3.2. Synthesis
Pentachloro-2-nitro-1,3-butadiene (
1) was prepared from 2
H-pentachloro-1,3-butadiene in 53% yield (b.p. 69–71 °C/1 mbar) according to the literature [
3,
4]. (
Z)- and (
E)
-4-Bromotetrachloro-2-nitrobuta-1,3-diene (
2) was obtained from (
Z)- and (
E)
-1-bromotetrachlorobuta-1,3-diene in 56% yield (b.p. 84–86 °C/1.3 mbar) [
5]. (
Z)-1,1,4-Trichloro-2,4-dinitrobuta-1,3-diene (
3) was synthetized from the (
Z)- and (
E)
-1-bromo-1,4,4-trichlorobuta-1,3-diene in 18% yield, m.p. 70–71 °C [
6].
Synthesis of 4-methyl-2-(2,3,3-trichloro-1-nitroprop-2-en-1-ylidene)-2,3-dihydro-1,3-benzoxazole (4a) (General method). At −40 °C, a solution of nitrodiene 1 (2.71 g, 10.0 mmol) in 5 mL methanol (MeOH) was added dropwise to a suspension of 2-amino-3-methylphenol (3.94 g, 32.0 mmol) in 30 mL MeOH within 5 min. The resulting mixture was kept for 1 h at this temperature, and was then allowed to reach room temperature (r.t.). After 5 h stirring, the mixture was poured into a cold solution (0 °C) of 5 mL conc. HCl in 250 mL of water. After 20 min, the precipitate was filtered off, washed with cold water (3 × 40 mL) and diethyl ether (2 × 10 mL). Drying in vacuo gave 2.57 g of oxazole 4a, yield 80%, yellowish solid m.p. 149–151 °C; IR (KBr) νmax = 3089, 1612, 1382, 1079, 968, 520 cm−1; 1H NMR (200 MHz, CDCl3), δ = 2.61 (3H, s, CH3 Ph), 7.21–7.33 (2H, m, H Ph), 7.36–7.41 (1H, m, H Ph), 11.78 (1H, s, NH) ppm; 13C NMR (50 MHz, CDCl3) δ = 16.6 (CH3), 107.0 (C NO2), 108.7 (CH), 120.2 (CCl2), 123.4 (C Me), 125.4 (CH), 127.4, 127.6 (CH), 128.2, 146.6, 159.0 ppm; MS m/z (Irel, %): 320 [M+] (4), 285 [M − Cl]+ (10), 274 [M-NO2]+ (65), 239 [M-Cl-NO2]+ (100); HRMS (ESI−) m/z calcd for C11H6N2O3Cl3 [M − H]−: 318.9444; found: 318.9446.
5-Methyl-2-(2,3,3-trichloro-1-nitroprop-2-en-1-ylidene)-2,3-dihydro-1,3-benzoxazole (4b). Same procedure as for 4a, but using 2-amino-4-methylphenol (3.94 g, 32.0 mmol). Yield 2.18 g (68%), yellowish solid, m.p. 168–170 °C. IR (KBr) νmax = 3380, 1612, 1434, 1375, 1056, 922 cm−1. 1H NMR (400 MHz, DMSO-d6) δ = 2.42 (3H, s, CH3 Ph), 7.20 (1H, d, J = 8.3 Hz, H Ph), 7.36 (1H, s, H Ph), 7.69 (1H, d, J = 8.3 Hz, H Ph), 13.64 (1H, s, NH) ppm; 13C NMR (100 MHz, DMSO-d6) δ = 21.2 (CH3), 105.4 (C NO2), 111.0 (CH), 113.6 (CH), 122.1 (CCl2), 126.0 (CH), 126.6, 129.8, 136.4, 144.8, 158.0 (NCO) ppm. MS m/z (Irel, %): 320 [M+] (2), 285 [M-Cl]+ (4), 274 [M-NO2]+ (55), 239 [M-Cl-NO2]+ (100), 204 [M-2Cl-NO2]+ (14); HRMS (ESI−) m/z calcd for C11H6N2O3Cl3 [M − H]−: 318.9453; found: 318.9444.
Synthesis of [{1-(1,3-benzoxazol-2-yl)-3,3-dichloro-2-[4-(dimethylamino)pyridinium-1-yl]prop-2-en-1-yli- dene}(oxido)-l5-azanyl]oxidanide (5a) (General method). To a suspension of benzoxazole 4b (0.322 g, 1.00 mmol) in 15 mL MeOH at 0 °C, a solution of 4-dimethylaminopyridine (DMAP) (0.257 g, 2.1 mmol) in 3 mL MeOH was added dropwise. The mixture was stirred for 1 h at 0 °C and at r.t. for 12 h. Subsequently, the precipitate was filtered off at 0 °C and washed with water (2 × 10 mL) and cold MeOH (5 mL). Finally, the product 5a was dried in vacuo. Yield 0.301 g (74%), yellowish solid, m.p. 189–190 °C. IR (ATR) νmax = 1641, 1531, 1355, 1139, 1055, 790 cm−1; 1H NMR (400 MHz, DMSO-d6) δ = 2.38 (3H, s, CH3 Ph), 3.23 (6H, s, N(CH3)2), 6.99 (1H, d, J = 7.8 Hz, H Ph), 7.06 (2H, d, J = 6.7 Hz, H pyr), 7.33 (1H, s, H Ph), 7.43 (1H, d, J = 7.9 Hz, H Ph), 8.29 (2H, d, J = 6.7 Hz, H pyr) ppm; 13C NMR (100 MHz, DMSO-d6) δ = 39.7 (CH3), 40.2 (NCH3), 103.7, 107.3 (CH), 109.3 (CH), 117.8 (CH), 122.7, 123.6 (CH), 133.2, 135.8, 142.3 (CH), 142.5, 147.8, 156.4, 160.9 ppm; MS m/z (Irel, %): 406 [M+] (1), 273 [M-benzoxazole-H]+ (5), 238 [M-benzoxazole-HCl]+ (3), 122 [DMAP]+ (73), 100 (100); HRMS (ESI+) m/z calcd for C18H17N4O3Cl2 [M + H]+: 407.0672; found: 407.0675.
[{1-(1,3-Benzoxazol-2-yl)-3,3-dichloro-2-[4-(morpholin-4-yl)pyridinium-1-yl]prop-2-en-1-ylidene} (oxido)-l5-azanyl]oxidanide (5b). Following the typical procedure for 5a, using 4b (0.322 g, 1.00 mmol) and 4-(4-morpholinyl)pyridine (0.345 g, 2.1 mmol) at −18 °C and holding at this temperature for 3 h. Yield 0.270 g (62%), yellowish solid, m.p. 180–181 °C. IR (ATR) νmax = 1640, 1548, 1348, 1253, 1156, 792 cm−1. 1H NMR (400 MHz, DMSO-d6) δ = 2.38 (3H, s, CH3 Ph), 3.73 (8H, s, H morph), 7.00 (1H, dd, J = 8.1 Hz, J = 1.3 Hz, H Ph), 7.25 (2H, d, J = 7.8, H pyr), 7.34 (1H, s, H Ph), 7.43 (1H, d, J = 8.2, H Ph), 8.34 (2H, d, J = 7.6, H pyr) ppm; 13C NMR (100 MHz, DMSO-d6) δ = 21.3 (CH3), 46.6 (N(CH2)2), 65.6 (O(CH2)2), 103.8, 107.5 (CH), 109.3 (CH), 117.8 (CH), 122.9, 123.9 (CH), 133.2, 135.7, 142.5, 143.0 (CH), 147.8, 156.1, 160.8 ppm; MS m/z (Irel, %): 448 [M+] (1), 325 [M-HCl-morpholine]+ (1), 316 [M-methylbenzoxazole]+ (1), 269 [M-pyridine+H]+ (4), 165 (100), 132 [methylbenzoxazole]+ (12); HRMS (ESI+) m/z calcd for C20H19N4O4Cl2 [M + H]+: 449.0778; found: 449.0780.
1-(Pyridin-4-yl)pyrrolidin-2-one as starting material for the synthesis of azinate
5c was obtained from 4-aminopyridine and 4-chlorobutanoyl chloride according to the literature [
88]. Yield 65%, colorless liquid.
1H NMR (600 MHz, CDCl
3) δ = 2.17 (2H, t,
J = 7.7 Hz, CH
2 pyrro), 2.61 (2H, t,
J = 8.2 Hz, CH
2 pyrro), 3.82 (2H, t,
J = 7.1 Hz, CH
2 pyrro), 7.57 (2H, d,
J = 4.9 Hz, H pyr), 8.50 (2H, d,
J = 4.9 Hz, H pyr) ppm;
13C NMR (150 MHz, CDCl
3) δ = 17.6 (CH
2), 32.8 (CH
2), 47.4 (CH
2), 112.8 (CH), 145.8, 150.4 (CH), 175.2 (CO) ppm; MS
m/z (
Irel, %): 162 [M
+] (31), 147 [M-OH]
+ (25), 119 [M-CH
2CHO]
+ (10), 107 [M-CH
2CH
2CO+H]
+ (100).
[{1-(1,3-Benzoxazol-2-yl)-3,3-dichloro-2-[4-(2-oxopyrrolidin-1-yl)pyridinium-1-yl]prop-2-en-1-ylidene}- (oxido)-l5-azanyl]oxidanide (5c). Same procedure as for 5a, using 1-(pyridin-4-yl)pyrrolidin-2-one (0.341 g, 2.1 mmol) at −18 °C and holding this temperature for 2 h. Stirring was continued for 24 h at r.t. Yield 67%, yellow solid, m.p. 157–158 °C. IR (ATR) νmax = 1733, 1627, 1550, 1349, 1257, 1157 cm−1. 1H NMR (600 MHz, DMSO-d6) δ = 2.13 (2H, t, J = 7.6 Hz, CH2 pyrro), 2.39 (3H, s, CH3 Ph), 2.67 (2H, t, J = 8.0 Hz, CH2 pyrro), 3.99 (2H, t, J = 7.2 Hz, CH2 pyrro), 7.01 (1H, dd, J = 8.2 Hz, J = 0.8 Hz, H Ph), 7.35 (1H, s, H Ph), 7.45 (1H, d, J = 8.2 Hz, H Ph), 8.25 (2H, d, J = 7.0, H pyr), 8.99 (2H, d, J = 7.5 Hz, H pyr) ppm; 13C NMR (150 MHz, DMSO-d6) δ = 17.1 (CH2), 21.2 (CH3), 32.7 (CH2), 48.1 (CH2), 104.0, 109.4 (CH), 113.9 (CH), 117.9 (CH), 123.8 (CH), 124.0, 133.3, 136.2, 142.5, 145.7 (CH), 147.8, 152.6, 160.5, 177.5 ppm; 15N NMR (43.4 MHz, DMSO-d6, doped with nitromethane (0.0 ppm)) δ = −187.0 (N-pyridine), -235.2 (N-pyrrolidine) ppm, other N-atoms could not be detected; MS m/z (Irel, %): 446 [M+] (1), 411 [M-Cl]+ (1), 162 [pyridinyl–pyrrolidinone]+ (45), 133 [methylbenzoxazole+H]+ (6), 107 (100); HRMS (ESI+) m/z calcd for C20H17N4O4Cl2 [M + H]+: 447.0621; found: 447.0623.
