
Novel Convenient Approach to 6-, 7-, and 8-Numbered Nitrogen Heterocycles Incorporating Endocyclic Sulfonamide Fragment

Abstract
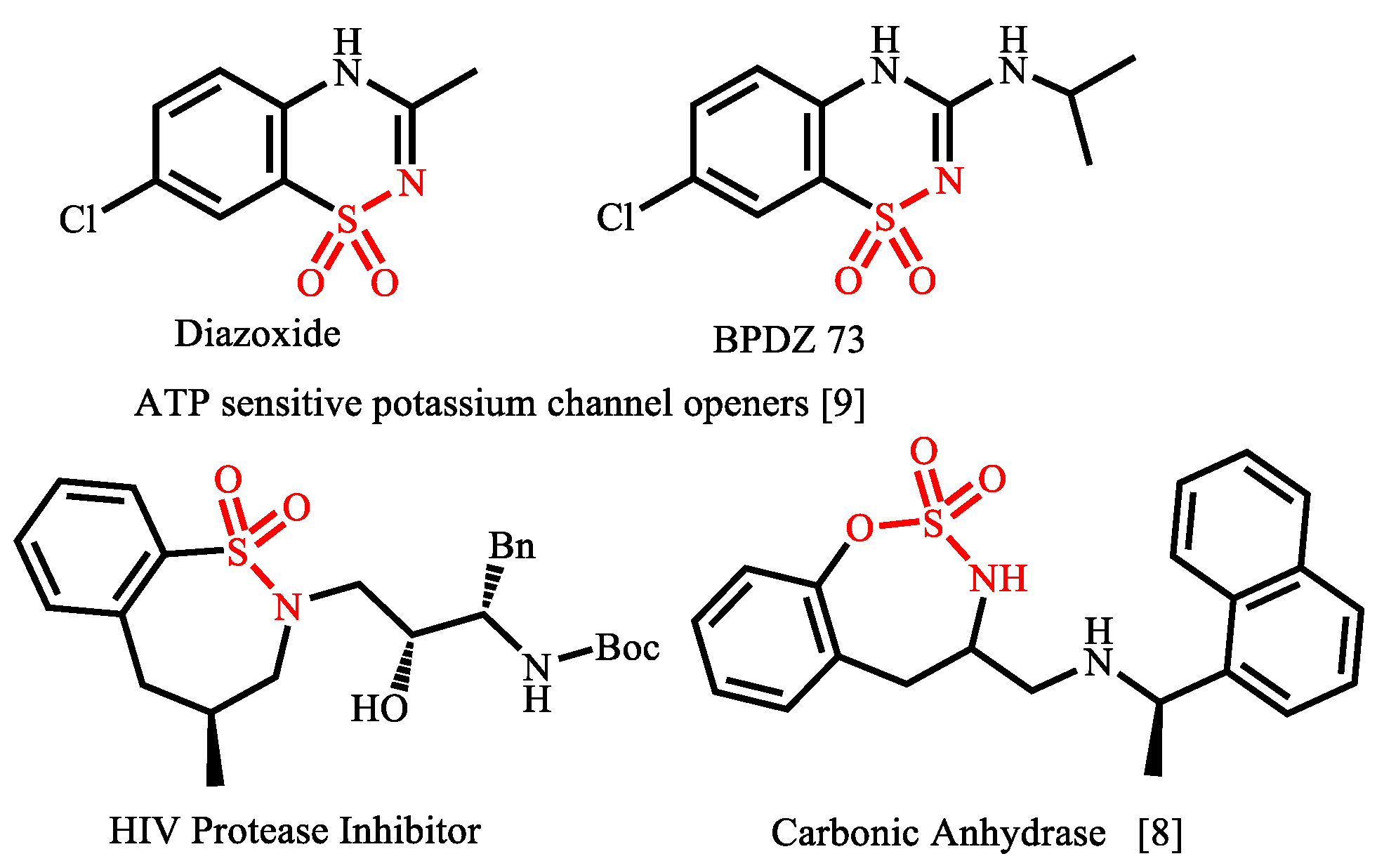
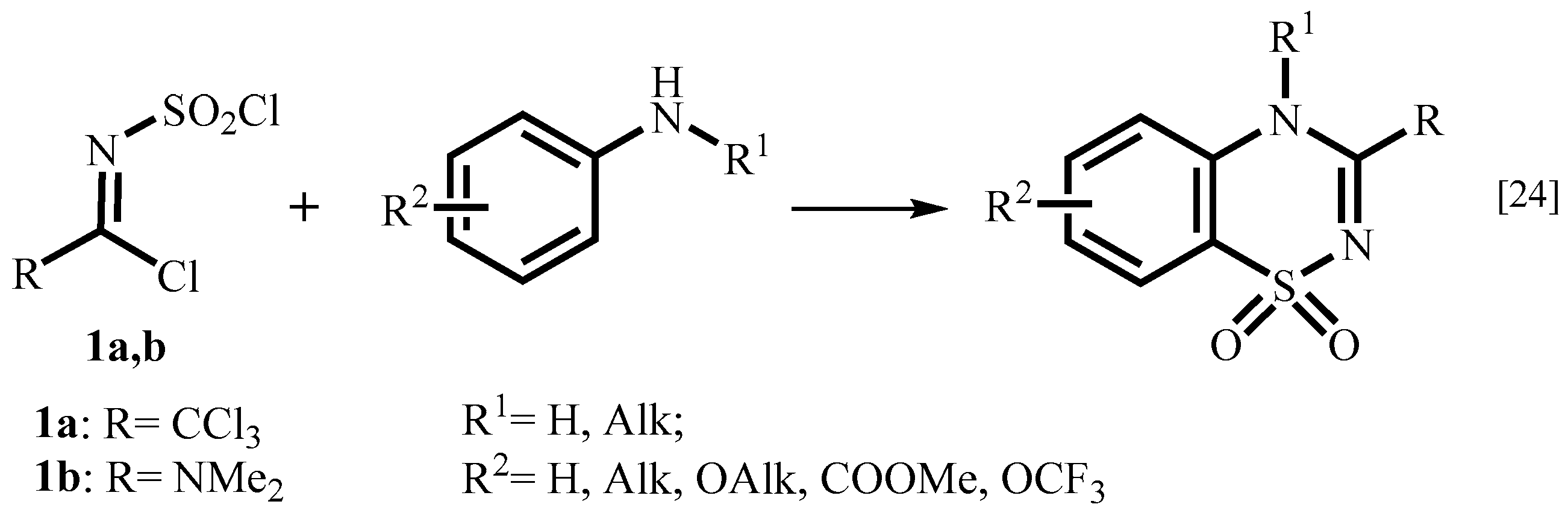
1. Introduction
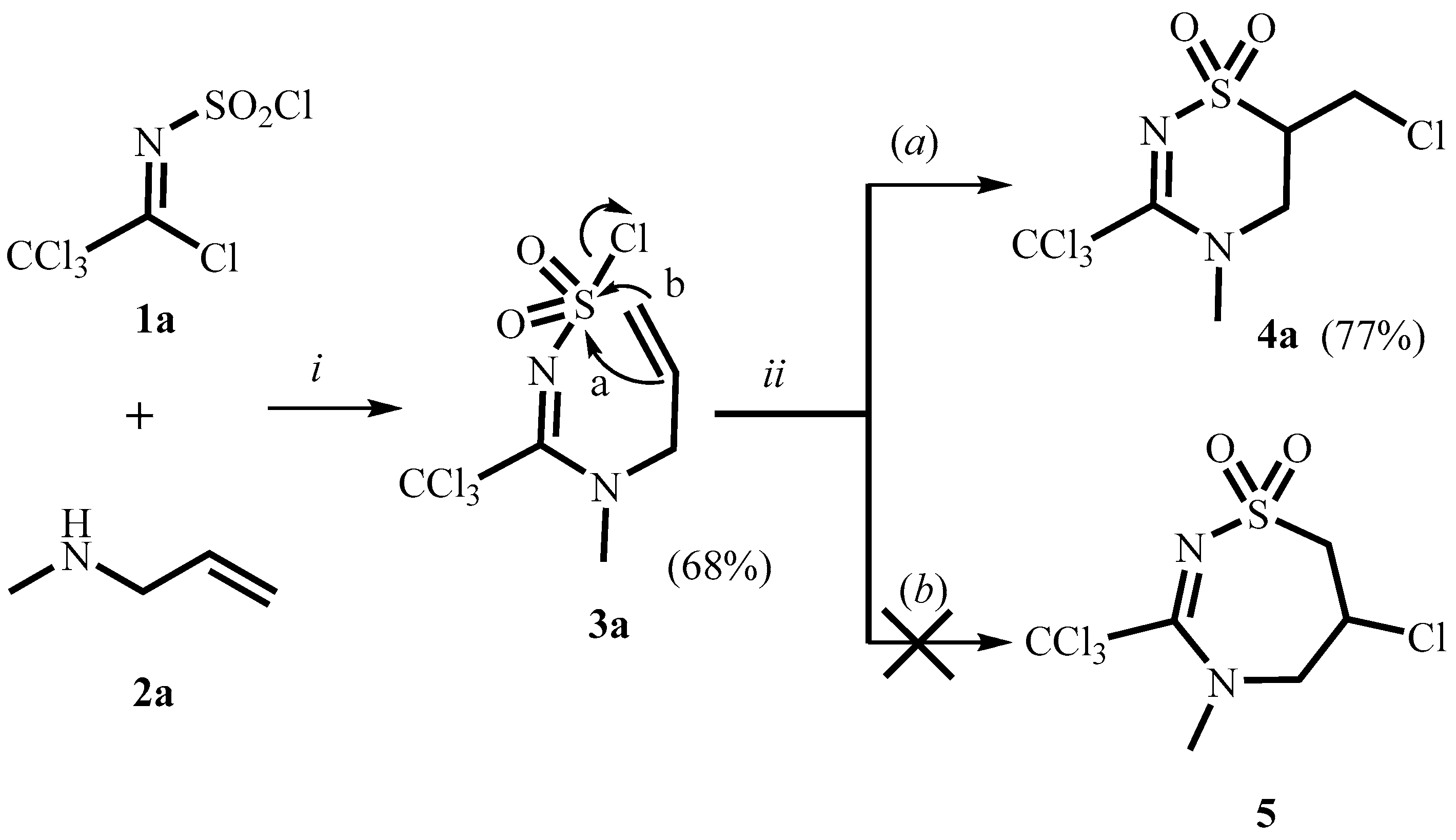
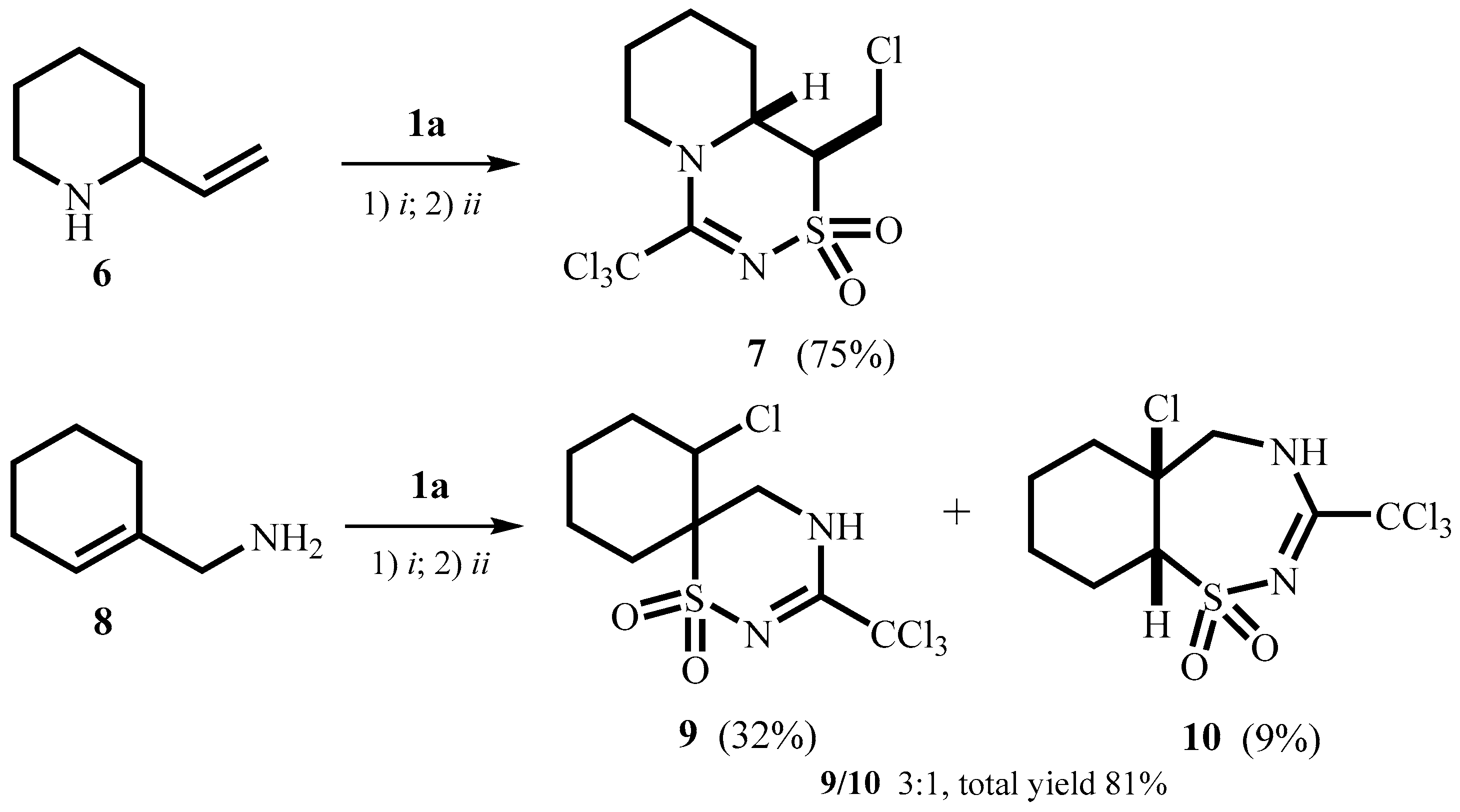
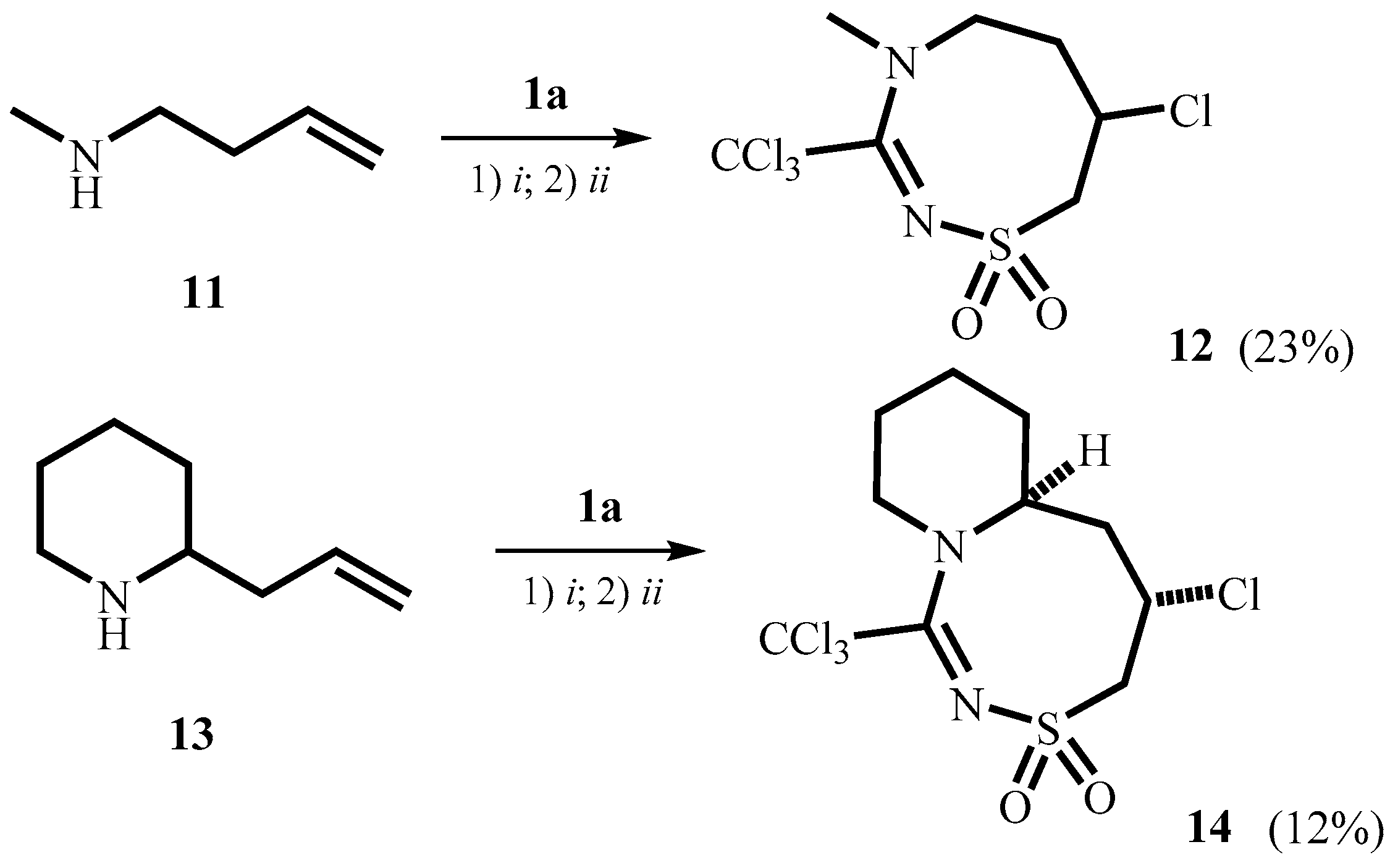
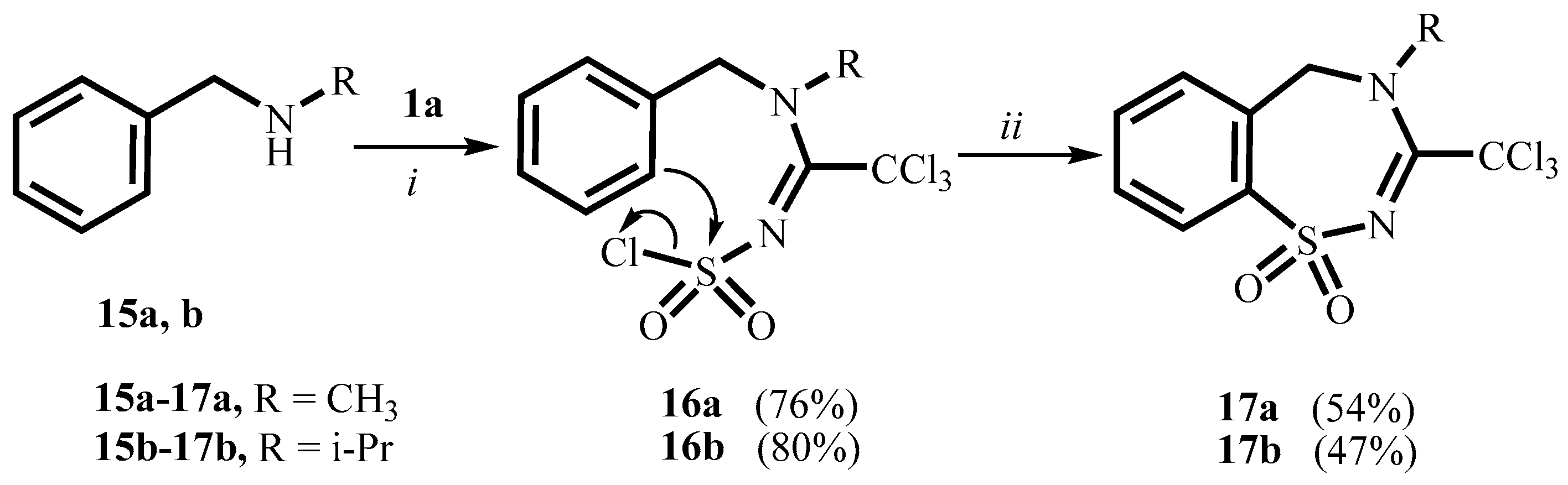
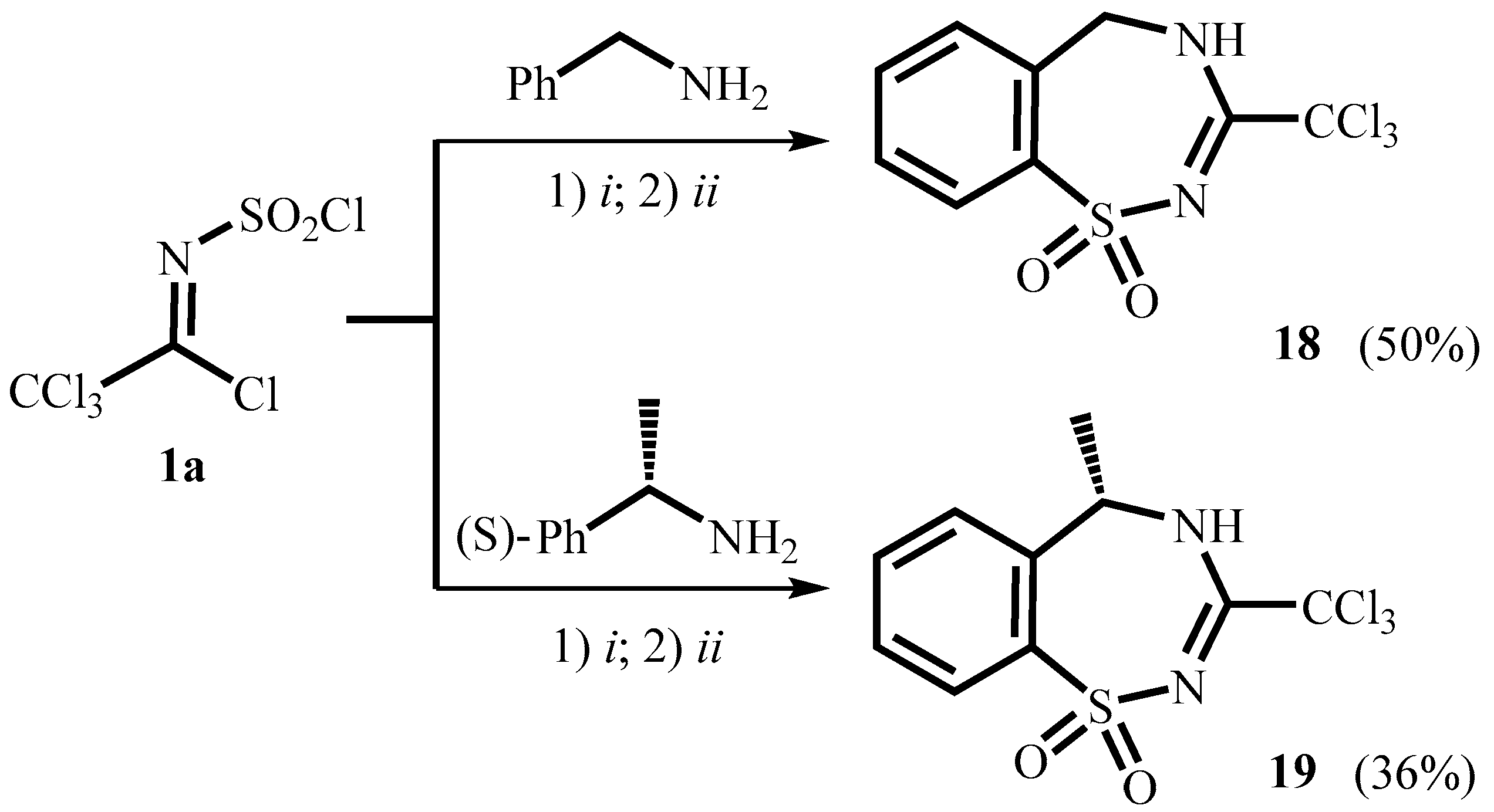
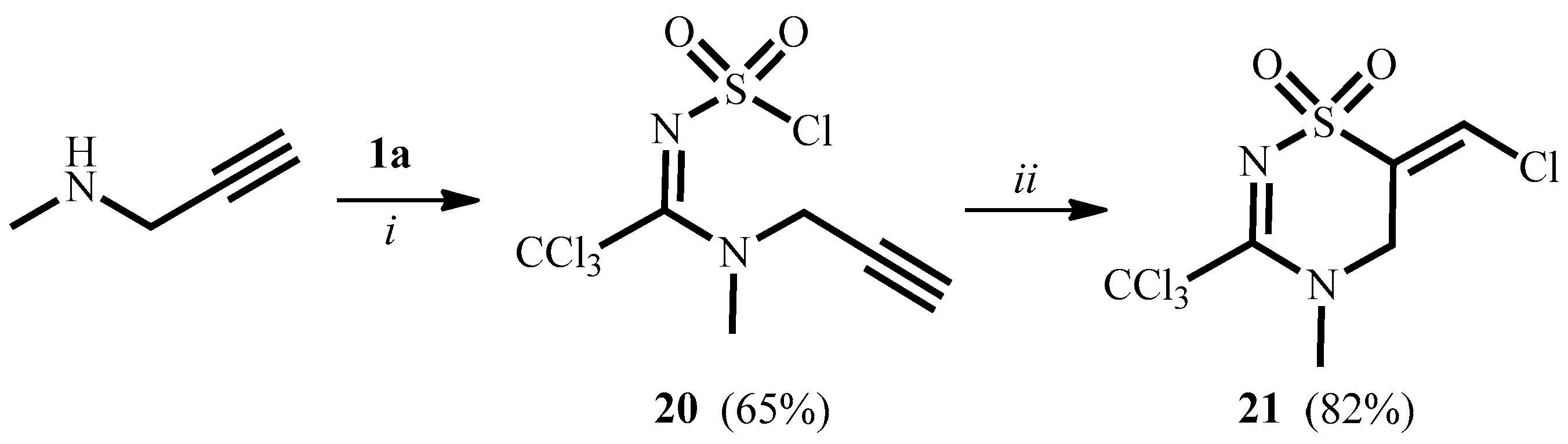
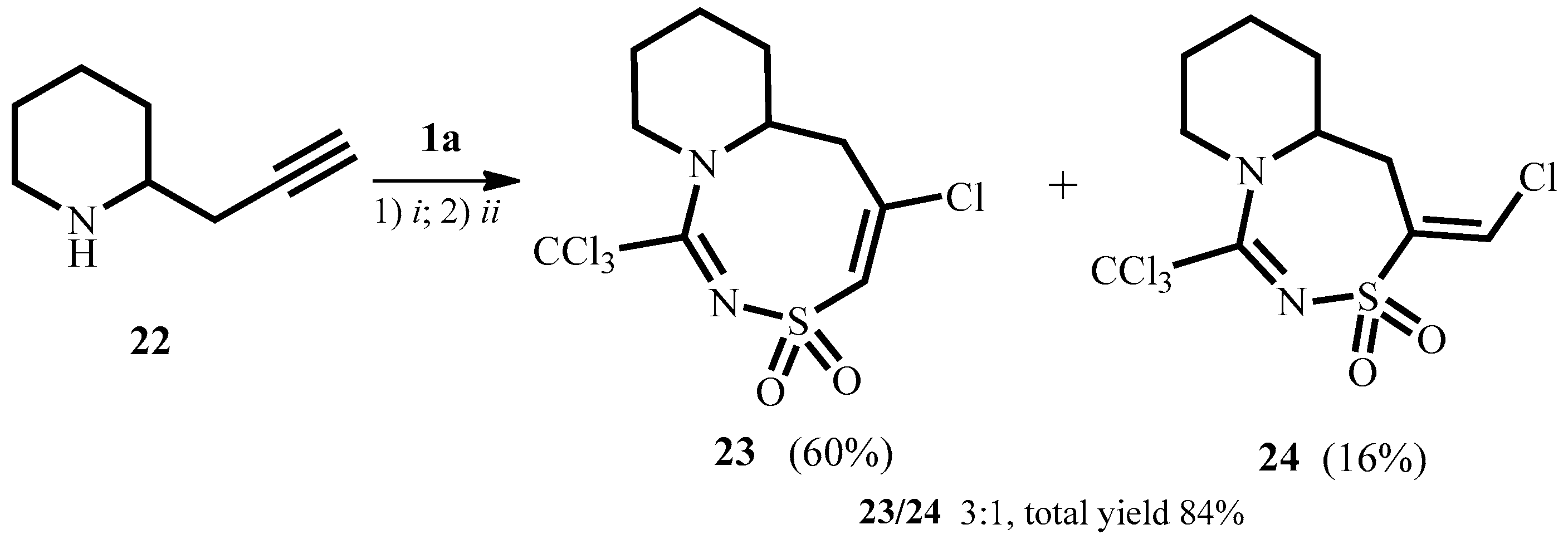
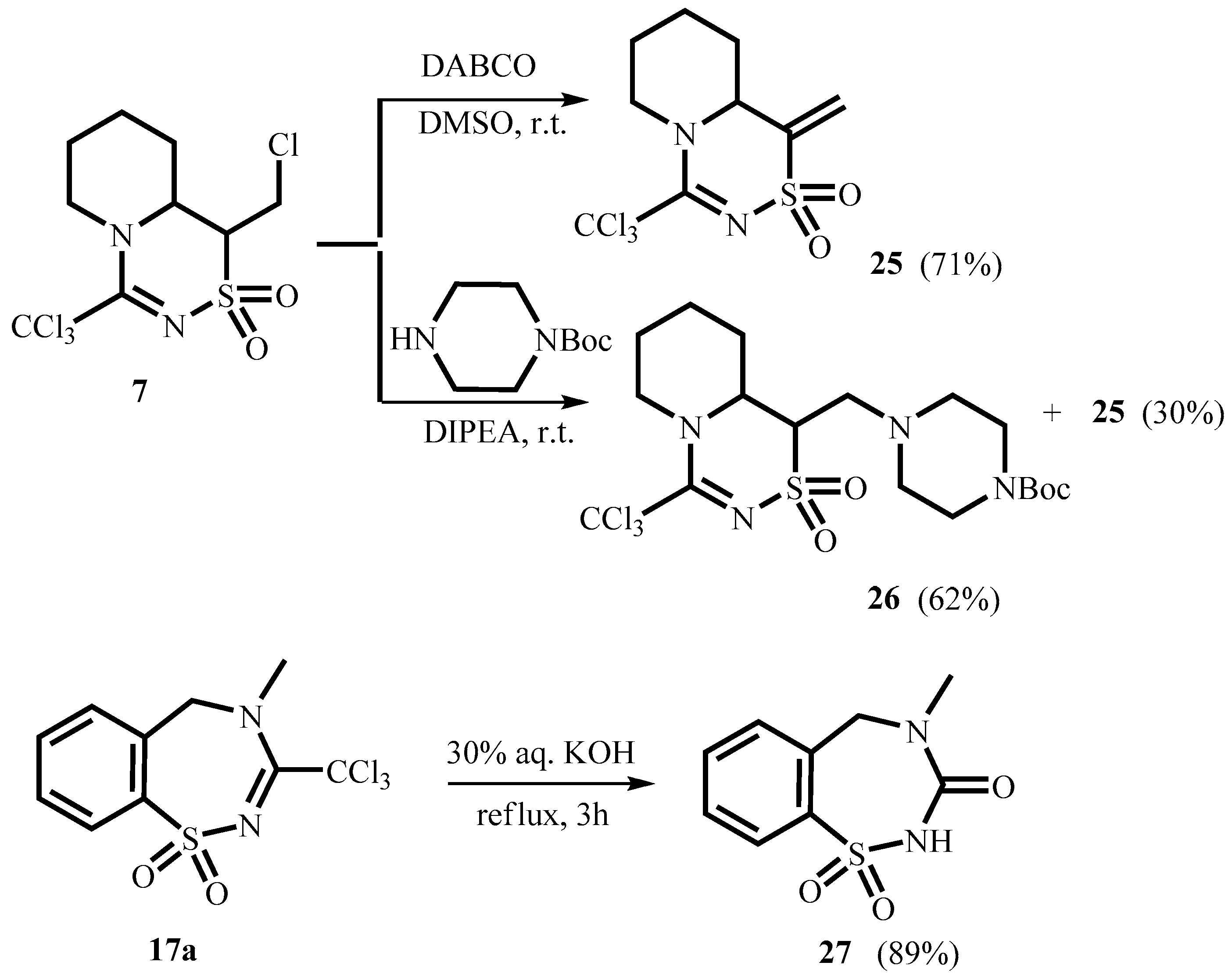
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. Typical Procedure for the Synthesis of Compounds 3a, 16a,b, 20
3.2.1. 1-(Allyl(methyl)amino)-2,2,2-trichloroethylidenesulfamoyl Chloride 3a
3.2.2. 1-(Benzyl(methyl)amino)-2,2,2-trichloroethylidenesulfamoyl Chloride 16a
3.2.3. 1-(Benzyl(isopropyl)amino)-2,2,2-trichloroethylidenesulfamoyl Chloride 16b
3.2.4. 1-(Propargyl(methyl)amino)-2,2,2-trichloroethylidenesulfamoyl Chloride 20
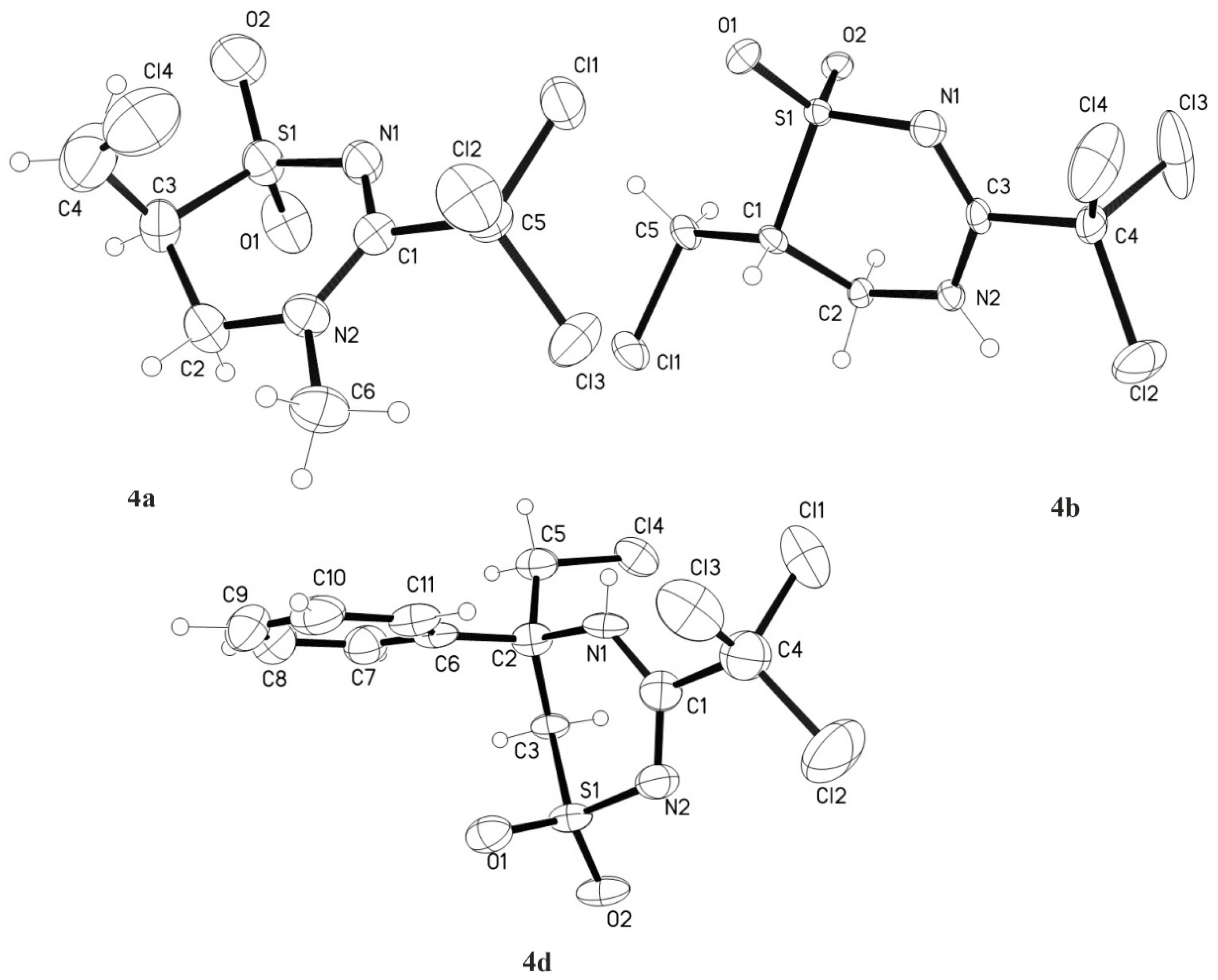
3.3. 6-Chloromethyl-4-methyl-3-trichloromethyl-5,6-dihydro-4H-1,2,4-thiadiazine-1,1-dioxide 4a
3.4. Typical Procedure for One-Pot Synthesis of Compounds 4a–e, 7, 12, 14
3.4.1. Compound 4a
3.4.2. 6-Chloromethyl-3-trichloromethyl-5,6-dihydro-4H-1,2,4-thiadiazine-1,1-dioxide 4b
3.4.3. 6-Chloromethyl-6-methyl-3-trichloromethyl-5,6-dihydro-4H-1,2,4-thiadiazine-1,1-dioxide 4c
3.4.4. 6-Chloromethyl-6-phenyl-3-trichloromethyl-5,6-dihydro-4H-1,2,4-thiadiazine-1,1-dioxide 4d
3.4.5. 6-Chloromethyl-5,5-dimethyl-3-trichloromethyl-5,6-dihydro-4H-1,2,4-thiadiazine-1,1-dioxide 4e
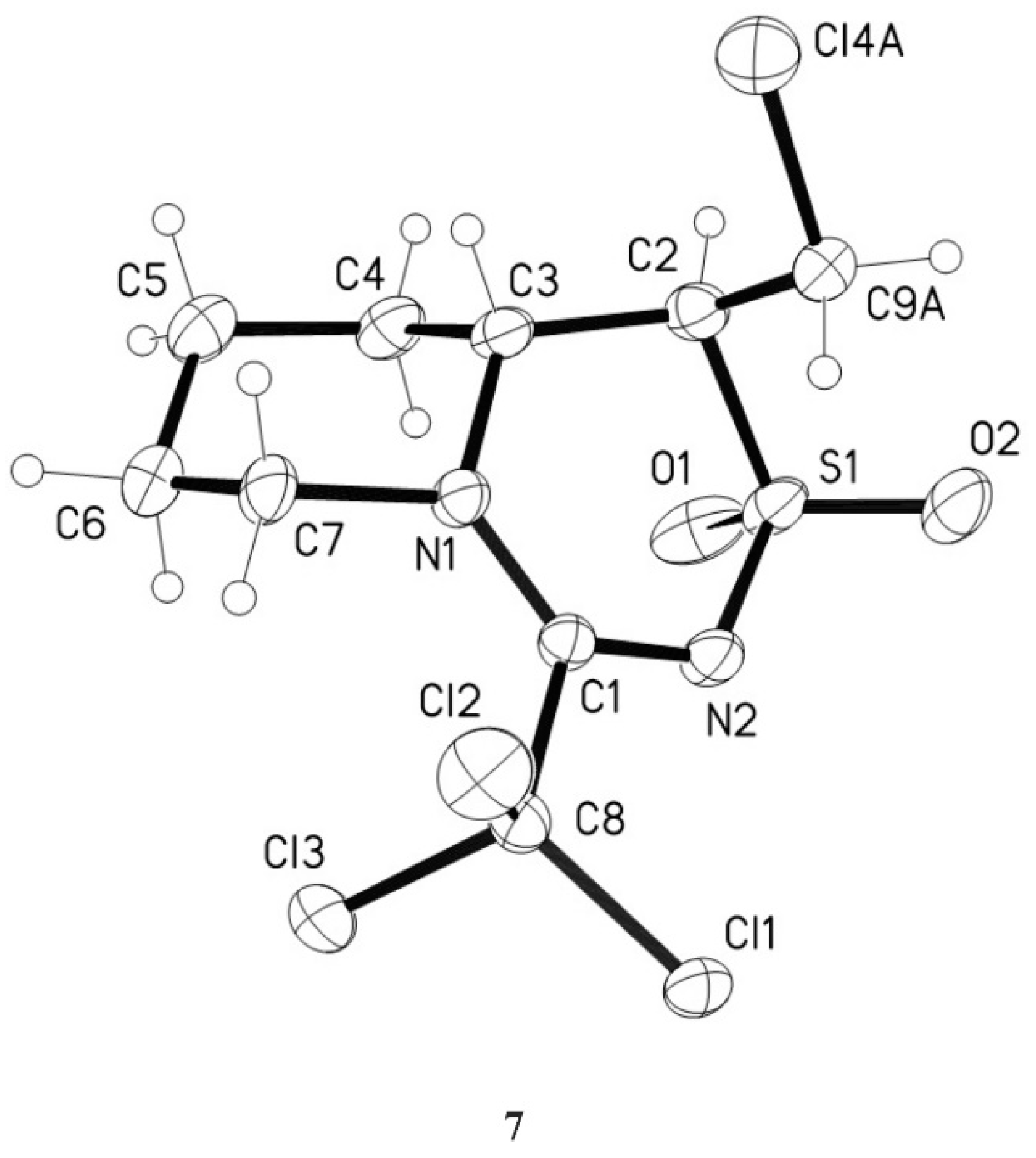
3.4.6. 1-Chloromethyl-4-trichloromethyl-1,6,7,8,9,9a-hexahydropyrido[1,2-d][1,2,4]thiadiazine-2,2-dioxide 7
