Development of an HPLC-MS/MS Method for the Determination of Silybin in Human Plasma, Urine and Breast Tissue

 ,
,  , , , , , and
, , , , , and
Abstract

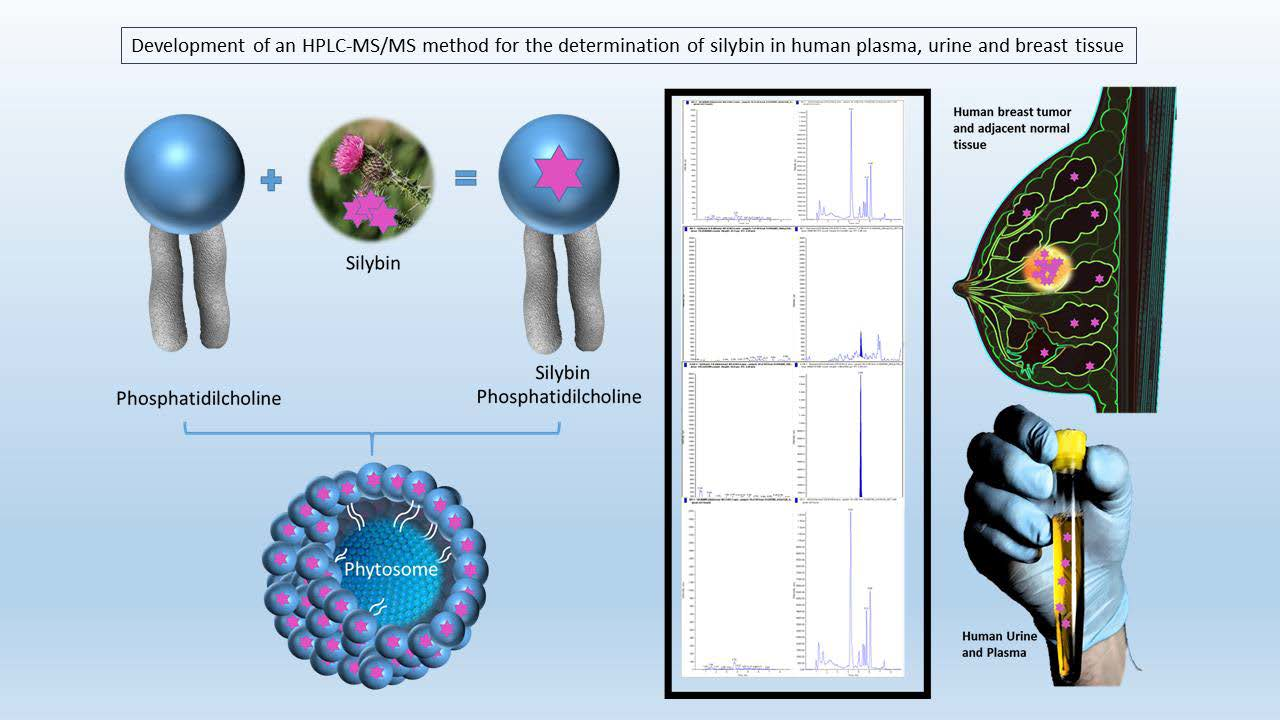
1. Introduction
2. Results and Discussion
2.1. Analytical Method Development
2.2. Method Validation
2.2.1. Selectivity
2.2.2. Calibration Curve
2.2.3. Limit of Quantification
2.2.4. Intra-Assay Precision and Accuracy
2.2.5. Carryover
3. Material and Methods
3.1. Description of Analytical Method
3.1.1. Working Solutions and Internal Standards
3.1.2. Calibration Standards, Quality Control Samples, Blank and Zero Samples
3.1.3. Sample Preparation
3.1.4. Chromatographic Method
HPLC Method
Mass Spectrometer
3.2. Method Validation
3.2.1. Selectivity
- Area response of the potential peak eluting with the same retention time, molecular weight and fragmentation transition as silybin <20% of the corresponding response of this compound in the lower limit of quantification (LLOQ).
- Area response of the potential peaks eluting with the same retention time, molecular weight and fragmentation transition as internal standard <5% of the corresponding response of this compound at the concentration used in the study.
3.2.2. Calibration Curve
- Correlation coefficient ≥ 0.99.
- Accuracy of the back-calculated concentrations ≤ 15% for all concentration levels
- Accuracy of the LLOQ ≤ 20%.