[{1-(1,3-Benzoxazol-2-yl)-3,3-dichloro-2-[3-(1-methylpyrrolidin-2-yl)pyridinium-1-yl]prop-2-en-1-ylidene}- (oxido)-λ5-azanyl]oxidanide (5d). To a suspension of benzoxazole 4b (0.322 g, 1.0 mmol) in MeOH (10 mL) at −18 °C, a solution of (-)-nicotine (0.324 g, 2.0 mmol) in MeOH (5 mL) was added dropwise. Subsequently, the mixture was stirred at −18 °C for 2 h and at r.t. for 18 h. After completion of the reaction water (10 mL), NaHCO3 (0.184 g, 2.2 mmol) were added and the mixture was stirred for 10 min, following extraction with chloroform (3 × 15 mL). The combined organic phases were dried over calcium chloride and purified by column chromatography using DCM–petroleum ether (1:1). The product 5d was dried in vacuo. Yield 0.237 g (53%), yellow solid, m.p. 96–98 °C. IR (ATR) νmax = 1622, 1516, 1354, 1259, 1141, 1062 cm−1. 1H NMR (400 MHz, CDCl3) δ = 1.64–1.77 (1H, m, CH2 pyrro), 1.83–2.01 (2H, m, NCH2), 2.26 (3H, s, NCH3 pyrro), 2.43 (3H, s, Ph-CH3), 2.43–2.49 (1H, m, CH2 pyrro), 3.24–3.34 (1H, m, CH2 pyrro), 3.42–3.57 (1H, m, CH2 pyrro), 3.63–3.89 (1H, m, CH2 pyrro), 7.00 (1H, dd, J = 1.0 Hz, J = 8.2 Hz, CH3C-CH), 7.37 (1H, s, H Ph), 7.39 (1H, d, J = 8.2 Hz, OCCH), 7.96 (1H, dd, J = 5.6 Hz, J = 7.8 Hz, H pyr), 8.48 (1H, d, J = 7.8 Hz, H pyr), 9.03 (1H, d, J = 5.6 Hz, H pyr), 9.15 (1H, s, H pyr) ppm; 13C NMR (100 MHz, CDCl3) δ = 21.5 (CH3 Ph), 23.1 (CH2), 35.8 (CH2), 40.3 (CH3 pyrro), 56.6 (CH2), 67.1 (CH pyrro), 104.6 (CNO2), 109.8 (CH Ph), 118.0 (CH Ph), 124.0 (CH Ph), 127.0 (CH pyr), 128.5 (CCl2), 133.7 (CCH3), 136.3, 142.2, 143.6 (CH pyr), 145.2 (CH pyr), 145.5 (CH pyr), 145.7, 148.3 (C-pyrro), 160.1 (NCO) ppm; MS (ESI+) m/z calcd for C21H21N4O3Cl2 [M + H]+: 447.1; found 447.1.
2-[3-Bromo-2,3-dichloro-1-nitroprop-2-en-1-ylidene]-4-methyl-2,3-dihydro-1,3-benzoxazole (6). Same proce-dure as for 4a, but starting from 2 (316 mg, 1.00 mmol, a 47: 53 mixture of isomers) using 2-amino-3-methylphenol (394 mg, 3.20 mmol). A 1:1 mixture of isomers was obtained. Yield 73%, yellowish solid, m.p. 145–146 °C. IR (KBr) νmax = 3329, 1620, 1571 (NO2), 1376, 1320, 1047 cm−1; 1H NMR (200 MHz, CDCl3) δ = 2.61 (3H, s, CH3 Ph), 7.22–7.41 (3H, m, H Ph), 11.77 (1H, s, NH) ppm; 13C NMR (50 MHz,CDCl3) δ = 16.6 (CH3); 106.9 (CNO2); 108.7 (CH); 114.9, 115.8 (CClBr); 121.8, 123.7 (CCl); 123.4 (C Me); 125.4 (CH), 127.4, 127.6 (CH), 146.7, 158.7, 158.9 (NCO) ppm; MS m/z (Irel, %): 364 [M+] (2), 329 [M − Cl]+ (4), 318 [M-NO2]+ (19), 285 [M − Br]+ (25), 283 [M-Cl-NO2]+ (50), 239 [M-Br-NO2]+ (100); HRMS (ESI−) m/z calcd for C11H6N2O3Cl2Br [M − H]−: 362.8944; found: 362.8948.
2-((Z)-3-chloro-1,3-dinitroallylidene)-5-methyl-2,3-dihydro-1H-benzo[d]imidazole (7a). Same procedure as for 4a, but starting from 3 (247 mg, 1.00 mmol, only Z-isomer) using 4-methylbenzene-1,2-diamine (257 mg, 2.10 mmol). Yield 93%, red solid, m.p. 163–165 °C. IR (KBr) νmax = 3330, 1602, 1577 (NO2), 1410, 1268, 987 cm−1. 1H NMR spectrum (200 MHz, DMSO-d6) δ = 2.49 (3H, s, CH3 Ph), 7.36 (1H, d, J = 8.5 Hz, H Ph), 7.58 (1H, s, H Ph), 7.67 (1H, d, J = 8.5 Hz, H Ph), 9.14 (1H, s, CH), 14.45 (2H, s, NH) ppm; 13C NMR (50 MHz, DMSO-d6) δ = 21.4 (CH3), 106.9 (C NO2), 113.4 (CH), 113.6 (CH), 120.1 (CClNO2), 127.5 (CH), 129.1 (C Me), 129.2 (CH), 131.2, 136.1, 142.3 (NCN) ppm; MS m/z (Irel, %): 296 [M+] (5), 250 [M-NO2]+ (55), 233 [M-NO2-OH]+ (12), 204 [M-2NO2]+ (60), 169 [M-2NO2-Cl]+ (48), 157 (100); HRMS (ESI−) m/z calcd for C11H8N4O4Cl [M − H]−: 295.0240; found: 295.0248.
2-(3-Chloro-1,3-dinitroprop-2-en-1-ylidene)-5-methyl-2,3-dihydro-1,3-benzoxazole (7b). Same procedure as for 4a, but using 3 (247 mg, 1.00 mmol, only Z-isomer) and 2-amino-4-methylphenol (394 mg, 3.20 mmol). Yield 67%, yellow solid, m.p. 110–112 °C. IR (KBr) νmax = 3250, 1602, 1545 (NO2), 1289, 1065, 871 cm−1. 1H NMR (200 MHz, DMSO-d6) δ = 2.45 (3H, s, CH3 Ph), 7.25 (1H, d, J = 8.3 Hz, H Ph), 7.55 (1H, s, H Ph), 7.63 (1H, d, J = 8.5 Hz, H Ph), 9.09 (1H, s, CH), 10.66 (1H, s, NH); 13C NMR (50 MHz, DMSO-d6) δ = 21.2 (CH3), 110.8 (CH), 112.5 (CNO2), 118.5 (CClNO2), 119.1 (CH), 126.8 (CH), 129.5 (CH), 134.5 (C Me), 139.5, 148.2, 157.2 (NCO) ppm; MS m/z (Irel, %): 297 [M+] (3), 251 [M − NO2]+ (45), 234 [M-NO2-OH]+ (13), 158 (100); HRMS (ESI−) m/z calcd for C11H7N3O5Cl [M − H]−: 296.0072; found: 296.0070.
2-(3-Chloro-1,3-dinitroprop-2-en-1-ylidene)-4-methyl-2,3-dihydro-1,3-benzoxazole (7c). Same procedure as for 4a, using 3 (247 mg, 1.00 mmol, only Z-isomer) and 2-amino-3-methylphenol (394 mg, 3.20 mmol). Yield 85%, yellow solid, m.p. 102–103 °C. IR (KBr) νmax = 3254, 1625, 1579 (NO2), 1520, 1077, 646 cm−1. 1H NMR (200 MHz, DMSO-d6) δ = 2.53 (3H, s, CH3 Ph), 7.19 (1H, d, J = 7.5 Hz, H Ph), 7.30 (1H, t, J = 7.5 Hz, H Ph), 7.52 (1H, d, J = 8.0 Hz, H Ph), 9.20 (1H, s, CH), 10.19 (1H, s, NH) ppm; 13C NMR (50 MHz, DMSO-d6) δ = 16.5 (CH3), 108.5 (CH), 113.6 (CNO2), 115.9 (CClNO2), 125.0 (CH), 125.5 (CH), 130.3 (CH), 140.3, 150.3, 156.3 (NCO) ppm; MS m/z (Irel, %): 297 [M+] (3), 251 [M-NO2]+ (40), 234 [M-NO2-OH]+ (12), 158 (100); MS (ESI−) m/z calcd for C11H7N3O5Cl [M − H]−: 296.0; found: 296.0.
Synthesis of 2-(3-bromo-2,3-dichloro-1-nitroprop-2-en-1-ylidene)-1,3-diphenylimidazolidine (8b) (General method). To a solution of N,N’-diphenylethane-1,2-diamine (0.446 g, 2.1 mmol) in 10 mL MeOH at −40 °C a solution of nitrodiene 2 (0.316 g, 1.0 mmol) in 5 mL MeOH was added dropwise. After 1 h of stirring at −40 °C, the solution was allowed to reach r.t. and stirred for another 5 h. The precipitate was filtered off, washed with water (3 × 20 mL), MeOH (1 × 10 mL), diethyl ether (2 × 10 mL) and dried in vacuo. A 1:1 mixture of isomers was obtained. Yield 0.410 g (90%), yellow solid, m.p. 222–223 °C. IR (KBr) νmax = 3455, 3059, 1594, 1523, 1295, 761 cm−1. 1H NMR (200 MHz, DMSO-d6) δ = 4.37 (4H, s, CH2), 7.25–7.47 (10H, m, H Ph) ppm; 13C NMR (50 MHz, DMSO-d6) δ = 51.0, 50.9 (CH2), 104.4, 105.9 (CNO2), 107.2, 107.7 (CBrCl), 122.4, 122.5 (CH), 127.0 (CH), 127.8, 129.5, 129.6 (CH), 139.9, 140.0 (NC Ph), 155.6, 155.7 (NCN) ppm; MS m/z (Irel, %): 453 [M+] (3), 418 [M − Cl]+ (3), 374 [M-Br]+ (45), 339 [M-Cl-Br]+ (8), 279 [M-C2Cl2Br]+ (100); HRMS (ESI+) m/z calcd for C18H15N3O2Cl2Br [M + H]+: 453.9719; found: 453.9728.
2-(3-Chloro-1,3-dinitroprop-2-en-1-ylidene)-1,3-diphenylimidazolidine (8c). Same procedure as for 8b, using dinitrodiene 3 (0.247 g, 1.0 mmol). Yield 86%, yellow solid, m.p. 236–237 °C. IR (KBr) νmax = 3054, 1570, 1491, 1349, 1193, 999 cm−1. 1H NMR (200 MHz, CDCl3) δ = 4.58 (4H, s, CH2), 7.29–7.33 (4H, m, H Ph), 7.39–7.45 (6H, m, H Ph), 8.69 (1H, s, CH) ppm; 13C NMR (50 MHz, DMSO-d6) δ = 50.6 (CH2), 105.1, 117.8 (CClNO2), 123.2 (CH), 128.4 (CH), 129.0 (CH), 130.0 (CH), 136.7 (NC Ph), 158.7 (NCN) ppm; MS m/z (Irel, %): 386 [M+] (20), 340 [M − NO2]+ (60), 305 [M-NO2-Cl]+ (3), 281 [M-(CH=CClNO2)+H]+ (20), 247 [M-(CH=CClNO2)-2OH]+ (100); HRMS (ESI+) m/z calcd for C18H16N4O4Cl [M + H]+: 387.0855; found: 387.0865.