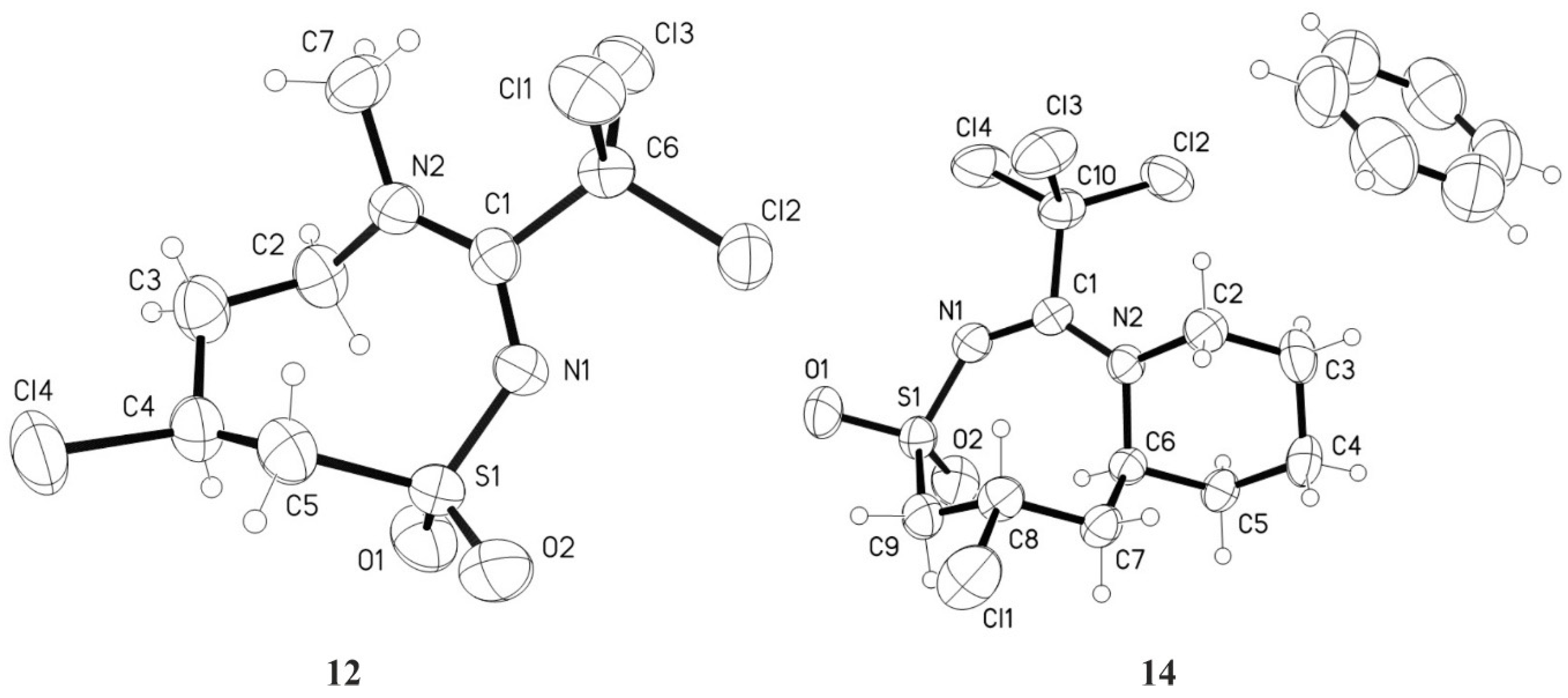
3.4.7. 7-Chloro-4-methyl-3-trichloromethyl-5,6,7,8-tetrahydro-4H-1,2,4-thiadiazocine-1,1-dioxide 12
3.4.8. 5-Chloro-1-trichloromethyl-4,5,6,6a,7,8,9,10-octahydropyrido[1,2-d][1,2,4]thiadiazocine-3,3-dioxide 14
3.5. 7-Chloro-3-trichloromethyl-1-thia-2,4-diazaspiro[5.5]undec-2-ene-1,1-dioxide 9 and 5a-Chloro-3-trichloromethyl-4,5,5a,6,7,8,9,9a-octahydro-1,2,4-benzothiadiazepine-1,1-dioxide 10
3.5.1. 7-Chloro-3-trichloromethyl-1-thia-2,4-diazaspiro[5.5]undec-2-ene-1,1-dioxide 9
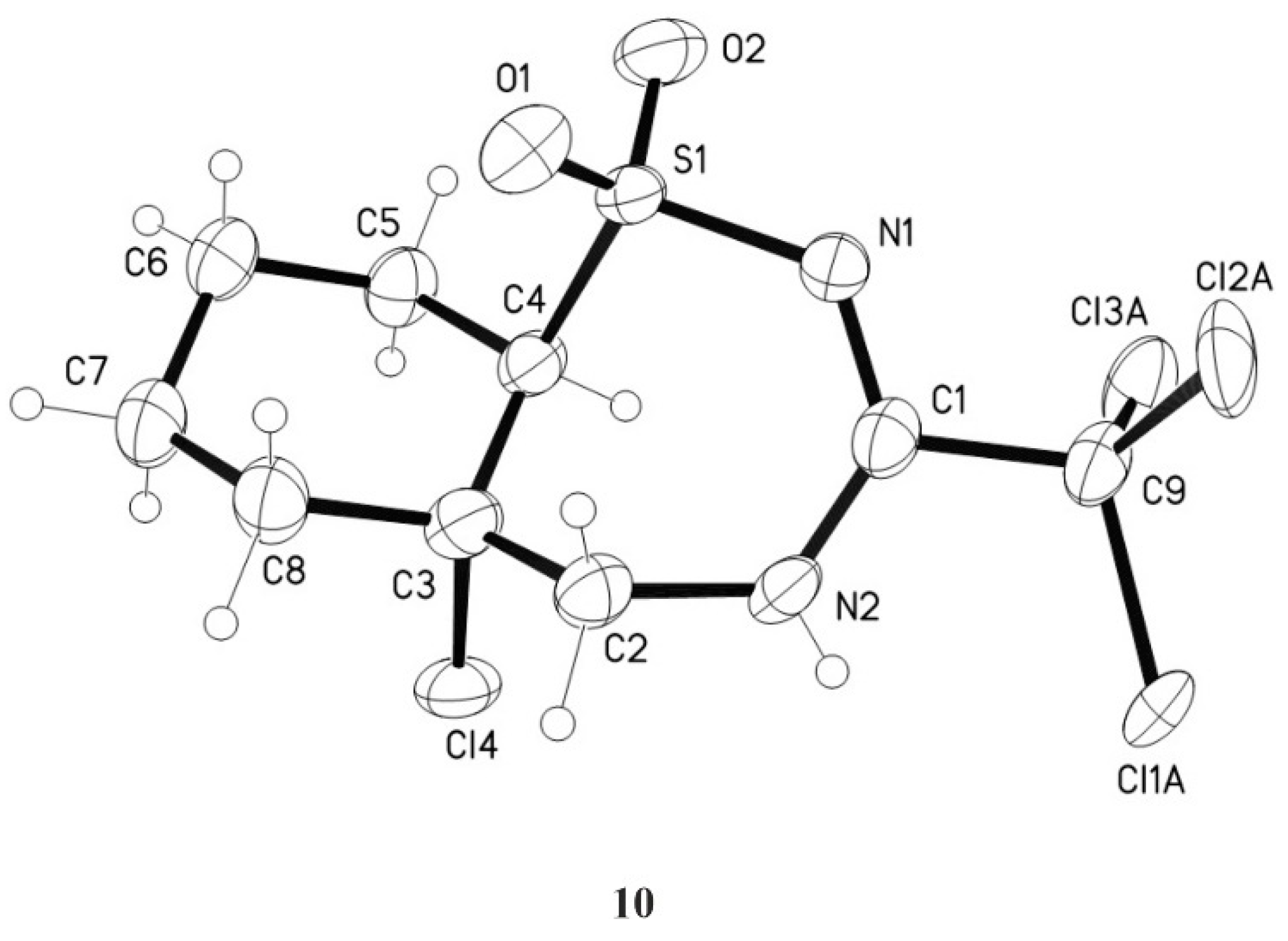
3.5.2. 5a-Chloro-3-trichloromethyl-4,5,5a,6,7,8,9,9a-octahydro-1,2,4-benzothiadiazepine-1,1-dioxide 10
3.6. General Procedure for the Synthesis of Compounds 17a,b
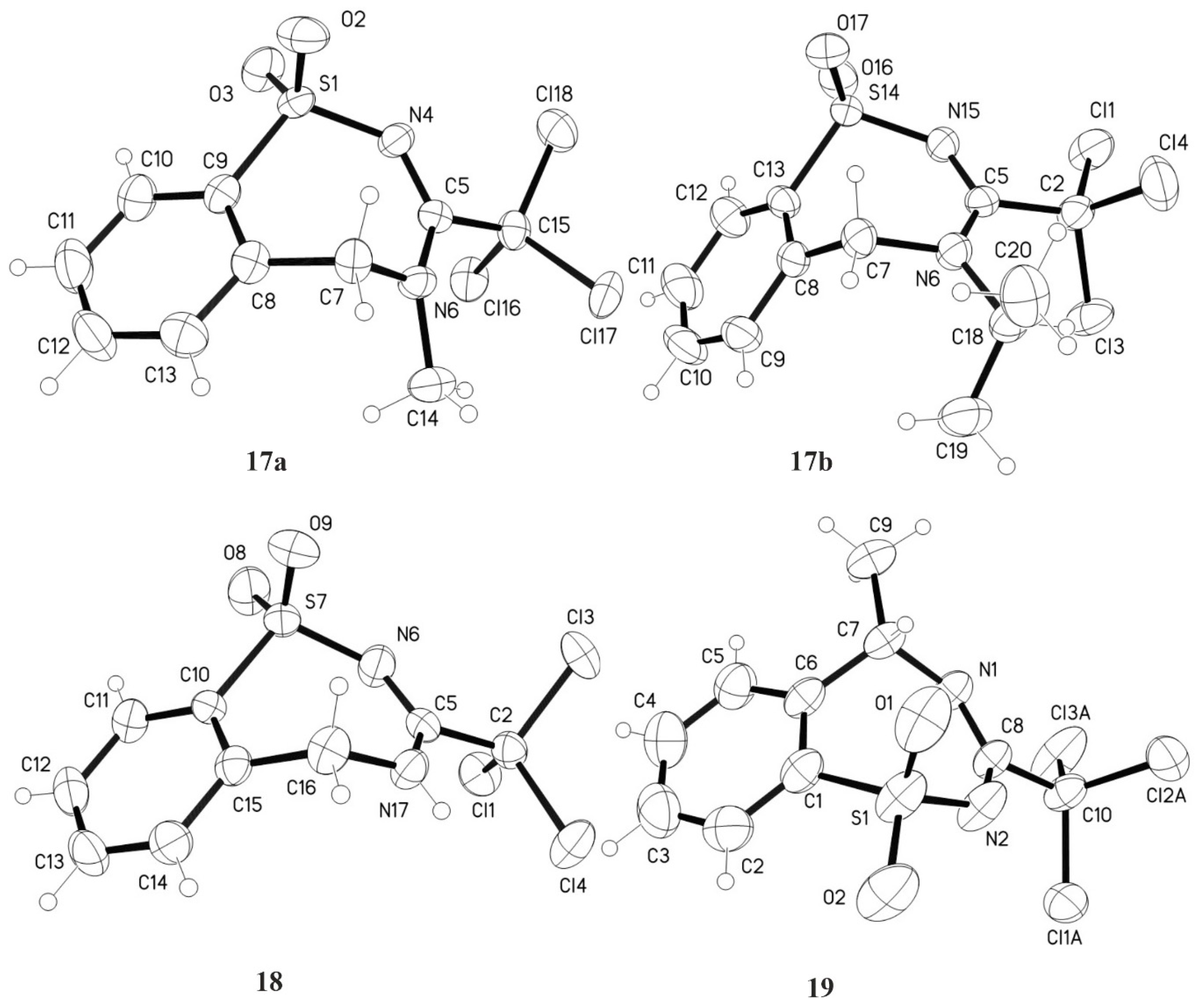
3.6.1. 4-Methyl-3-trichloromethyl-4,5-dihydro-1,2,4-benzothiadiazepine-1,1-dioxide 17a
3.6.2. 4-Isopropyl-3-trichloromethyl-4,5-dihydro-1,2,4-benzothiadiazepine-1,1-dioxide 17b
3.7. General Procedure for the Synthesis of Compounds 18, 19
3.7.1. 3-Trichloromethyl-4,5-dihydro-1,2,4-benzothiadiazepine-1,1-dioxide 18
3.7.2. (S)-5-Methyl-3-trichloromethyl-2,3,4,5-tetrahydro-1,2,4-benzothiadiazepine-1,1-dioxide 19
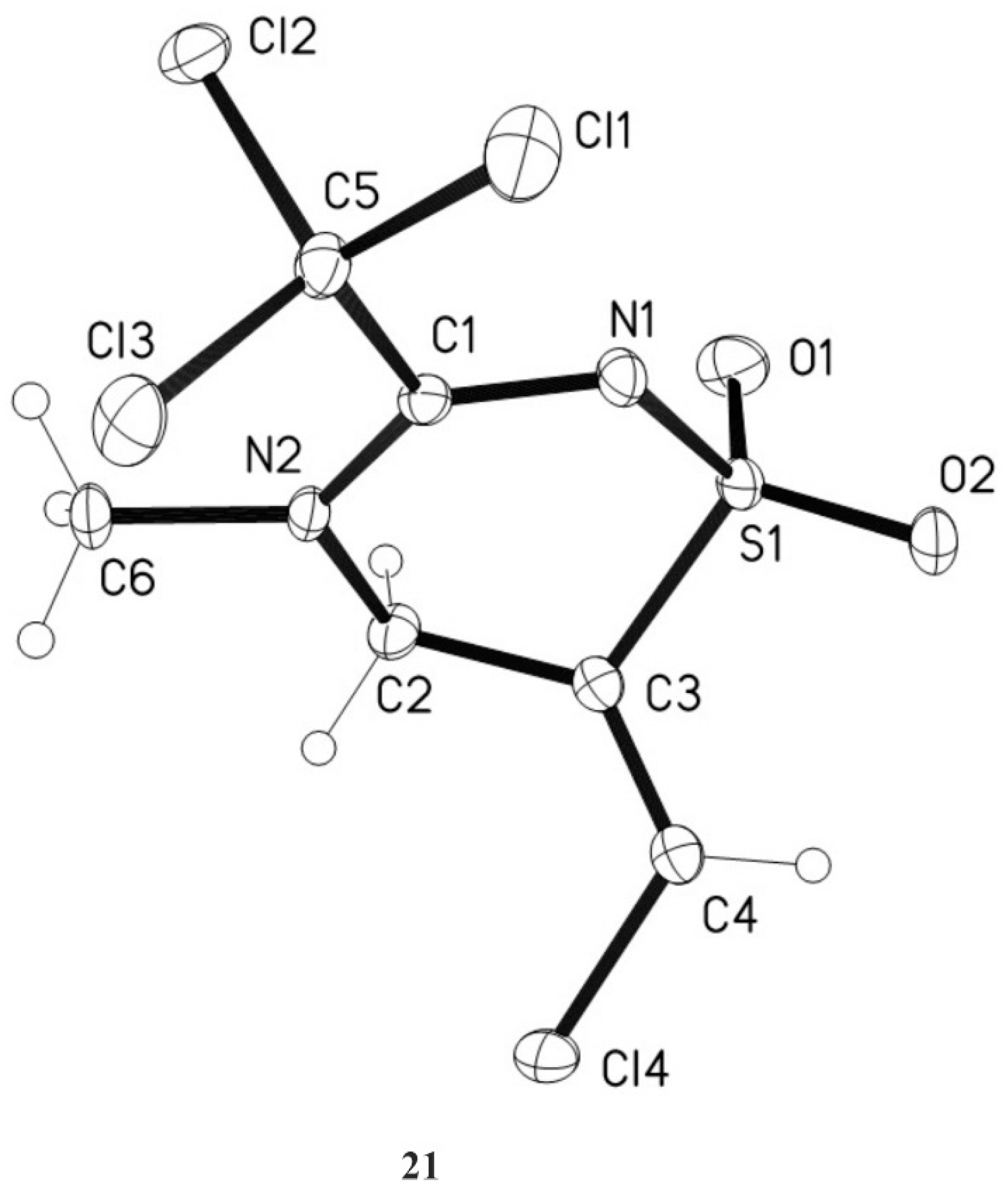
3.8. (E)-6-Chloromethylene-4-methyl-3-trichloromethyl-5,6-dihydro-4H-1,2,4-thiadiazine-1,1-dioxide 21
3.9. 5-Chloro-1-trichloromethyl-6,6a,7,8,9,10-hexahydropyrido[1,2-d][1,2,4]thiadiazocine 3,3-dioxide 23 and (E)-4-Chloromethylene-1-trichloromethyl-5,5a,6,7,8,9-hexahydro-4H-pyrido[1,2-d][1,2,4]thiadiazepine-3,3-dioxide 24
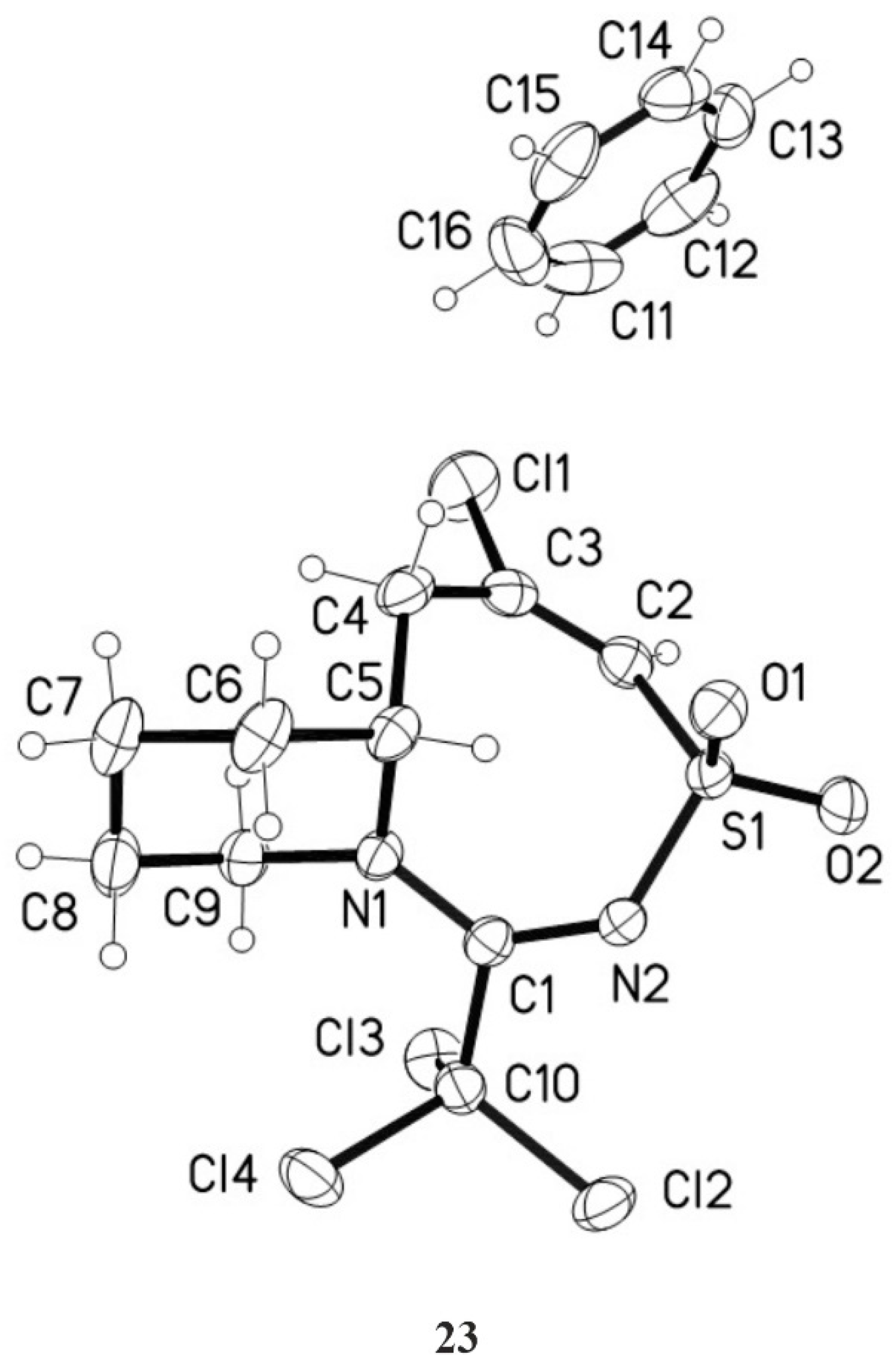
3.9.1. 5-Chloro-1-trichloromethyl-6,6a,7,8,9,10-hexahydropyrido[1,2-d][1,2,4]thiadiazocine 3,3-dioxide 23