3.2.3. Limit of Quantification
- Intra-assay precision: coefficient of variation of the LLOQ concentrations in the five replicates ≤ 20%
- Intra-assay accuracy: mean percentage deviation of the concentrations of the five replicates within ± 20% of the nominal value.
3.2.4. Determination of Precision and Accuracy
- Intra-assay precision: coefficient of variation of the analyte concentrations in the five replicates ≤ 15%
- Intra-assay accuracy: mean percentage deviation of the concentrations of the five replicates within ± 15% of the nominal value.
3.2.5. Carryover
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Surh, Y. Cancer chemoprevention with dietary phytochemicals. Nat. Rev. Cancer 2003, 3, 768. [Google Scholar] [CrossRef]
- Kaur, M.; Agarwal, R. Silymarin and epithelial cancer chemoprevention: How close we are to bedside? Toxicol. Appl. Pharmacol. 2007, 224, 350–359. [Google Scholar] [CrossRef]
- Ferenci, P.; Dragosics, B.; Dittrich, H.; Frank, H.; Benda, L.; Lochs, H.; Meryn, S.; Base, W.; Schneider, B. Randomized controlled trial of silymarin treatment in patients with cirrhosis of the liver. J. Hepatol. 1989, 9, 105–113. [Google Scholar] [CrossRef]
- Feher, J.; Lengyel, G. Silymarin in the prevention and treatment of liver diseases and primary liver cancer. Curr. Pharm. Biotechnol. 2012, 13, 210–217. [Google Scholar] [CrossRef]
- Comelli, M.C.; Mengs, U.; Schneider, C.; Prosdocimi, M. Toward the definition of the mechanism of action of silymarin: Activities related to cellular protection from toxic damage induced by chemotherapy. Integr. Cancer Ther. 2007, 6, 120–129. [Google Scholar] [CrossRef]
- Tyagi, A.K.; Agarwal, C.; Chan, D.C.; Agarwal, R. Synergistic anti-cancer effects of silibinin with conventional cytotoxic agents doxorubicin, cisplatin and carboplatin against human breast carcinoma MCF-7 and MDA-MB468 cells. Oncol. Rep. 2004, 11, 493–499. [Google Scholar] [CrossRef]
- Zheng, D.; Wang, Y.; Zhang, D.; Liu, Z.; Duan, C.; Jia, L.; Wang, F.; Liu, Y.; Liu, G.; Hao, L. In Vitro antitumor activity of silybin nanosuspension in PC-3 cells. Cancer Lett. 2011, 307, 158–164. [Google Scholar] [CrossRef]
- Kaur, M.; Velmurugan, B.; Tyagi, A.; Deep, G.; Katiyar, S.; Agarwal, C.; Agarwal, R. Silibinin suppresses growth and induces apoptotic death of human colorectal carcinoma LoVo cells in culture and tumor xenograft. Mol. Cancer. Ther. 2009, 8, 2366–2374. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, K.; Agarwal, R. Multitargeted therapy of cancer by silymarin. Cancer Lett. 2008, 269, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.P.; Agarwal, R. Prostate cancer prevention by silibinin. Curr. Cancer Drug Targets 2004, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.P.; Agarwal, R. Mechanisms and preclinical efficacy of silibinin in preventing skin cancer. Eur. J. Cancer 2005, 41, 1969–1979. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.P.; Agarwal, R. Prostate cancer chemoprevention by silibinin: Bench to bedside. Mol. Carcino. 2006, 45, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Forghani, P.; Khorramizadeh, M.R.; Waller, E.K. Silibinin inhibits accumulation of myeloid—derived suppressor cells and tumor growth of murine breast cancer. Cancer Med. 2014, 3, 215–224. [Google Scholar] [CrossRef]
- National Toxicology Program. Toxicology and carcinogenesis studies of milk thistle extract (CAS No. 84604-20-6) in F344/N rats and B6C3F1 mice (Feed Studies). Natl. Toxicol. Program Tech. Rep. Ser. 2011, 1, 177. [Google Scholar]