Synthesis of 2-[3,3-dichloro-2-nitro-1-(pyrrolidin-1-yl)prop-2-en-1-ylidene]-1,3-diphenylimidazolidine (9). To a suspension of imidazolidine 8a (0.387 g, 1.0 mmol) in 10 mL MeOH at r.t. a solution of pyrrolidine (0.356 g, 5.0 mmol) in 5 mL MeOH was added. Subsequently, the mixture was stirred for 3 h at r.t. and 4 h at reflux. After cooling to 10 °C, the pH was adjusted to 6–7 with HCl (5%). The resulting precipitate was filtered off and washed with water (3 × 20 mL), MeOH (2 × 10 mL) and dried in vacuo. Yield 0.223 g (50%), yellow solid, m.p. 156–159 °C. IR (KBr) νmax = 2968, 2868, 1597, 1497, 1347, 908 cm−1. 1H NMR (200 MHz, DMSO-d6) δ = 1.52–1.56 (4H, m, CH2 pyr), 2.68–2.73 (4H, m, CH2 pyr), 4.29 (4H, s, CH2 im), 7.21–7.44 (10H, m, H Ph) ppm; 13C NMR (50 MHz, DMSO-d6) δ = 25.0 (CH2), 50.1 (CH2), 51.0 (CH2), 98.6 (CCl2), 104.7 (CNO2), 122.8 (CH), 126.7 (CH), 129.2 (CH), 140.2, 140.6, 157.1 (NCN) ppm; MS m/z (Irel, %): 444 [M+] (2), 427 [M-OH]+ (2), 408 [M-HCl]+ (3), 373 [M-pyrrolidine]+ (22), 281 [M-(CCl2=C)-pyrrolidine+H]+ (20), 264 [M-(CCl2=C)-pyrrolidine-O]+ (85), 248 [M-(CCl2=C)-pyrrolidine-2O]+ (100); HRMS (ESI+) m/z calcd for C22H23N4O2Cl2 [M + H]+: 445.1193; found: 445.1190.
Synthesis of ethyl 1-(1,1-dichloro-3-{1-[(6-chloropyridin-3-yl)methyl]imidazolidin-2-ylidene}-3- nitroprop-1-en-2-yl)piperidine-4-carboxylate (12a) (General method). To a suspension of compound 11a (0.384 g, 1.0 mmol) and piperidine-4-carboxylic acid ethyl ester (0.393 g, 2.5 mmol) in 15 mL MeOH at r.t., a solution of sodium ethanolate (0.177 g, 2.6 mmol) in 5 mL MeOH was added, and stirred for 24 h at 35 °C. At r.t. 3 drops of conc. HCl were added before adding 50 mL cold water. The resulting precipitate was filtered off and washed with cold MeOH (2 × 7 mL) and water (3 × 10 mL). The product was dried in vacuo. Yield 0.303 g (60%), yellow solid, m.p. 60–62 °C. IR (KBr) νmax = 3319, 1726, 1561, 1319, 1177, 1043 cm−1. 1H NMR (200 MHz, DMSO-d6) δ = 1.16 (3H, t, J = 6.9 Hz, CH3 Et), 1.53–1.76 (4H, m, CH2 pip), 2.28–2.42 (1H, m, CH pip), 2.73 (4H, s, CH2 pip), 3.35 (2H, s, CH2 imi), 3.67 (2H, s, CH2 imi), 4.06 (2H, q, J = 6.9 Hz, CH2 Et), 4.37–4.47 (2H, m, pyr-CH2-imi), 7.56 (1H, d, J = 8.1 Hz, H pyr), 7.81 (1H, d, J = 8.2 Hz, H pyr), 8.36 (1H, s, H pyr), 9.43 (1H, s, NH) ppm; 13C NMR (50 MHz, DMSO-d6) δ = 14.3 (CH3), 28.4 (CH2), 40.5 (CH), 42.2 (CH2), 48.0 (CH2 pip), 48.4 (CH2), 49.8 (CH2), 60.1 (CH2), 102.8 (CNO2), 103.6 (CCl2), 124.4 (CH), 131.4, 138.8 (CH), 141.3, 148.9 (CH), 149.7, 160.1, 174.4 ppm; MS m/z (Irel, %): 503 [M+] (3), 467 [M − HCl]+ (3), 377 [M-CH2pyridine]+ (2), 254 [M-C2Cl2-piperidine]+ (18), 126 [CH2pyridine]+ (100); HRMS (ESI−) m/z calcd for C20H23N5O4Cl3 [M − H]−: 502.0821; found: 502.0818.
2-(1,1-Dichloro-3-{1-[(6-chloropyridin-3-yl)methyl]imidazolidin-2-ylidene}-3-nitroprop-1-en-2-yl)- 1,2,3,4-tetrahydroisoquinoline (12b). Same procedure as for 12a, using 11a (0.384 g, 1.00 mmol) and 1,2,3,4-tetrahydroisoquinoline (0.368 g, 2.50 mmol) and stirring for 4 h at 50 °C. Yield 85%, orange solid, m.p. 146–148 °C. IR (KBr) νmax = 3318, 2895, 1559, 1460, 1320, 749 cm−1. 1H NMR spectrum (200 MHz, CDCl3) δ = 2.70–3.16 (2H, m, CH2 pip), 3.30–3.55 (2H, m, CH2 pip), 3.59–3.90 (4H, m, CH2 imi), 4.08 (1H, d, J = 15.2 Hz, NCH2); 4.26–4.38 (2H, m, CH2 pip), 4.67 (1H, d, J = 15.2 Hz, NCH2), 6.91–6.97 (1H, m, H Ph), 7.07–7.15 (3H, m, H Ph), 7.32 (1H, d, J = 8.2 Hz, H pyr), 7.60 (1H, dd, J = 8.3 Hz, J = 2.5 Hz, H pyr), 8.30 (1H, d, J = 2.4 Hz, H pyr), 9.56 (1H, s, NH) ppm. 13C NMR (50 MHz, CDCl3) δ = 29.7 (CH2), 41.7 (CH2), 48.7 (CH2), 49.6 (CH2), 49.9 (CH2), 51.3 (CH2), 105.1, 105.7, 124.7 (CH), 125.7 (CH), 126.7 (CH), 126.3 (CH), 128.8 (CH), 129.6, 133.9, 134.9, 138.2 (CH), 139.7, 148.9 (CH), 151.6, 160.2 ppm; MS m/z (Irel, %): 479 [M+] (2), 443 [M − HCl]+ (3), 407 [M-2(HCl)]+ (5), 254 [M-C2Cl2-isoquinoline + H]+ (7), 126 [Cl-pyr-CH2]+ (100); HRMS (ESI−) m/z calcd for C21H19N5O2Cl3 [M − H]−: 478.0610; found: 478.0606.
2-[(1,1-Dichloro-3-{1-[(6-chloropyridin-3-yl)methyl]imidazolidin-2-ylidene}-3-nitroprop-1-en-2-yl)sulfanyl]- ethanol (12c). Same procedure as for 12a, using 11a (0.384 g, 1.00 mmol), 2-mercaptoethanol (0.086 g, 1.1 mmol) and sodium ethanolate (0.082 g, 1.2 mmol) in EtOH. The mixture was stirred for 12 h at 40 °C. Yield 0.268 g (63%), orange solid, m.p. 185–187 °C. IR (KBr) νmax = 3220, 2974, 1575, 1299, 1125, 844 cm−1. 1H NMR spectrum (200 MHz, DMSO-d6) δ = 2.77 (2H, t, J = 6.4 Hz, SCH2), 3.53 (2H, t, J = 6.4 Hz, CH2OH), 3.73 (4H, s, CH2), 4.30 (2H, s, NCH2), 4.70 (1H, s, OH), 7.53 (1H, d, J = 8.3 Hz), 7.75 (1H, dd, J = 8.3 Hz, J = 2.5 Hz), 8.33 (1H, d, J = 2.3 Hz), 9.55 (1H, s, NH) ppm; 13C NMR (50 MHz, DMSO-d6) δ = 34.9 (CH2), 42.4 (CH2), 48.9 (CH2), 50.8 (CH2), 60.1 (CH2), 102.8 (CNO2), 115.1 (CCl2), 124.3 (CH), 131.6, 131.9, 138.1 (CH), 148.0 (CH), 149.5, 159.7 ppm; MS m/z (Irel, %): 424 [M+] (1), 254 [M-C2Cl2-mercaptoethanol + H]+ (27), 235 [M − C2Cl2S-H2O]+ (12), 126 [Cl-pyr-CH2]+ (100); HRMS (ESI−) m/z calcd for C14H14N4O3SCl3 [M − H]−: 422.9858; found: 422.9854.
1,1-Dichloro-3-{3-[(6-chloropyridin-3-yl)methyl]-1,3-oxazolidin-2-ylidene}-N,N-dimethyl-3-nitroprop-1-en-2-amine (12d). Same procedure as for 12a, using pyridine 11b (0.385 g, 1.00 mmol) and dimethylamine (0.366 g, 5.00 mmol) and stirring at r.t. for 5 h. Yield 70%, yellow solid, m.p. 127–129 °C. IR (KBr) νmax = 2868, 1597, 1561, 1298, 1118, 903 cm−1. 1H NMR (400 MHz, DMSO-d6) δ = 2.59 (6H, s, NCH3), 3.96–4.10 (2H, m, CH2 oxa), 4.52–4.81 (4H, m, CH2 oxa, NCH2), 7.59 (1H, d, J = 7.7 Hz, H pyr), 7.91 (1H, d, J = 7.5 Hz, H pyr), 8.41 (1H, s, H pyr) ppm; 13C NMR (100 MHz, DMSO-d6) δ = 42.0 (CH3), 49.3 (CH2), 50.8 (CH2), 68.2 (CH2), 104.0, 105.1, 124.4 (CH), 130.0, 139.8 (CH), 142.1, 149.8 (CH), 150.1, 165.4 ppm; MS m/z (Irel, %): 392 [M+] (1), 311 [M − Cl-NO2]+ (1), 254 [M − C2Cl2-N(CH3)2]+ (2), 126 [Cl-pyr-CH2]+ (100); HRMS (ESI+) m/z calcd for C14H16N4O3Cl3 [M + H]+: 393.0283; found: 393.0280.
Synthesis of 1-[(4-bromo-1,3,4-trichloro-2-nitrobuta-1,3-dien-1-yl)sulfanyl]-4-fluorobenzene (13). To a solution of nitrodiene 2 (0.316 g, 1.0 mmol) in dry DCM (15 mL) at 0 °C, a solution of 4-fluorothiophenole (0.128 g, 1.0 mmol) in 5 mL of dry DCM was added and stirred for 3 h. After reaching r.t. and further stirring for 24 h the mixture was concentrated and water (20 mL) was added before extraction with chloroform (3 × 10 mL). The product was purified by column chromatography using petroleum ether-ethyl acetate (10:1) and dried in vacuo. A 1:1 mixture of isomers was obtained. Yield 0.301 g (74%), yellow solid, m.p. 98–99 °C. IR (ATR) νmax = 1587, 1527, 1292, 1223, 815, 526 cm−1. 1H NMR (400 MHz, CDCl3) δ = 7.11–7.25 (2H, m, H Ph), 7.48–7.64 (2H, m, H Ph) ppm; 13C NMR (101 MHz, CDCl3) δ = 115.2, 116.4 (CClBr), 117.0, 117.2 (CH), 124.0, 124.1, 124.2, 124.5, 138.3, 138.4 (CH), 157.3, 157.8 (CClS), 163.4, 165.9 (CF) ppm, CNO2 could not be detected; MS m/z (Irel, %): 405 [M+] (1), 370 [M − Cl]+ (1), 326 [M − Br]+ (6), 291 [M − Br-Cl]+ (3), 127 [S-Ph-F]+ (100); HRMS (ESI+) m/z calcd for C10H5NO2Cl3SBrF [M + H]+: 405.8269; found: 405.8286.