3.9.2. (E)-4-Chloromethylene-1-(trichloromethyl)-5,5a,6,7,8,9-hexahydro-4H-pyrido[1,2-d][1,2,4]thiadiazepine-3,3-dioxide 24
3.10. 1-Methylene-4-(trichloromethyl)-1,6,7,8,9,9a-hexahydropyrido[1,2-d][1,2,4]thiadiazine-2,2-dioxide 25
3.11. 1-(4-N-Boc-piperazin-1-yl)methyl-4-trichloromethyl-1,6,7,8,9,9a-hexahydropyrido[1,2-d][1,2,4]thiadiazine-2,2-dioxide 26
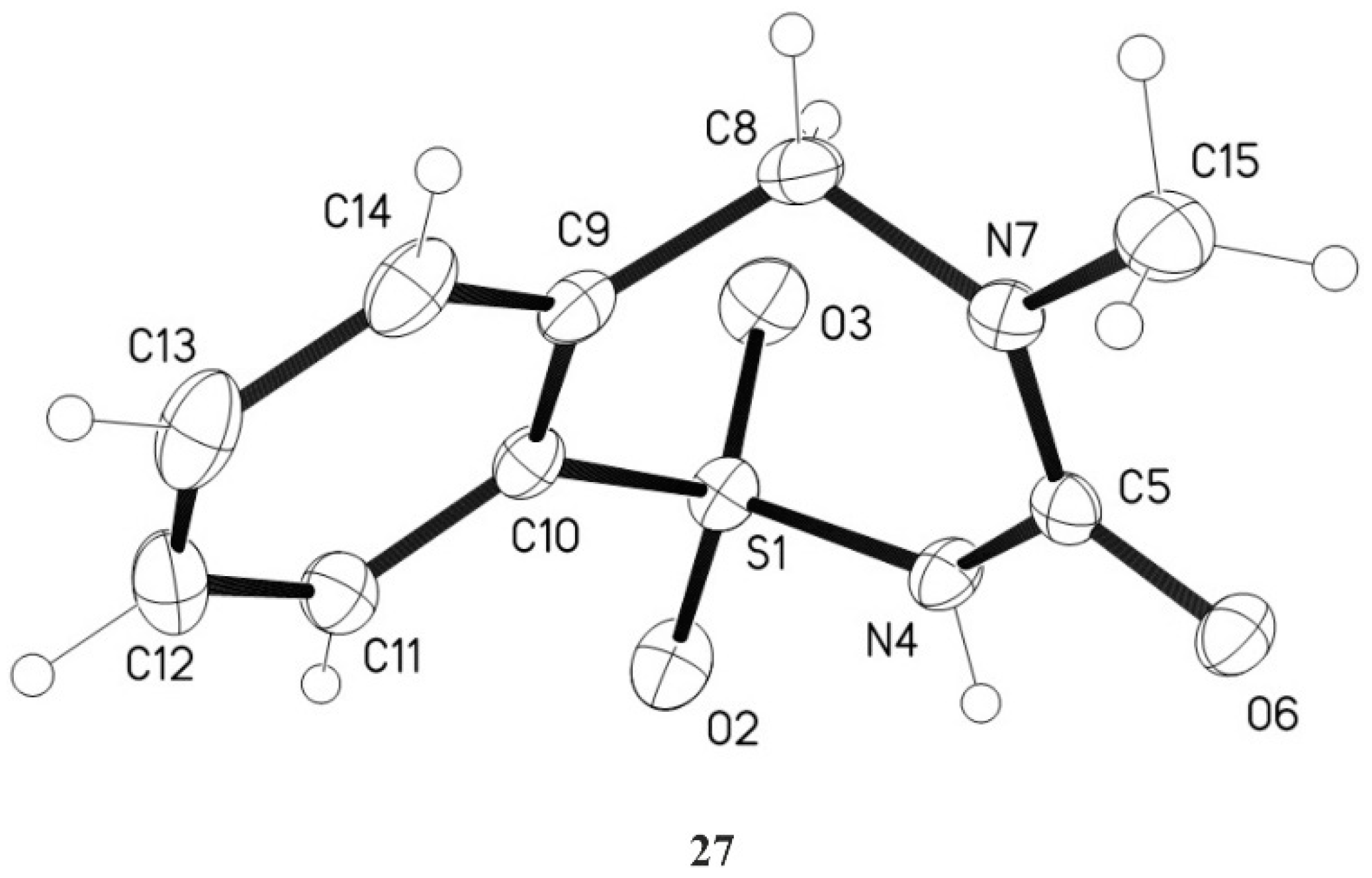
3.12. 4-Methyl-4,5-dihydro-1,2,4-benzothiadiazepin-3(2H)-one-1,1-dioxide 27
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Drews, J. Drug discovery: A historical perspective. Science 2000, 287, 1960–1964. [Google Scholar] [CrossRef]
- Cherepakha, A.; Kovtunenko, V.O.; Tolmachev, A.; Lukin, O.; Nazarenko, K.G. Facile synthesis of 4H-1,2,4-benzothiadiazine-1,1-dioxides. Synth. Commun. 2011, 41, 1977–1989. [Google Scholar] [CrossRef]
- Topiliss, J.G.; Yudis, M.D. Correlation of antihypertensive activity with structure in a series of 2H-1,2,4-benzothiadiazine 1,1-dioxides using the substituent constant approach. J. Med. Chem. 1972, 15, 394–400. [Google Scholar] [CrossRef]
- Tait, A.; Luppi, A.; Franchini, S.; Preziosi, E.; Parenti, C.; Buccioni, M.; Marucci, G.; Leonardi, A.; Poggessi, E.; Brasili, L. 1,2,4-Benzothiadiazine derivatives as α1 and 5-HT1A receptor ligands. Bioorg. Med. Chem. Lett. 2005, 15, 1185–1188. [Google Scholar] [CrossRef] [PubMed]
- Dibella, M.; Rinaldi, M.; Fabio, U.; Manicard, G. Antimicrobial action of 1,2,4-benzothiadiazine-1,1-dioxide derivatives. Farmaco 1973, 28, 777–783. [Google Scholar]
- Zhan, P.; Liu, X.; De Clercq, E. Recent advances in antiviral activity of benzo/heterothiadiazine dioxide derivatives. Curr. Med. Chem. 2008, 15, 1529–1540. [Google Scholar] [CrossRef]
