Roles of Solution NMR Spectroscopy in Drug Discovery. Molecules. 2020; 25(13):2974. https://doi.org/10.3390/molecules25132974
Chicago/Turabian StyleLi, Qingxin, and CongBao Kang. 2020. "A Practical Perspective on the Roles of Solution NMR Spectroscopy in Drug Discovery" Molecules 25, no. 13: 2974. https://doi.org/10.3390/molecules25132974
APA StyleLi, Q., & Kang, C. (2020). A Practical Perspective on the Roles of Solution NMR Spectroscopy in Drug Discovery. Molecules, 25(13), 2974. https://doi.org/10.3390/molecules25132974