Synthesis of 5-(((2E)-2-(3-bromo-2,3-dichloro-1-nitroallylidene)imidazolidin-1-yl)methyl)-2-chloropyridine (14) and 1,1’-[(4-bromo-3,4-dichloro-2-nitrobuta-1,3-diene-1,1-diyl)disulfanediyl]bis(4-fluorobenzene) (15). A solution of N-[(6-chloropyridin-3-yl)methyl]ethane-1,2-diamine 10a (0.515 g, 3.0 mmol) in MeOH (5 mL) was added to a suspension of diene 13 (0.407 g, 1.0 mmol, a 1: 1 mixture of isomers ) in MeOH (10 mL) at –10 °C and stirred for 1 h at the same temperature. The precipitated bisthiodiene 15 was filtered off, washed with water and cold MeOH (2 × 3 mL), and dried under reduced pressure to yield diene 15. The collected filtrates were carefully neutralized by means of hydrochloric acid and stirred with additional 50 mL of water. Again, the solid was filtered off, and then washed with water and diethyl ether (3 × 5 mL). Recrystallization from methanol gave imidazolidine 14.
Imidazolidine14. A 1: 1 mixture of isomers was obtained. Yield 0.189 g (44%), white solid, m.p. 172–173 °C. IR (KBr) νmax = 3312, 3055, 2917, 1588, 1420, 1137, 824 cm−1. 1H NMR (200 MHz, DMSO-d6) δ = 3.67–3.90 (4H, m, CH2 imi), 4.46-4.60 (2H, m, NCH2-pyr), 7.48–7.60 (1H, m, H pyr), 7.71–7.81 (1H, m, H pyr), 8.24–8.39 (1H, m, H pyr), 9.41 (1H, s, NH) ppm; 13C NMR (50 MHz, DMSO-d6) δ = 42.7 (CH2 imi), 49.4 (NCH2-pyr), 50.8 (CH2 imi), 103.4, 105.2 (CNO2), 113.0, 113.8 (CClBr), 124.3, 124.4 (CH), 126.6, 128.7, 131.4, 131.5, 137.9, 138.0 (CH), 147.9, 148.0, 149.6, 159.6, 159.7 (NCN) ppm; MS m/z (Irel, %): 426 [M+] (1), 380 [M-NO2]+ (1), 347 [M-Br]+ (20), 314 [M-pyr]+ (3), 312 [M-Br-Cl]+ (3), 254 [M-C2Cl2Br+H]+ (35), 126 [Cl-pyr-CH2]+ (100); HRMS (ESI+) m/z calcd for C12H9N4O2Cl3Br [M-H]−: 424.8980; found: 424.8967.
Bisthiodiene15. A 1: 1 mixture of isomers was obtained. Yield 0.150 g (30%), yellow solid, m.p. 141–142 °C. IR (ATR) νmax = 1589, 1489, 1293, 1230, 823, 505 cm−1. 1H NMR (400 MHz, CDCl3) δ = 6.84–7.01 (6H, m, H Ph), 7.05–7.17 (2H, m, H Ph) ppm; 13C NMR (101 MHz, CDCl3) δ = 115.1, 117.2 (CClBr), 116.3–116.7 (CH Ph), 123.4, 125.5, 126.1 (CPh-S), 133.4, 133.5, 136.5, 136.6, 136.7 (C Ph), 138.3, 138.4 (CNO2), 159.1, 159.5 (CSS), 163.1 (CF, JC,F = 251.5 Hz), 163.1 (CF, JC,F = 252.3 Hz) ppm; MS, m/z (Irel, %): 497 [M+] (2), 462 [M − Cl]+ (2), 418 [M − Br]+ (30), 383 [M − Br-Cl]+ (3), 372 [M − Br-NO2]+ (4), 127 [S-Ph-F]+ (100); HRMS (ESI+) m/z calcd for C16H9NO2BrCl2F2S2 [M + H]+: 497.8598; found 497.8588.
Alternative synthesis of diene15: To a solution of bromonitrodiene 2 (0.316 g, 1.0 mmol, a 47: 53 mixture of isomers) and 4-fluorobenzenethiol (0.256 g, 2.0 mmol) in MeOH (10 mL) at 0 °C, a solution of sodium methanolate (0.108 g, 2.0 mmol) in MeOH (5 mL) was added. The solution was stirred for 3 h at 0 °C and at r.t. for 1 d. Subsequently, the solution was concentrated and the precipitate filtered off and washed with diluted HCl (5 mL) and cold MeOH (2 × 2 mL). A mixture of both isomers was obtained. Yield of bisthiodiene 15 is 0.414 g (83%, a 1:1 mixture of isomers).
N,N’-Bis[(2-chloro-1,3-thiazol-5-yl)methyl]ethane-1,2-diamine (
16) was prepared according to the literature [
13] in 70% yield.
Synthesis of 5,5’-{[2-(3-bromo-2,3-dichloro-1-nitroprop-2-en-1-ylidene)imidazolidine-1,3-diyl] dimethanediyl}-bis(2-chloro-1,3-thiazole) (17). To a solution of diene 2 (0.166 g, 0.5 mmol) in a mixture of MeOH and water (10 mL, 10: 1) at 0 °C N,N’-bis[(2-chloro-1,3-thiazol-5-yl)methyl]ethane-1,2-diamine (16) (0.170 g, 0.5 mmol) and Na2CO3 (0.112 g, 2.0 mmol) were added carefully. After 1 h at 0 °C, the mixture was allowed to reach r.t. and stirred for another 5 h. Subsequently, the mixture was concentrated in vacuo and the resulting precipitate was filtered off and washed with cold MeOH (2 × 5 mL), water (2 × 5 mL) and again MeOH (1 × 5 mL). A 1: 1 mixture of isomers was obtained. Yield 0.252 g (89%), white solid, mp 162–163 °C. IR (ATR) νmax = 1569, 1533, 1325, 1143, 1046, 770 cm−1. 1H NMR (400 MHz, DMSO-d6) δ = 3.73–3.96 (4H, m, CH2), 4.58–4.80 (4H, m, imi-CH2), 7.69 (2H, s, CH) ppm; 13C NMR (100 MHz, DMSO-d6) δ = 45.2, 45.4 (CH2 imi), 45.2, 45.4 (CH2 imi), 47.2, 47.3 (imi-CH2), 98.1, 99.7 (CNO2), 109.7, 110.6 (CClBr), 127.0, 128.8 (CCl), 133.6, 133.7 (SC thiaz), 141.8, 141.9 (NC thiaz), 151.7, 151.8 (NCS), 161.6, 161.8 (NCN) ppm; MS m/z (Irel, %): 563 [M+] (1), 527 [M − Cl]+ (1), 484 [M − Br]+ (1), 132 [thiazole-Cl-CH2]+ (100); HRMS (ESI+) m/z calcd for C14H11N5O2Cl4BrS2 [M + H]+: 563.8286; found: 563.8284.
Ethyl [(1,3,4,4-tetrachloro-2-nitrobuta-1,3-dien-1-yl)sulfanyl]acetate (
18) was prepared according to the literature [
18] in 80% yield.
Synthesis of 3-[4-(propan-2-yl)phenyl]-2-(2,3,3-trichloro-1-nitroprop-2-en-1-ylidene)-1,3-thiazolidin-4-one (19a) (General method). To a suspension of the acetate 18 (0.355 g, 1.0 mmol) in MeOH (3 mL) at −18 °C a solution of 4-(propan-2-yl)aniline (0.297 g, 2.2 mmol) in MeOH (3 mL) was added dropwise within 10 min. The mixture was stirred for 3 h at −18 °C and 12 h at r.t. before it was concentrated. The resulting precipitate was filtered off and washed with cold MeOH (1 × 5 mL), water (2 × 5 mL) and again MeOH (2 × 5 mL). The product was dried in vacuo. Yield 0.298 g (73%), beige solid, m.p. 208–209 °C. IR (ATR) νmax = 2962, 1758, 1520, 1289, 1167, 686 cm−1. 1H NMR (400 MHz, DMSO-d6) δ = 1.22 (3H, d, J = 6.9 Hz, i-Pr), 1.23 (3H, d, J = 6.9 Hz, i-Pr), 2.95 (1H, sep, J = 6.8 Hz, i-Pr), 4.13 (1H, d, J = 18.7 Hz, SCH2), 4.17 (1H, d, J = 18.7 Hz, SCH2), 7.28–7.33 (2H, m, H Ar), 7.34–7.40 (2H, m, H Ar) ppm; 13C NMR (100 MHz, DMSO-d6) δ = 23.6, 23.8 (CH3), 32.5 (CH2), 33.6 (CH i-Pr), 121.3 (CCl2), 121.4 (CNO2), 126.9 (CH), 127.3 (CH), 127.4 (CH), 128.4, 128.8 (CH), 132.1, 150.6, 165.8 (NCS), 174.1 (C=O) ppm; MS m/z (Irel, %): 406 [M+] (10), 389 [M-OH]+ (5), 371 [M-Cl]+ (5), 278 [M-C2Cl3+H]+ (15), 261 [M-C2Cl3-O]+ (100); MS (ESI−) m/z calcd for C15H12N2O3Cl3S 405.0; found: 405.0.
2-(2,3,3-Trichloro-1-nitroprop-2-en-1-ylidene)-3-[4-(trifluoromethyl)phenyl]-1,3-thiazolidin-4-one (19b). Same procedure as for 19a, using 4-(trifluoromethyl)aniline (0.355 g, 2.2 mmol) at 0 °C for 3 h and 15 h at r.t.. Yield 0.330 g (76%), yellowish solid, m.p. 228–229 °C. IR (ATR) νmax = 3058, 1737, 1515, 1285, 1170, 691 cm−1. 1H NMR (400 MHz, DMSO-d6) δ = 4.13 (1H, d, J = 18.7 Hz, SCH2), 4.20 (1H, d, J = 18.7 Hz, SCH2), 7.67–7.74 (2H, m, H Ar), 7.91–7.98 (2H, m, H Ar) ppm; 13C NMR (400 MHz, DMSO-d6) δ = 32.8 (CH2), 121.2 (CNO2), 121.3 (CCl2), 123.9 (JC,F = 272.4 Hz, CF3), 126.2 (JC,F = 3.9 Hz, CH), 126.6 (JC,F = 4.2 Hz, CH), 128.9 (CH), 129.1, 130.2 (CH), 130.7 (JC,F = 32.3 Hz, CF3-C), 138.1, 165.4 (NCS), 173.9 (C=O) ppm; MS m/z (Irel, %): 432 [M+] (3); 397 [M − Cl]+ (12); 304 [M-C2Cl3+H]+ (30); 287 [M-C2Cl3-O]+ (93); 145 (100); HRMS (ESI−) m/z calcd for C13H5N2O3Cl3SF3 [M − H]−: 430.9044; found: 430.9062.