- Castro, A.M.; Abasolo, I.; Gil, C.; Segarra, V.; Martinez, A. CoMFA of benzyl derivatives of 2,1,3-benzo and benzothieno[3,2-a]thiadiazine 2,2-dioxides: Clues for the design of phosphodiesterase 7 inhibitors. Eur. J. Med. Chem. 2001, 36, 333–338. [Google Scholar] [CrossRef][Green Version]
- Shi, L.; Bao, R.; Li, Y.; Zheng, L.; Zhao, R. B(C6F5)3-Catalyzed reduction of cyclic N-sulfonyl ketimines. Eur. J. Org. Chem. 2019, 2019, 6550–6556. [Google Scholar] [CrossRef]
- Boverie, S.; Antoine, M.-H.; Somers, F.; Becker, B.; Sebille, S.; Ouedraogo, R.; Counerotte, S.; Pirotte, B.; Lebrun, P.; de Tullio, P. Effect on KATP channel activation properties and tissue selectivity of the nature of the substituent in the 7- and the 3-position of 4H-1,2,4-benzothiadiazine 1,1-dioxides. J. Med. Chem. 2005, 48, 3492–3503. [Google Scholar] [CrossRef]
- Martinez, A.; Gil, C.; Abasolo, M.I.; Castro, A.; Bruno, A.M.; Perez, C.; Prieto, C.; Otero, J. Benzothiadiazine dioxide dibenzyl derivatives as potent human cytomegalovirus inhibitors: Synthesis and comparative molecular field analysis. J. Med. Chem. 2000, 43, 3218–3225. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chern, J.-W.; Tao, P.-L.; Wang, K.-C.; Gutcait, A.; Liu, S.-W.; Yen, M.-H.; Chien, S.-L.; Rong, J.-K. Studies on quinazolines and 1,2,4-benzothiadiazine 1,1-dioxides. 8. Synthesis and pharmacological evaluation of tricyclic fused quinazolines and 1,2,4-benzothiadiazine 1,1-dioxides as potential α1-adrenoceptor antagonists. J. Med. Chem. 1998, 41, 3128–3141. [Google Scholar] [CrossRef] [PubMed]