- Provinciali, M.; Papalini, F.; Orlando, F.; Pierpaoli, S.; Donnini, A.; Morazzoni, P.; Riva, A.; Smorlesi, A. effect of the silybin-phosphatidylcholine complex (IdB 1016) on the development of mammary tumors in HER-2/Neu transgenic mice. Cancer Res. 2007, 67, 2022–2029. [Google Scholar] [CrossRef] [PubMed]
- Barzaghi, N.; Crema, F.; Gatti, G.; Pifferi, G.; Perucca, E. Pharmacokinetic studies on IdB 1016, a silybin-phosphatidylcholine complex, in healthy human subjects. Eur. J. Drug Metab. Pharmacokinet. 1990, 15, 333–338. [Google Scholar] [CrossRef]
- Lazzeroni, M.; Guerrieri-Gonzaga, A.; Gandini, S.; Johansson, H.; Serrano, D.; Cazzaniga, M.; Aristarco, V.; Puccio, A.; Mora, S.; Caldarella, P.; et al. A presurgical study of oral silybin-phosphatidylcholine in patients with early breast cancer. Cancer. Prev. Res. 2016, 9, 89–95. [Google Scholar] [CrossRef]
- Gunaratna, C.; Zhang, T. Application of liquid chromatography–electrospray Ionization-Ion trap mass spectrometry to investigate the metabolism of silibinin in human liver microsomes. J. Chromatogr. B 2003, 794, 303–310. [Google Scholar] [CrossRef]
- Venisetty, R.; Keshetty, S.; Ciddi, V. Biotransformation of silibinin (silybin) using fungal organisms. Indian J. Pharm. Educ. Res. 2011, 45, 384–391. [Google Scholar]
- Li, W.; Han, J.; Li, Z.; Li, X.; Zhou, S.; Liu, C. Preparative chromatographic purification of diastereomers of silybin and their quantification in human plasma by liquid chromatography–tandem mass spectrometry. J. Chromatogr. B 2008, 862, 51–57. [Google Scholar] [CrossRef]
- Hoh, C.S.; Boocock, D.J.; Marczylo, T.H.; Brown, V.; Cai, H.; Steward, W.P.; Berry, D.P.; Gescher, A.J. Quantitation of silibinin, a putative cancer chemopreventive agent derived from milk thistle (silybum marianum), in human plasma by high-performance liquid chromatography and identification of possible metabolites. J. Agric. Food Chem. 2007, 55, 2532–2535. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Lin, L.; Hung, S.; Chi, C.; Tsai, T. Analysis of silibinin in rat plasma and bile for hepatobiliary excretion and oral bioavailability application. J. Pharm. Biomed. Anal. 2007, 45, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Dumas, T.E.; Schrieber, S.J.; Hawke, R.L.; Fried, M.W.; Smith, P.C. Pharmacokinetics and metabolic profile of free, conjugated, and total silymarin flavonolignans in human plasma after oral administration of milk thistle extract. Drug Metab. Dispos. 2008, 36, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Guideline on Bioanalytical Method Validation; European Medicines Agency: London, UK, 2011; Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf (accessed on 23 June 2020).
- Bioanalytical Method Validation. Guidance for Industry; U.S. Food and Drug Administration: Rockville, MD, USA, 2018. Available online: https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf (accessed on 23 June 2020).
Sample Availability: Samples of the compounds are not available from the authors. |


{kind=link}
{kind=link}
| Calibration Sample | Accuracy (RE, %) | ||
|---|---|---|---|
| Human Plasma | Human Urine | Pig Muscle | |
| PL1 | −1.87 | −11.54 | −1.18 |
| PL2 | −0.83 | −5.25 | −2.12 |
| PL3 | 1.30 | −0.18 | 3.08 |
| PL4 | −0.18 | 1.84 | 1.80 |
| PL5 | 1.03 | 4.57 | 6.26 |
| PL6 | 2.29 | 3.32 | −3.42 |
| PL7 | −1.51 | 4.74 | −2.69 |
| PL8 | 0.27 | −6.53 | 1.40 |
| Sample | Mean | RE (%) | C.V. (%) |
|---|---|---|---|
| Human Plasma | |||
| PQ | 0.59 | 18.40 | 13.61 |
| PE3 | 1.69 | 12.54 | 6.38 |