Synthesis of 3-(hydrazinylidenemethyl)-4-nitro-N-[4-(propan-2-yl)phenyl]-1H-pyrazol-5-amine (20a). To a stirred suspension of thiazolidinone 19a (0.408 g, 1.0 mmol) in MeOH (10 mL) at −18 °C, a solution of hydrazine hydrate (0.400 g, 8.0 mmol) in MeOH (5 mL) was added dropwise within 5 min. Subsequently, the mixture was allowed to reach 0 °C. After 5 h, and 18 h at r.t., the solution was concentrated and neutralized to pH 7 with HCl (5%), then extracted with ethyl acetate (3 × 10 mL). The combined organic phases were purified over a short column chromatography using ethyl acetate-petroleum ether (1:1). Subsequently, the obtained solution was concentrated and the resulting precipitate was filtered and washed with diethyl ether (2 × 3 mL). Yield 0.150 g (52%), red solid, m.p. 194–195 °C. IR (ATR) νmax = 3365, 2956, 1597, 1562, 1461, 1350, 579 cm−1. 1H NMR (400 MHz, DMSO-d6) δ = 1.18 (6H, d, J = 6.9 Hz, CH3 i-Pr), 2.83 (1H, sep, J = 6.8 Hz, CH i-Pr), 7.16 (2H, d, J = 8.5 Hz, H Ar), 7.61 (2H, d, J = 8.4 Hz, H Ar), 8.07 (1H, s, NCH), 8.15 (2H, s, N-NH2), 8.53 (1H, s, NH anil), 13.29 (1H, s, NH pyr) ppm; 13C NMR (100 MHz, DMSO-d6) δ =24.2 (CH3), 32.9 (CH i-Pr), 117.4 (CNO2), 118.0 (2 × CH Ph), 122.7 (NCH), 126.7 (2 × CH Ph), 138.3, 138.7, 141.4, 147.4 (NNHC) ppm; MS m/z (Irel, %): 288 [M+] (60), 273 [M − CH3]+ (100), 256 [M-CH3-OH]+ (5), 245 [M-i-Pr]+ (8), 169 [M-NH-Ph-i-Pr]+ (5); HRMS (ESI−) m/z calcd for C13H15N6O2 [M − H]−: 287.1261; found: 287.1272.
Synthesis of 3-(hydrazinylidenemethyl)-4-nitro-N-[4-(trifluoromethyl)phenyl]-1H-pyrazol-5-amine (20b). To a stirred suspension of thiazolidinone 19b (0.434 g, 1.0 mmol) in MeOH (10 mL) at −18 °C, a solution of hydrazine hydrate (0.250 g, 5.0 mmol) in MeOH (5 mL) was added dropwise within 5 min. Subsequently, the mixture was allowed to reach 0 °C. After 5 h with stirring at 0 °C and 18 h at r.t., the solution was concentrated and the resulting precipitate filtered off. Washing the product with cold MeOH (2 × 5 mL) and drying in vacuo yielded the pyrazole 20b. Yield 0.145 g (46%), yellow solid, m.p. 248–250 °C. IR (ATR) νmax = 3440, 1602, 1516, 1327, 1160, 832 cm−1. 1H NMR (400 MHz, DMSO-d6) δ = 7.63 (2H, d, J = 8.7 Hz, H Ph), 7.91 (2H, d, J = 8.6 Hz, H Ph), 8.07 (1H, s, NCH), 8.21 (2H, s, NH2), 8.97 (1H, s, Ph-NH), 13.45 (1H, s, NH Pyr) ppm; 13C NMR (100 MHz, DMSO-d6) δ = 117.6 (2 × CH Ph), 117.8 (CNO2), 121.0 (JC,F = 32.1 Hz, CCF3), 122.2 (JC,F = 270.7 Hz, CF3), 122.5 (NCH), 126.2 (JC,F = 3.7 Hz, 2 × CH Ph), 139.0, 144.2, 146.4 (NNHC) ppm;. MS m/z (Irel, %): 314 [M+] (2), 270 [M-CH2=N-NH2]+ (3), 254 [M-CH2=N-NH2-O]+ (14), 161 [CF3-Ph-NH2]+ (60), 142 [CF2-Ph-NH2]+ (27), 96 (100); HRMS (ESI−) m/z calcd for C11H8N6O2F3 [M − H]−: 313.0665; found: 313.0666.
Synthesis of 5-(furan-2-ylmethylidene)-3-[4-(propan-2-yl)phenyl]-2-(2,3,3-trichloro-1-nitroprop-2-en-1- ylidene)-1,3-thiazolidin-4-one (21a) (General method). To a suspension of thiazolidinone 19a (0.408 g, 1.0 mmol) in acetic acid (15 mL) furan-2-carbaldehyde (0.115 g, 1.2 mmol) and triethylamine (0.152 g, 1.5 mmol) were added. The resulting mixture was refluxed for 4 h. After concentration of this mixture in vacuo to a volume of about 3 mL and cooling to r.t., the resulting precipitate was filtered off and washed with water (2 × 10 mL) and cold MeOH (1 × 2 mL). The product was dried in vacuo. Yield 0.466 g (96%), orange solid, m.p. 228–229 °C. IR (ATR) νmax = 2958, 1718, 1598, 1519, 1167, 764 cm−1. 1H NMR (400 MHz, DMSO-d6) δ = 1.24 (3H, d, J = 6.9 Hz, CH3 i-Pr), 1.25 (3H, d, J = 6.9 Hz, CH3 i-Pr), 2.97 (1H, sep, J = 6.8 Hz, CH i-Pr), 6.86 (1H, dd, J = 1.8 Hz, J = 3.6 Hz, H fur), 7.34 (1H, d, J = 3.5 Hz, H fur), 7.37–7.45 (4H, m, H Ar), 7.90 (1H, s, =CH), 8.30 (1H, d, J = 1.7 Hz, H fur) ppm; 13C NMR (100 MHz, DMSO-d6) δ = 23.6 (CH3), 23.9 (CH3), 33.6 (CH i-Pr), 114.4 (CH), 116.7, 120.8 (CNO2), 121.0 (CCl2), 121.8 (CH), 122.8 (CH), 126.9 (CH), 127.3 (CH), 127.4 (CH), 128.8 (CH), 128.8 (CCl-CCl2), 132.1, 149.4 (CH), 149.9, 150.8, 158.5 (NCS), 166.5 (C=O) ppm; MS m/z (Irel, %): 484 [M+] (6), 449 [M-Cl]+ (2), 339 [M-C2Cl3-O]+ (75), 325 [M-furan-CH=C=S-Cl]+ (7), 124 [furan-CH=C=S]+ (100). HRMS (ESI+) m/z calcd for C20H15N2O4SCl3Na [M + Na]+: 506.9716; found: 506.9712.
3-[4-(Propan-2-yl)phenyl]-5-(thiophen-2-ylmethylidene)-2-(2,3,3-trichloro-1-nitroprop-2-en-1-ylidene) -1,3-thiazolidin-4-one (21b). Same procedure as for 21a, using thiophene-2-carbaldehyde (0.135 g, 1.2 mmol). After filtration of the product, it was further purified by column chromatography using chloroform. Yield 0.462 g (92%), dark yellow solid, m.p. 221–223 °C. IR (ATR) νmax = 2963, 1717, 1586, 1525, 1258, 729 cm−1. 1H NMR (400 MHz, CDCl3) δ = 1.29 (6H, d, J = 6.7 Hz, CH3 i-Pr), 2.99 (1H, sep, J = 6.9 Hz, CH i-Pr), 7.16–7.20 (1H, m, H Ar), 7.24–7.27 (1H, m, H thien), 7.27–7.30 (1H, m, H Ar), 7.35–7.39 (2H, m, H Ar), 7.56 (1H, d, J = 3.7 Hz, H thien), 7.78 (1H, d, J = 5.0 Hz, H thien), 8.12 (1H, s, =CH) ppm; 13C NMR (100 MHz, CDCl3) δ = 23.5 (CH3), 23.7 (CH3), 34.1 (CH i-Pr), 117.6, 120.6 (CCl2), 122.6 (CNO2), 126.8 (CH), 127.4 (CH), 127.8 (CH), 127.9 (CH), 129.1 (CH), 129.6 (CH), 129.7 (CClCCl2), 131.6, 133.6 (CH), 134.8 (CH), 137.6, 151.4 (i-Pr-C), 155.3 (NCS), 166.7 (C=O) ppm; MS m/z (Irel, %): 500 [M+] (3), 465 [M-Cl]+ (2), 372 [M-C2Cl3+H]+ (4), 355 [M-C2Cl3-O]+ (73), 140 [thienyl-C=CS]+ (100). HRMS (ESI+) m/z calcd for C20H15N2O3Cl3S2Na [M + Na]+: 522.9487; found: 522.9482.
5-[(1-Methyl-1H-pyrrol-2-yl)methylidene]-3-[4-(propan-2-yl)phenyl]-2-(2,3,3-trichloro-1-nitroprop-2 -en-1-ylidene)-1,3-thiazolidin-4-one (21c). Same procedure as for 21a, using 1-methyl-1H-pyrrole-2-carbaldehyde (0.328 g, 3.0 mmol) and triethylamine (0.304 g, 3.0 mmol). The mixture was refluxed for 6 h. Yield 0.459 g (92%), red solid, m.p. 244–246 °C. IR (ATR) νmax = 2986, 1700, 1584, 1520, 1273, 745 cm−1. 1H NMR (400 MHz, DMSO-d6) δ = 1.24 (3H, d, J = 6.9 Hz, CH3 i-Pr), 1.25 (3H, d, J = 6.9 Hz, CH3 i-Pr), 2.98 (1H, sep, J = 6.9 Hz, CH i-Pr), 3.83 (3H, s, NCH3), 6.47 (1H, dd, J = 2.6 Hz, J = 4.2 Hz, H pyrr), 6.88 (1H, dd, J = 1.1 Hz, J = 4.2 Hz, H pyrr), 7.36–7.44 (5H, m, 4H Ph, H pyrr), 7.84 (1H, s, =CH) ppm; 13C NMR (100 MHz, DMSO-d6) δ = 23.6 (CH3), 23.9 (CH3 i-Pr), 33.6 (NCH3), 34.3 (CH i-Pr), 111.5 (CCl2), 111.9 (CH), 118.0 (CH), 120.2 (CNO2), 121.2, 124.9 (CH), 126.9 (CH), 127.4 (2 × CH), 128.0, 128.6, 128.7 (CH), 131.8 (CH), 132.2, 150.7 (i-Pr-C), 157.5 (NCS), 166.5 (C=O) ppm; MS m/z (Irel, %): 497 [M+] (10), 462 [M-Cl]+ (1), 361 [M-pyrrol-CH=C=S+H]+ (3), 352 [M-C2Cl3-CH4]+ (25), 137 [Me-pyrrol-CH=C=S]+ (100); HRMS (ESI+) m/z calcd for C21H18N3O3Cl3SNa [M + Na]+: 520.0032; found: 520.0018.
5-(Furan-2-ylmethylidene)-2-(2,3,3-trichloro-1-nitroprop-2-en-1-ylidene)-3-[4-(trifluoromethyl) phenyl]-1,3-thiazolidin-4-one (21d). Same procedure as for 21a, using thiazolidinone 21b (0.433 g, 1.0 mmol), furan-2-carbaldehyde (0.115 g, 1.2 mmol) and triethylamine (0.152 g, 1.5 mmol). Yield 0.374 g (73%), orange solid, m.p. 227–229 °C. IR (ATR) νmax = 3130, 1720, 1525, 1286, 1162, 622 cm−1. 1H NMR (400 MHz, CDCl3) δ = 6.67 (1H, dd, J = 1.8 Hz, J = 3.6 Hz, H fur), 6.98 (1H, d, J = 3.6 Hz, H fur), 7.41–7.45 (1H, m, H Ar), 7.50–7.55 (1H, m, H Ar), 7.72 (1H, s, =CH), 7.78–7.84 (2H, m, H Ar), 7.85 (1H, d, J = 1.8 Hz, H fur) ppm; 13C NMR (100 MHz, CDCl3) δ = 113.9 (CH), 116.9, 120.4 (CH), 120.6 (CCl2), 123.4 (JC,F = 272.8 Hz, CF3), 122.5 (CNO2), 122.9 (CH), 126.4 (JC,F = 3.7 Hz, CH), 126.9 (JC,F = 3.7 Hz, CH), 127.8 (CH), 128.8 (CH), 130.2 (CClCCl2), 132.6 (JC,F = 33.0 Hz, CF3C), 137.2, 148.0 (CH), 149.9, 156.0, 166.4 (C=O)ppm; MS m/z (Irel, %): 510 [M+] (3), 475 [M-Cl]+ (1), 429 [M-Cl-NO2]+ (1), 382 [M-C2Cl3+H]+ (5), 365 [M-C2Cl3-O]+ (37), 145 [Ph-CF3]+ (24), 124 [furan-CH=C=S]+ (100); HRMS (ESI+) m/z calcd for C18H8N2O4Cl3SF3Na [M + Na]+: 532.9120; found: 532.9117.