- Nematpour, M.; Rezaee, E.; Jahani, M.; Tabatabai, S.A. Synthesis of functionalized benzothiadiazine 1,1-dioxide derivatives via intramolecular C-H activation reactions of trichloroacetamidine and benzenesulfonyl chloride. Tetrahedron Lett. 2018, 59, 2054–2056. [Google Scholar] [CrossRef]
- Mert, B.D.; Elattar, K.M. Seven-membered rings with three heteroatoms: Chemistry of 1,2,5- and 1,4,5-thiadiazepines. Curr. Org. Chem. 2018, 22, 386–410. [Google Scholar] [CrossRef]
- Szostak, R.; Szostak, M. Chemistry of bridged lactams: Recent developments. Molecules 2019, 24, 274. [Google Scholar] [CrossRef]
- Szostak, M.; Aubé, J. Chemistry of Bridged Lactams and Related Heterocycles. Chem. Rev. 2013, 113, 5701–5765. [Google Scholar] [CrossRef]
- Étienne, A.; Le Berre, A.; Giorgetti, J.-P. Thiadiazine-1,2,4 dioxydes-1,1. I.—Synthèse à partir du chlorure de chloro-2 éthanesulfonyle. Bull. Soc. Chim. Fr. 1973, 3, 985–991. [Google Scholar]
- Wu, Q.; Yang, Z.; Xu, J. Temperature-dependent annuloselectivity and stereochemistry in the reactions of methanesulfonyl sulfene with imines. Org. Biomol. Chem. 2016, 14, 7258–7267. [Google Scholar] [CrossRef]
- Toldy, L.; Sohar, P. Uber die Synthese und Oxidation von 2-Imino-3-R-Thiazolidinen und ihrer Thiazin-analogen. Tetrahedron Lett. 1970, 2, 181–185. [Google Scholar] [CrossRef]
- Majumdar, K.C. Regioselective formation of medium-ring heterocycles of biological relevance by intramolecular cyclization. RSC Adv. 2011, 1, 1152–1170. [Google Scholar] [CrossRef]
- Palmisano, G.; Danieli, B.; Lesma, G.; Fiori, G. Electrochemical heterocyclization of o-tolenesulfonamides to 3-alkyl-4,5-dihydro-1,2,4-benzothiadiazepine-1,1-dioxides. Tetrahedron 1988, 44, 1545–1552. [Google Scholar] [CrossRef]