| PE2 | 16.80 | 12.03 | 1.66 |
| PE1 | 404.85 | 1.21 | 3.74 |
| Human urine | |||
| PQ | 1.196 | 19.64 | 5.14 |
| PE3 | 3.015 | 0.51 | 3.07 |
| PE2 | 32.533 | 8.44 | 1.29 |
| PE1 | 728.992 | −8.88 | 2.95 |
| Pig Muscle | |||
| PQ | 1.987 | −0.64 | 9.60 |
| PE3 | 6.583 | 9.71 | 7.89 |
| PE2 | 26.007 | 8.36 | 3.20 |
| PE1 | 394.834 | −1.29 | 3.76 |
| Calibration Standard | Human Plasma | Human Urine | Pig Muscle |
|---|---|---|---|
| Silybin Concentration (ng/mL) | Silybin Concentration (ng/mL) | Silybin Concentration (ng/g) | |
| PL1 | 500 | 1000 | 500 |
| PL2 | 250 | 500 | 250 |
| PL3 | 100 | 200 | 100 |
| PL4 | 50 | 100 | 50 |
| PL5 | 10 | 20 | 25 |
| PL6 | 2.5 | 5 | 10 |
| PL7 | 1 | 2 | 5 |
| PL8 | 0.5 | 1 | 2 |
| Quality Control Sample | Human Plasma | Human Urine | Pig Muscle |
|---|---|---|---|
| Silybin Concentration (ng/mL) | Silybin Concentration (ng/mL) | Silybin Concentration (ng/g) | |
| PQ | 0.5 | 1 | 2 |
| PE3 | 1.5 | 3 | 6 |
| PE2 | 15 | 30 | 24 |
| PE1 | 400 | 800 | 400 |
| Human Plasma | Human Urine | Pig Muscle | |||
|---|---|---|---|---|---|
| Time (Minutes) | Mobile Phase B (%) | Time (Minutes) | Mobile Phase B (%) | Time (Minutes) | Mobile Phase B (%) |
| 0.00 | 15 | 0.00 | 25 | 0.00 | 25 |
| 3.00 | 80 | 3 | 75 | 2.50 | 90 |
| 4.50 | 80 | 6 | 75 | 3.50 | 90 |
| 4.60 | 15 | 6.6 | 25 | 3.60 | 25 |
| 6.00 | 15 | 9 | 25 | 7.50 | 25 |
| Compound | Q1 Mass | Q3 Mass | Time (msec) | DP(v) | FP (v) | EP (v) | CE (eV) | CXP(v) |
|---|---|---|---|---|---|---|---|---|
| Human Plasma | ||||||||
| Silybin | 481.0 | 301.0 | 200 | −120 | - | −11 | −28 | −7 |
| Naringenin | 270.8 | 151.0 | 200 | −100 | - | −11 | −26 | −9 |
| Human Urine | ||||||||
| Silybin | 481.2 | 301.0 | 200 | −75 | - | −10 | −28 | −10 |
| Naproxen | 229.0 | 184.9 | 200 | −23 | - | −2 | −11 | −4 |
| Pig Muscle | ||||||||
| Silybin | 481.2 | 301.0 | 200 | −80 | −210 | −10 | −29 | −18 |
| Narigenin | 271.0 | 150.9 | 200 | −70 | −150 | −8 | −24 | −11 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lazzeroni, M.; Petrangolini, G.; Legarreta Iriberri, J.A.; Pascual Avellana, J.; Tost Robusté, D.; Cagnacci, S.; Macis, D.; Aristarco, V.; Bonanni, B.; Morazzoni, P.; et al. Development of an HPLC-MS/MS Method for the Determination of Silybin in Human Plasma, Urine and Breast Tissue. Molecules 2020, 25, 2918. https://doi.org/10.3390/molecules25122918
Lazzeroni M, Petrangolini G, Legarreta Iriberri JA, Pascual Avellana J, Tost Robusté D, Cagnacci S, Macis D, Aristarco V, Bonanni B, Morazzoni P, et al. Development of an HPLC-MS/MS Method for the Determination of Silybin in Human Plasma, Urine and Breast Tissue. Molecules. 2020; 25(12):2918. https://doi.org/10.3390/molecules25122918
Chicago/Turabian StyleLazzeroni, Matteo, Giovanna Petrangolini, José Antonio Legarreta Iriberri, Jaume Pascual Avellana, Digna Tost Robusté, Sara Cagnacci, Debora Macis, Valentina Aristarco, Bernardo Bonanni, Paolo Morazzoni, and et al. 2020. "Development of an HPLC-MS/MS Method for the Determination of Silybin in Human Plasma, Urine and Breast Tissue" Molecules 25, no. 12: 2918. https://doi.org/10.3390/molecules25122918
APA StyleLazzeroni, M., Petrangolini, G., Legarreta Iriberri, J. A., Pascual Avellana, J., Tost Robusté, D., Cagnacci, S., Macis, D., Aristarco, V., Bonanni, B., Morazzoni, P., Johansson, H., & Riva, A. (2020). Development of an HPLC-MS/MS Method for the Determination of Silybin in Human Plasma, Urine and Breast Tissue. Molecules, 25(12), 2918. https://doi.org/10.3390/molecules25122918