5-(Thiophen-2-ylmethylidene)-2-(2,3,3-trichloro-1-nitroprop-2-en-1-ylidene)-3-[4-(trifluoromethyl)- phenyl]-1,3-thiazolidin-4-one (21e). Same procedure as for 21a, using thiazolidinone 21b (0.433 g, 1.0 mmol), thiophene-2-carbaldehyde (0.135 g, 1.2 mmol) and triethylamine (0.152 g, 1.5 mmol). After filtration, the product was purified by column chromatography using chloroform. The fraction containing the product was dried, suspended in MeOH (2 mL), filtered, and the solid washed with cold MeOH (1 × 2 mL). Yield 0.449 g (85%), orange solid, m.p. 245–246 °C. IR (ATR) νmax = 3076, 1699, 1522, 1281, 1126, 730 cm−1. 1H NMR (400 MHz, CDCl3) δ = 7.28 (1H, dd, J = 3.8 Hz, J = 5.0 Hz, H thien), 7.43–7.45 (1H, m, H Ar), 7.52–7.54 (1H, m, H Ar), 7.58 (1H, d, J = 3.7 Hz, H thien), 7.79–7.84 (2H, m, H Ar), 7.84–7.86 (1H, m, H thien), 8.15 (1H, s, =CH) ppm; 13C NMR (100 MHz, CDCl3) δ = 116.9, 120.4 (CCl2), 123.3 (JC,F = 271.9 Hz, CF3), 126.4 (JC,F = 3.9 Hz, CH), 126.9 (JC,F = 4.2 Hz, CH), 127.8 (CH), 128.8 (CH), 129.3 (CH), 130.3 (CH), 130.4 (CClCCl2), 132.3 (JC,F = 32.0 Hz, CF3-C), 134.1 (CH), 135.3 (CH), 137.2, 137.4, 154.5 (NCS), 166.2 (C=O) ppm, CNO2 could not be detected; MS m/z (Irel, %): 526 [M+] (4), 491 [M-Cl]+ (1), 398 [M-C2Cl3+H]+ (5), 381 [M-C2Cl3-O]+ (48), 140 [thienyl-C=CS]+ (100); HRMS (ESI+) m/z calcd for C18H8N2O3Cl3S2F3Na [M + Na]+: 548.8892; found, m/z: 548.8888.
5-[(1-Methyl-1H-pyrrol-2-yl)methylidene]-2-(2,3,3-trichloro-1-nitroprop-2-en-1-ylidene)-3- [4-(tri fluoro-methyl)-phenyl]-1,3-thiazolidin-4-one (21f). Same procedure as for 21a, using thiazolidinone 21b (0.433 g, 1.0 mmol), 1-methyl-1H-pyrrole-2-carbaldehyde (0.262 g, 2.4 mmol) and triethylamine (0.152 g, 1.5 mmol). The mixture was refluxed for 8 h. Yield 0.289 g (55%), red solid, shows fluorescence, m.p. 241–242 °C. IR (ATR) νmax = 1688, 1589, 1521, 1269, 1064, 620 cm−1. 1H NMR (400 MHz, DMSO-d6) δ = 3.84 (3H, s, NCH3), 6.48 (1H, dd, J = 2.3 Hz, J = 4.1 Hz, H pyrr), 6.90 (1H, dd, J = 1.3 Hz, J = 4.1 Hz, H pyrr), 7.39 (1H, dd, J = 1.3 Hz, J = 2.3 Hz, H pyrr), 7.78–7.86 (2H, m, H Ar), 7.87 (1H, s, =CH), 7.94–8.00 (2H, m , H Ph) ppm; 13C NMR (100 MHz, DMSO-d6) δ = 34.3 (NCH3), 111.3, 112.1 (CH), 118.2 (CH), 120.0 (CNO2), 121.2 (CCl2), 123.9 (JC,F = 273.1 Hz, CF3), 125.1 (CH), 126.2 (CH), 126.7 (CH), 128.0, 129.0 (CH), 129.2, 130.1 (CH), 130.8 (JC,F = 32.3 Hz, CCF3), 132.0 (CH), 138.2, 157.2 (NCS), 166.2 (C=O) ppm; MS m/z (Irel, %): 523 [M+] (5), 458 [M-F-NO2]+ (2), 378 [M-Ph-CF3]+ (12), 287 (100), 145 [Ph-CF3]+ (90); HRMS (ESI+) m/z calcd for C19H11N3O3Cl3SF3Na [M + Na]+: 545.9436; found: 545.9433.
1-Methyl-4-nitro-3-(morpholin-4-yl)-1H-pyrazole-5-carbaldehyde (
22) was prepared according to the literature [
26] in 48% yield.
Synthesis of 5-{[3-(Morpholin-4-yl)-4-nitro-1H-pyrazol-5-yl]methylidene}-3-[4-(propan-2-yl)phenyl]-2-(2,3,3-trichloro-1-nitropropylidene)-1,3-thiazolidin-4-one (23a) (General method). To a suspension of thiazolidinone 19a (0.408 g, 1.0 mmol) in acetic acid (10 mL), a solution of 1-methyl-3-(morpholin-4-yl)-4-nitro-1H-pyrazole-5-carbaldehyde (0.240 g, 1.0 mmol) in acetic acid (5 mL) and triethylamine (0.152 g, 1.5 mmol) was added. Subsequently, the mixture was refluxed for 4 h. After cooling, the solvent was removed and MeOH (5 mL) added. The resulting precipitate was filtered off and washed with cold MeOH (2 × 5 mL). The product was dried in vacuo. Yield 0.592 g (94%), orange solid, mixture of two isomers (100: 16), m.p. 212–213 °C. IR (ATR) νmax = 2961, 1735, 1535, 1290, 1178, 683 cm−1. 1H NMR major isomer (400 MHz, DMSO-d6) δ = 1.21 (6H, d, J = 6.9 Hz, CH3 i-Pr), 2.99 (1H, sep, J = 6.8 Hz, CH i-Pr), 3.22–3.28 (4H, m, NCH2), 3.72–3.78 (4H, m, OCH2), 3.85 (3H, s, NCH3), 7.33–7.53 (4H, m, H Ar), 7.90 (1H, s, =CH) ppm; 13C NMR major isomer (100 MHz, DMSO-d6) δ = 23.5, 23.8 (CH3 i-Pr), 33.6 (CH i-Pr), 39.0 (NCH3), 49.9 (NCH2), 65.9 (OCH2), 120.1, 121.4 (CH), 121.8, 123.5 (CNO2), 127.0 (CH), 127.4 (2 × CH), 128.9 (CH), 129.5, 130.9, 131.7, 137.4, 151.1 (C-i-Pr), 153.2 (N-N-C-N), 156.5 (NCS), 165.1 (C=O) ppm; MS m/z (Irel, %): 628 [M+] (55), 613 [M-CH3]+ (83), 598 [M-2(CH3)]+ (22), 582 [M-NO2]+ (20), 499 [M-C2Cl3]+ (10), 146 (100); HRMS (ESI+) m/z calcd for C24H23N6O6Cl3SNa [M + Na]+: 651.0363; found: 651.0360.
5-{[3-(Morpholin-4-yl)-4-nitro-1H-pyrazol-5-yl]methylidene}-2-(2,3,3-trichloro-1-nitropropylidene) -3-[4-(trifluoromethyl)phenyl]-1,3-thiazolidin-4-one (23b). Same procedure as for 23a, using thiazolidinone 19b (0.434 g, 1.0 mmol), 1-methyl-3-(morpholin-4-yl)-4-nitro-1H-pyrazole-5-carbaldehyde (0.240 g, 1.0 mmol) and triethylamine (0.152 g, 1.5 mmol). The mixture was refluxed for 2 h. Yield 0.636 g (97%), mixture of two isomers (100: 17), orange solid, m.p. 226–227 °C. IR (ATR) νmax = 1733, 1538, 1291, 1171, 1065, 630 cm−1. 1H NMR major isomer (400 MHz, DMSO-d6) δ = 3.22–3.29 (4H, m, NCH2), 3.72–3.79 (4H, m, OCH2), 3.85 (3H, s, NCH3), 7.81–8.04 (4H, m, 4H Ar), 7.93 (1H, s, =CH) ppm; 13C NMR major isomer (100 MHz, DMSO-d6) δ = 39.1 (NCH3), 49.9 (NCH2), 65.9 (OCH2), 120.1, 121.6 (pyr-CH), 121.7 (CH), 123.9 (JC,F = 272.7 Hz, CF3), 123.6 (CNO2), 126.3 (JC,F = 3.9 Hz, CH), 126.8 (JC,F = 3.9 Hz, CH), 129.0 (CH), 130.2, 130.3 (CH), 130.8, 131.2 (JC,F = 32.1 Hz, CCF3), 137.3, 137.7, 153.2, 156.2 (NCS), 164.9 (C=O) ppm; MS m/z (Irel, %): 654 [M+] (35), 639 [M − CH3]+ (60), 619 [M − Cl]+ (8), 145 [Ph-CF3]+ (50), 127 (100); HRMS (ESI+) m/z calcd for C22H16N6O6Cl3SF3Na [M + Na]+: 676.9767; found: 676.9769.
1,1’-(3,4,4-Trichloro-2-nitrobuta-1,3-diene-1,1-diyl)bis(1H-benzotriazole) (
24) was prepared according to the literature [
28] in 76% yield.
Synthesis of N-[1-(1H-Benzotriazol-1-yl)-3,4,4-trichloro-2-nitrobuta-1,3-dien-1-yl]-2-methoxyaniline (25a) (General method). To a suspension of azole 24 (0.437 g, 1.0 mmol) in MeOH (10 mL) at 0 °C, 2-methoxyaniline (0.129 g, 1.05 mmol) was slowly added. The mixture was allowed to reach r.t. and stirred for another 3 h. After evaporation of the solvent, HCl (10%, 10 mL) was added and the resulting sludge was stirred for 20 min. The precipitate was then filtered off and washed with HCl (10%, 5 mL), cold water (5 mL) and cold Et2O (2 × 5 mL). The product was dried in vacuo. Yield 0.392 g (89%), yellow solid, m.p. 128–129 °C. IR (KBr) νmax = 1621, 1580, 1463, 1252, 1023, 753 cm−1. 1H NMR (200 MHz, CDCl3) δ = 3.91 (3H, s, OCH3), 6.15 (1H, dd, J = 1.3 Hz, J = 8.0 Hz, H Ar), 6.46 (1H, ddd J = 7.7 Hz, J = 1.2 Hz, J = 7.5 Hz, H Ar), 6.82 (1H, dd, J = 1.5 Hz, J = 8.3 Hz, H Ar), 7.00 (1H, ddd, J = 8.3 Hz, J = 1.4 Hz, J = 7.5 Hz, H Ar), 7.27–7.42 (3H, m, H Bzt), 8.02–8.06 (1H, m, H Bzt), 11.65 (1H, s, NH) ppm; 13C NMR (50 MHz, CDCl3) δ = 55.9 (OCH3), 109.8 (CH Bzt), 111.3 (CH Ar), 120.6 (CH Ar), 120.9 (CH Bzt), 122.2 (CH Ar), 123.8 (CNH), 125.3 (CH Bzt), 128.6 (CH Ar), 129.5 (CH Bzt), 131.9, 145.3 (NCN), 145.9, 151.4 (COCH3) ppm, three C signals from butadiene-chain due to their low intensity could not be detected; MS m/z (Irel, %): 439 [M+] (4), 403 [M-HCl]+ (3), 358 [M-Cl-NO2]+ (3), 331 [M-Ph-OCH3]+ (6), 122 [H2NPhOCH3]+ (100); HRMS (ESI+) m/z calcd for C17H13N5O3Cl3 [M + H]+: 440.0079; found: 440.0077.