- Tan, D.; Friscic, T. Carbodiimide insertion into sulfonimides: One-step route to azepine derivatives via a two-atom saccharin ring expansion. Chem. Commun. 2017, 53, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Ramana, P.V.; Reddy, A.R. Synthesis of a few cyclothiadiazanones and aminosulfonyl benzamides from saccharin. J. Sulfur Chem. 2010, 31, 71–81. [Google Scholar] [CrossRef]
- Hassan, A.K.; Abd El-Aal, H.A.K.; Khalaf, A.A. Design and diversity-oriented synthesis of benzo- and pyrido-annulated medium-sized N,S-heterocycles via thio-Michael and Friedel-Crafts approaches. Arkivoc 2019, 4, 212–227. [Google Scholar]
- Shalimov, A.A.; Chudakova, T.I.; Vlasenko, Y.G.; Sinitsa, A.D.; Onys’ko, P.P. Heterocyclization of N-(chlorosulfonyl)imidoyl chlorides with anilines, a new method of synthesis of 1,2,4-benzothiadiazine 1,1-dioxides. Chem. Heterocycl. Comp. 2016, 52, 267–274. [Google Scholar] [CrossRef]
- Niu, T.-F.; Lin, D.; Xue, L.-S.; Jiang, D.-Y.; Ni, B.-Q. Visible-light-induced chemoselective synthesis of α-chloro and vinyl sulfones by sulfonylation of alkenes. Synlett 2018, 29, 364–368. [Google Scholar]
- Wallentin, C.-J.; John, D.; Nguyen, J.D.; Finkbeiner, P.; Stephenson, C.R.J. Visible light-mediated atom transfer radical addition via oxidative and reductive quenching of photocatalysts. J. Am. Chem. Soc. 2012, 134, 8875–8884. [Google Scholar] [CrossRef]
- Dneprovskii, A.S.; Kasatochkin, A.N.; Boyarskii, V.P.; Ermoshkin, A.A.; Yakovlev, A.A. Application of copper(I) halides to modifying reactivity of polyhalomethanes and arenesulfonyl chlorides in free-radical addition. “Cross-halogenation” reaction. Russ. J. Org. Chem. 2006, 42, 1120–1130. [Google Scholar] [CrossRef]
- Kameyama, M.; Shimezava, H.; Satoh, T.; Kamigata, N. Synthesis of substituted 1,3-dienes by the reaction of alkenesulfonyl chlorides with olefins catalyzed by a ruthenium(II) complex. Bull. Chem. Soc. Jpn. 1988, 61, 1231–1235. [Google Scholar] [CrossRef]
- Kamigata, N.; Sawada, H.; Kobayashi, M. Reactions of arenesulfonyl chlorides with olefins catalyzed by a ruthenium(II) complex. J. Org. Chem. 1983, 48, 3793–3796. [Google Scholar] [CrossRef]
- Culshaw, P.N.; Walton, J.C. Sulphonate esters as sources of sulphonyl radicals; ring-closure reactions of alk-4- and -5-enesulphonyl radical. J. Chem. Soc. Perkin Trans. 2 1991, 8, 1201–1208. [Google Scholar] [CrossRef]
- Alkan-Zambada, M.; Hu, X. Cu-Catalyzed photoredox chlorosulfonation of alkenes and alkynes. J. Org. Chem. 2019, 84, 4525–4533. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Ilies, L.; Nakamura, E. Iron-catalyzed regio- and stereoselective chlorosulfonylation of terminal alkynes with aromatic sulfonyl chlorides. Org. Lett. 2012, 14, 954–956. [Google Scholar] [CrossRef] [PubMed]
- Chakrasali, P.; Kim, K.; Jung, Y.-S.; Kim, H.; Han, S.B. Visible-light-mediated photoredox-catalyzed regio- and stereoselective chlorosulfonylation of alkynes. Org. Lett. 2018, 20, 7509–7513. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Betteridge, P.W.; Carruthers, J.R.; Cooper, R.I.; Prout, K.; Watkin, D.J. CRYSTALS version 12: Software for guided crystal structure analysis. J. Appl. Cryst. 2003, 36, 1487. [Google Scholar] [CrossRef]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Cryst. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- van der Sluis, P.; Spek, A.L. BYPASS: An effective method for the refinement of crystal structures containing disordered solvent regions. Acta Cryst. Sect. A 1990, 46, 194–201. [Google Scholar] [CrossRef]