N-[1-(1H-Benzotriazol-1-yl)-3,4,4-trichloro-2-nitrobuta-1,3-dien-1-yl]-2-ethoxyaniline (25b). Same procedure as for 25a, using 2-ethoxyaniline (0.144 g, 1.05 mmol). Yield 0.355 g (78%), orange solid, m.p. 145–146 °C. IR (ATR) νmax = 1620, 1583, 1471, 1241, 1147, 741 cm−1. 1H NMR (400 MHz, CDCl3) δ = 1.56 (3H, t, J = 7.0 Hz, OCH2CH3), 4.15 (2H, q, J = 6.7 Hz, OCH2CH3), 5.99 (1H, dd, J = 1.0 Hz, J = 8.0 Hz, H Ar), 6.41 (1H, ddd, J = 7.6 Hz, J = 1.0 Hz, J = 7.9 Hz, H Ar), 6.82 (1H, dd, J = 8.3 Hz, J = 0.9 Hz, H Ar), 6.97 (1H, ddd, J = 8.0 Hz, J = 1.2 Hz, J = 7.8 Hz, H Ar), 7.31 (1H, d, J = 8.0 Hz, H Bzt), 7.37 (ddd, J = 7.0 Hz, J = 1.1 Hz, J = 8.1 Hz, H Bzt), 7.43 (1H, t, J = 7.36 Hz, H Bzt), 8.06 (1H, d, J = 8.2 Hz, H Bzt) ppm; 13C NMR (100 MHz, CDCl3) δ = 14.7 (CH3), 64.7 (OCH2), 109.8 (CH), 112.2 (CH), 120.6 (CH), 120.7 (CH), 121.5 (CH), 124.0, 125.3 (CH), 128.3 (CH), 129.5 (CH), 131.9, 145.3, 145.6, 150.6 ppm, three C signals from butadiene-chain due to their low intensity could not be detected; MS m/z (Irel, %): 453 [M+] (1), 417 [M − HCl]+ (1), 390 [M-Cl-N2]+ (1), 135 [NPhOEt2]+ (38), 100 (100); HRMS (ESI+) m/z calcd for C18H15N5O3Cl3 [M + H]+: 454.0235; found: 454.0234.
N’-[1-(1H-Benzotriazol-1-yl)-3,4,4-trichloro-2-nitrobuta-1,3-dien-1-yl]-N,N-dimethylbenzene-1,4-di- amine (25c). Same procedure as for 25a, using N,N-dimethylbenzene-1,4-diamine (0.143 g, 1.05 mmol) keeping the reaction mixture at 0 °C until completion. Yield 0.413 g (91%), dark red solid, m.p. 147–148 °C. IR (ATR) νmax = 2891, 1620, 1491, 1358, 1168, 818 cm−1. 1H NMR (400 MHz, CDCl3) δ = 2.81 (6H, s, NCH3), 6.31 (2H, d, J = 9.0 Hz, H Ar), 6.60 (2H, d, J = 9.0 Hz, H Ar), 7.35–7.44 (3H, m, H Bzt), 8.04 (1H, d, J = 8.4 Hz, H Bzt), 11.88 (1H, s, NH) ppm; 13C NMR (100 MHz, CDCl3) δ = 40.1 (CH3), 109.8 (CH), 112.2 (CH), 117.0, 120.6 (CH), 120.9, 123.8 (CH), 125.2 (CH), 128.0, 129.3 (CH), 129.5, 145.3, 146.8, 149.4 ppm, three C signals from butadiene-chain due to their low intensity could not be detected; MS m/z (Irel, %): 452 [M+] (2), 416 [M − Cl]+ (1), 388 [M-HCl-N2]+ (1), 119 [Bzt + H]+ (100); HRMS (ESI+) m/z calcd for C18H16N6O2Cl3 [M + H]+: 453.0395; found: 453.0395.
1-[3,4,4-Trichloro-1-(2,3-dihydro-1H-indol-1-yl)-2-nitrobuta-1,3-dien-1-yl]-1H-benzotriazole (25f). Same procedure as for 25a, using 1H-indoline (0.125 g, 1.05 mmol). Yield 0.354 g (87%), yellow solid, m.p. 178–179 °C. IR (ATR) νmax = 3065, 1533, 1295, 1023, 905, 655 cm−1. 1H NMR (400 MHz, CDCl3) δ = 3.34–3.46 (2H, m, CH2 ind), 3.89–4.25 (2H, m, CH2 ind), 5.06–6.38 (1H, s, H ind), 6.66–7.00 (1H, m, H Bzt), 7.06 (1H, s, H ind), 7.29–7.36 (1H, m, H ind), 7.40–7.88 (3H, m, 2H Bzt, 1H ind), 8.16 (1H, d, J = 8.1 Hz, H Bzt) ppm; 13C NMR (100 MHz, CDCl3) δ = 29.2 (CH2), 54.5 (CH2), 113.9 (CNO2), 121.1 (2 × CH), 123.2, 125.9 (2 × CH), 126.0 (CH), 127.9 (CH), 128.0 (CH), 130.0 (CH), 131.8 (CCl2), 141.2 (NCN), 146.1, 146.2 ppm; MS m/z (Irel, %): 435 [M+] (12), 289 [M-indolin-N2]+ (34), 271 [M-Bzt-NO2]+ (79), 118 [benzotriazole]+ (100); HRMS (ESI+) m/z calcd for C18H13N5O2Cl3 [M + H]+: 436.0129; found: 436.0127.
Synthesis of 5-Methoxy-8-(2,3,3-trichloro-1-nitroprop-2-en-1-ylidene)-7-azabicyclo[4.2.0]octa-1,3,5- triene (26a) (General method). A suspension of azole 25a (0.441 g, 1.0 mmol) in MeOH (10 mL) was stirred at reflux for 5 h. Subsequently, the mixture was concentrated and cooled to 0 °C. The precipitate was filtered off and washed with diethyl ether (2 × 5 mL). Yield 0.119 g (37%), yellow solid, m.p. 168–169 °C. IR (KBr) νmax = 3112, 1637, 1536, 1395, 1083, 859 cm−1. 1H NMR (200 MHz, DMSO-d6) δ = 3.96 (3H, s, OCH3), 7.30–7.42 (2H, m, CH), 7.74 (1H, dd, J = 2.3 Hz, J = 7.6 Hz, CH), 12.40 (1H, s, NH) ppm; 13C NMR (50 MHz, DMSO-d6) δ = 57.0 (OCH3), 110.9 (CH), 114.3 (CH), 119.5, 123.9 (CH), 124.0, 125.9, 130.6, 134.0, 147.3, 153.9 (COCH3) ppm; MS m/z (Irel, %): 320 [M+] (1), 284 [M − HCl]+ (12), 256 [M-NO2-NH2+H]+ (40), 64 (100); HRMS (ESI+) m/z calcd for C11H8N2O3Cl3 [M + H]+: 320.9595; found: 320.9594.
5-Ethoxy-8-(2,3,3-trichloro-1-nitroprop-2-en-1-ylidene)-7-azabicyclo[4.2.0]octa-1,3,5-triene (26b). Same procedure as for 26a, using azole 25b (0.455, 1.0 mmol) and refluxing in EtOH for 10 h. Yield 0.091 g (27%), yellow solid, m.p. 204–205 °C. IR (KBr) νmax = 2992, 1647, 1533, 1378, 1234, 554 cm−1. 1H NMR (400 MHz, DMSO-d6) δ = 1.42 (3H, t, J = 7.0 Hz, OCH2CH3), 4.16–4.26 (2H, m, OCH2CH3), 7.30–7.38 (2H, m, CH), 7.71 (1H, dd, J = 1.5 Hz, J = 8.2 Hz, CH), 12.37 (1H, s, NH) ppm; 13C NMR (100 MHz, DMSO-d6) δ = 14.5 (CH3), 65.4 (OCH2), 110.7 (CH), 114.9 (CH), 119.5, 123.9 (CH), 124.0, 125.9, 130.6, 133.9, 146.5, 153.9 ppm; MS m/z (Irel, %): 334 [M+] (3), 299 [M − Cl]+ (52), 271 [M-Cl-EtO+H]+ (100); HRMS (ESI+) m/z calcd for C12H10N2O3Cl3 [M + H]+: 334.9752; found: 334.9750.
Synthesis of N,N-Dimethyl-8-(2,3,3-trichloro-1-nitroprop-2-en-1-ylidene)-7-azabicyclo[4.2.0]octa-1,3,5- trien-3-amine (26c). A suspension of azole 25c (0.454 g, 1.0 mmol) in THF (20 mL) was stirred at reflux for 10 h. After evaporation of the solvent, dilute HCl (10 mL) was added and the mixture stirred for 15 min. Subsequently, the mixture was extracted with chloroform and purified by column chromatography using petroleum ether - ethyl acetate (1:1). Yield 0.084 g (25%), red solid, m.p. 171–173 °C. IR (KBr) νmax = 2856, 1567, 1369, 1063, 846, 550 cm−1. 1H NMR (400 MHz, DMSO-d6) δ = 2.97 (6H, s, N(CH3)2), 7.22 (1H, d, J = 2.0 Hz, CH), 7.32 (1H, d, J = 9.0 Hz, CH), 7.36 (1H, dd, J = 2.1 Hz, J = 9.2 Hz, CH), 12.65 (1H, s, NH) ppm; 13C NMR (100 MHz, DMSO-d6) δ = 40.3 (Me), 98.6 (CH), 117.8 (CH), 119.9, 121.1 (CH), 124.0, 125.6, 130.9, 133.5, 147.5 (CNMe2), 153.1 ppm; MS m/z (Irel, %): 333 [M+] (27), 298 [M − Cl]+ (40), 270 [M-NO2-NH3]+ (100); HRMS (ESI+) m/z calcd for C12H11N3O2Cl3 [M + H]+: 333.9917; found: 333.9919.
Synthesis of N’-[6-(Dichloromethyl)-2-methyl-5-nitropyrimidin-4-yl]-N, N-dimethylbenzene-1,4- diamine (27c). To a solution of the nitrobutadiene 25c (0.454 g, 1.0 mmol) and acetamidine hydrochloride (0.284 g, 3.0 mmol) in 20 mL dry THF, sodium hydride (0.160 g, 4.0 mmol, 60%) was added at 0 °C. The solution was thoroughly stirred for 1 h at 0 °C and then at r.t. for 2 d. After evaporation of the solvent and addition of 10% HCl (5 mL) the precipitate was filtered off and washed with 10% HCl (2 × 10 mL), cold water (10 mL), cold MeOH (2 × 5 mL) and dried in vacuo. Yield 0.232 g (65%), yellow solid, m.p. 124–125 °C. IR (KBr) νmax = 3321, 1612, 1573 (NO2), 1517, 1204, 746 cm−1. 1H NMR (400 MHz, CDCl3) δ = 2.64 (3H, s, CCH3), 2.99 (6H, s, NCH3), 6.74 (2H, d, J = 9.0 Hz, H Ar), 7.40 (2H, d, J = 8.9 Hz, H Ar), 7.42 (1H, s, CHCl2), 9.73 (1H, s, NH) ppm; 13C NMR (100 MHz, CDCl3) δ = 26.5 (CH3), 40.5 (NCH3), 66.8 (CHCl2), 112.3 (CH), 122.8, 124.5 (CH), 125.3, 148.9, 153.2, 160.6, 171.0 ppm; MS m/z (Irel, %): 355 [M+] (39), 136 [phenylendiamine+H]+ (100); HRMS (ESI+) m/z calcd for C14H16N5O2Cl2 [M + H]+: 356.0676; found: 356.0674.