- Spek, A.L. PLATON, A Multipurpose Crystallographic Tool; Utrecht University: Utrecht, The Netherlands, 1998. [Google Scholar]
Sample Availability: Samples of the compounds 4a–e, 7, 17b, 21, 23, 25 are available from the authors. |




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
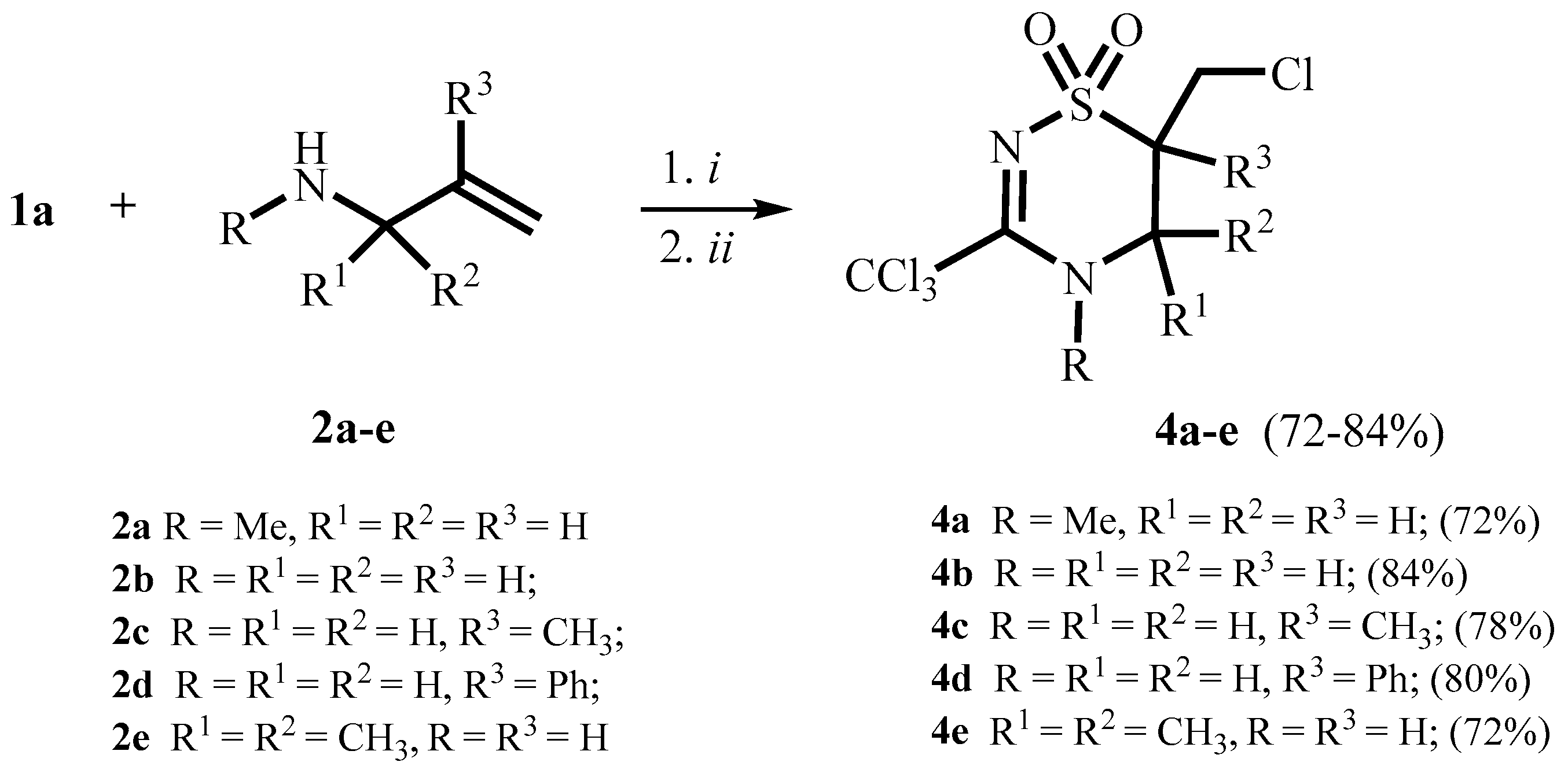{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Reagent and Conditions | Conversion of 3a to 4a (%) a |
|---|---|---|
| 1 | ClCH2CH2Cl, reflux, 7 h | NR b |
| 2 | AlCl3, CH2Cl2, r.t, 24 h | 100 |
| 3 | TiCl4, CH2Cl2, r.t, 24 h | 100 |
| 4 | BF3·Et2O, CH2Cl2, r.t, 24 h | NR b |
| 5 | ZnCl2, CH2Cl2, r.t, 24 h | NR b |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shalimov, O.; Rusanov, E.; Muzychka, O.; Onys’ko, P. Novel Convenient Approach to 6-, 7-, and 8-Numbered Nitrogen Heterocycles Incorporating Endocyclic Sulfonamide Fragment. Molecules 2020, 25, 2887. https://doi.org/10.3390/molecules25122887
Shalimov O, Rusanov E, Muzychka O, Onys’ko P. Novel Convenient Approach to 6-, 7-, and 8-Numbered Nitrogen Heterocycles Incorporating Endocyclic Sulfonamide Fragment. Molecules. 2020; 25(12):2887. https://doi.org/10.3390/molecules25122887
Chicago/Turabian StyleShalimov, Oleksandr, Eduard Rusanov, Oksana Muzychka, and Petro Onys’ko. 2020. "Novel Convenient Approach to 6-, 7-, and 8-Numbered Nitrogen Heterocycles Incorporating Endocyclic Sulfonamide Fragment" Molecules 25, no. 12: 2887. https://doi.org/10.3390/molecules25122887
APA StyleShalimov, O., Rusanov, E., Muzychka, O., & Onys’ko, P. (2020). Novel Convenient Approach to 6-, 7-, and 8-Numbered Nitrogen Heterocycles Incorporating Endocyclic Sulfonamide Fragment. Molecules, 25(12), 2887. https://doi.org/10.3390/molecules25122887