6-(Dichloromethyl)-N,N,2-trimethyl-5-nitropyrimidin-4-amine (27d). Same procedure as for 27c, using diene 25d (0.363 g, 1.0 mmol). The precipitate was further purified by column chromatography using petroleum ether - ethyl acetate, 5: 1. Yield 0.148 g (56%), yellowish solid, m.p. 82–83 °C. IR (ATR) νmax = 3048, 1579 (NO2), 1500, 1338 (NO2), 1195, 746 cm−1. 1H NMR (400 MHz, CDCl3) δ = 2.61 (3H, s, CCH3), 3.09 (6H, s, NCH3), 6.98 (1H, s, CHCl2); 13C NMR (100 MHz, CDCl3) δ = 26.1 (CH3), 39.3 (NCH3), 65.4 (CHCl2), 125.6 (CNO2), 154.7, 156.2, 168.1 ppm; MS m/z (Irel, %): 264 [M+] (55), 218 [M − NO2]+ (30), 182 [M-CHCl2+H]+ (63), 135 [M-CHCl2-NO2]+ (52). HRMS (ESI+) m/z calcd for C8H11N4O2Cl2 [M + H]+: 265.0254; found: 265.0255.
N-[6-(Dichloromethyl)-2-methyl-5-nitropyrimidin-4-yl]quinolin-8-amine (27e). Same procedure as for 27c, using azole 25e (0.462 g, 1.0 mmol) as starting material. Yield 0.178 g (49%), yellow solid, m.p. 217–219 °C. IR (KBr) νmax = 3262, 1573 (NO2), 1298 (NO2), 1208, 766, 590 cm−1. 1H NMR (400 MHz, CDCl3) δ = 2.82 (3H, s, CCH3), 7.37 (1H, s, CHCl2), 7.52 (1H, dd, J = 8.3 Hz, J = 4.2 Hz, H quin), 7.58–7.63 (2H, m, H quin), 8.21 (1H, dd, J = 8.3 Hz, J = 1.6 Hz, H quin), 8.94 (1H, dd, J = 4.2 Hz, J = 1.6 Hz, H quin), 9.04–9.07 (1H, m, H quin), 12.17 (1H, s, NH) ppm; 13C NMR (100 MHz, CDCl3) δ = 26.6 (CH3), 66.4 (CHCl2), 118.4 (CH), 122.0 (CH), 122.93 (CH), 124.6, 127.0 (CH), 128.1, 134.0, 136.4 (CH), 139.3, 149.0 (CH), 152.1, 159.7, 170.9 ppm; MS m/z (Irel, %): 363 [M+] (15), 317 [M-NO2]+ (100), 281 [M − CHCl2]+ (25), 235 [M − quin]+ (12), 128 [quinoline]+ (45); HRMS (ESI+) m/z calcd for C15H12N5O2Cl2 [M + H]+: 364.0368; found: 364.0368.
1-[6-(Dichloromethyl)-2-methyl-5-nitropyrimidin-4-yl]-2,3-dihydro-1H-indole (27f). Same procedure as for 27c, using diene 25f (0.437 g, 1.0 mmol), DMSO (25 mL) and NaOH (30%, 4 mmol). Yield 0.216 g (64%), yellow solid, m.p. 132–133 °C. IR (ATR) νmax = 3048, 1562 (NO2), 1529, 1340 (NO2), 1209, 768 cm−1. 1H NMR (400 MHz, CDCl3) δ = 2.72 (3H, s, CCH3), 3.20 (2H, t, J = 7.9 Hz, H ind), 3.86 (2H, t, J = 7.9 Hz, H ind), 7.04 (1H, s, CHCl2), 7.11 (1H, t, J = 7.4 Hz, H ind), 7.24–7.29 (2H, m, H ind), 7.93 (1H, d, J = 8.1 Hz, H ind) ppm; 13C NMR (100 MHz, CDCl3) δ = 26.0 (CH), 28.8 (CH2), 50.2 (CH2), 65.3 (CHCl2), 117.6 (CH), 124.8 (CH), 125.1 (CH), 126.4, 127.1 (CH), 132.3, 142.1, 151.6, 156.8, 168.7 ppm; MS m/z (Irel, %): 338 [M+] (100), 323 [M-CH3]+ (13), 257 [M-NO2-Cl] (48), 118 [indole]+ (24); HRMS (ESI+) m/z calcd for C14H13N4O2Cl2 [M + H]+: 339.0416; found: 339.0413.
Synthesis of 1-[6-(Dichloromethyl)-2-methyl-5-nitropyrimidin-4-yl]-1H-indole (28). A solution of pyrimidine 27f (0.400 g, 1.2 mmol) and DDQ (0.600 g, 2.7 mmol) in toluene (10 mL) was heated to reflux for 5 h, allowed to cool down to r.t., and the precipitate filtered off. The filtrate was purified by column chromatography using petroleum ether–ethyl acetate, 25: 1. The product was dried in vacuo. Yield 0.267 g (66%), m.p. 103–104 °C. IR (ATR) νmax = 3022, 1589 (NO2), 1342, 1261 (NO2), 1099, 745 cm−1. 1H NMR (600 MHz, CDCl3) δ = 2.92 (3H, s, CCH3), 6.79 (1H, dd, J = 3.7 Hz, J = 0.7 Hz, H ind), 7.01 (1H, s, CHCl2), 7.12 (1H, d, J = 3.7 Hz, H ind), 7.30 (1H, t, J = 8.0 Hz, H ind), 7.36 (1H, t, J = 8.4 Hz, H ind), 7.63 (1H, d, J = 8.2 Hz, H ind), 8.10 (1H, d, J = 9.1 Hz, H ind) ppm; 13C NMR (150 MHz, CDCl3) δ = 26.2 (CH3), 64.8 (CHCl2), 110.9 (CH), 114.1 (CH), 121.6 (CH), 123.8 (CH), 124.5 (CH), 124.9 (CH), 128.3, 130.4, 135.4, 150.5, 157.6, 170.5 ppm; MS m/z (Irel, %): 336 [M+] (100), 291 [M + H-NO2]+ (42), 238 [M-CH3-CHCl2]+ (48), 116 [indole]+ (62); HRMS (ESI+) m/z calcd for C14H11N4O2Cl2 [M + H]+: 337.0259; found: 337.0261.
Synthesis of 1-[1-(1H-Benzotriazol-1-yl)-3,4,4-trichloro-2-nitrobuta-1,3-dien-1-yl]-4-(4-chlorophenyl)- piperidin-4-ol (25g). Same procedure as for 25a, using 4-(4-chlorophenyl)piperidin-4-ol (0.223 g, 1.05 mmol). Yield 0.482 g (91%), yellowish solid, m.p. 149–150 °C. IR (ATR) νmax = 2931, 1565, 1498, 1290, 1005, 749 cm−1. 1H NMR (400 MHz, CDCl3) δ = 1.83–2.03 (2H, m, H pip), 2.33–2.61 (2H, m, H pip), 2.95–3.25 (2H, m, NCH2), 3.76–4.08 (3H, m, OH + NCH2), 7.35–7.38 (2H, m, H Ar), 7.45–7.47 (2H, m, H Ar), 7.47–7.79 (3H, m, H Bzt), 8.10–8.13 (1H, m, H Bzt) ppm; 13C NMR (100 MHz, CDCl3) δ = 38.3 (CH2), 47.3 (CH2), 70.4 (COH), 110.4 (CH), 111.89 (CNO2), 120.9 (CH), 124.8 (CCl), 125.9 (2 × CH), 126.0 (CH), 126.2 (CCl2), 128.8 (2 × CH), 130.6 (CH), 132.4, 133.6 (CCl), 144.8, 146.1, 148.0 (CNN) ppm; MS m/z (Irel, %): 527 [M+] (1), 499 [M-N2]+ (1), 453 [M-N2-NO2]+ (1), 416 [M-Ph-Cl]+ (1), 111 [PhCl]+ (20), 91 (100); HRMS (ESI+) m/z calcd for C21H18N5O3Cl4 [M + H]+: 538.0158; found: 528.0156.
{4-[1-(1H-Benzotriazol-1-yl)-3,4,4-trichloro-2-nitrobuta-1,3-dien-1-yl]piperazin-1-yl}(tetrahydrofuran-2-yl)-methanone (25h). Same procedure as for 25a, using piperazin-1-yl(tetrahydrofuran-2-yl)methanone (0.193 g, 1.05 mmol). Yield 0.426 g (85%), yellow solid, m.p. 170–172 °C. IR (KBr) νmax = 2871, 1657, 1560, 1290, 1006, 747 cm−1. 1H NMR (200 MHz, CDCl3) δ = 1.80–2.11 (3H, m, CH2 fur), 2.30–2.49 (1H, m, CH2 fur), 3.18–3.60 (4H, m, NCH2), 3.72–4.41 (6H, m, 2NCH2, OCH2), 4.52–4.67 (1H, m, OCH), 7.48–7.80 (3H, m, H Bzt), 8.17–8.20 (1H, m, H Bzt) ppm; 13C NMR (50 MHz, CDCl3) δ 25.7 (CH2), 27.7 (CH2), 41.8 (NCH2), 45.2 (NCH2), 50.6 (NCH2), 69.2 (OCH2), 76.1 (OCH), 110.2 (CH), 116.2 (CCl2), 120.6 (CNO2), 121.1 (CH), 125.5, 126.1 (CH), 129.9, 130.7 (CH), 146.3, 147.5 (CNN), 169.7 (C=O) ppm; MS m/z (Irel, %): 500 [M+] (1), 465 [M-Cl]+ (1), 426 [M-NO2-N2]+ (1), 337 [M-Bzt-NO2+H]+ (4), 119 [Bzt + H]+ (16), 92 (100); HRMS (ESI+) m/z calcd for C19H20N6O4Cl3 [M + H]+: 501.0606; found: 501.0605.
1-{3,4,4-Trichloro-1-[4-(3-chlorophenyl)piperazin-1-yl]-2-nitrobuta-1,3-dien-1-yl}-1H-benzotriazole (25i). Same procedure as for 25a, using 1-(3-methylphenyl)piperazine (0.207 g, 1.05 mmol). Yield 0.452 g (88%), yellow solid, m.p. 68–70 °C. IR (KBr) νmax = 2916, 1594, 1560, 1291, 942, 768 cm−1. 1H NMR (200 MHz, CDCl3) δ = 3.41–3.61 (8H, m, NCH2), 6.75–6.90 (1H, m, H Ar), 6.91–6.98 (2H, m, H Ar), 7.18–7.24 (1H, m, H Ar), 7.40–7.68 (3H, m, H Bzt), 8.16 (1H, d, J = 8.1 Hz, H Bzt) ppm; 13C NMR (50 MHz, CDCl3) δ = 49.2 (NCH2), 49.4 (NCH2), 110.3 (CH), 115.1 (CH), 117.2 (CH), 121.1 (CH), 121.6 (CH), 124.3 (CCl2), 125.3 (CCl), 126.1 (CH), 130.4 (CH), 130.7 (CH), 132.4, 135.2 (CCl), 146.3, 147.6 (CNN), 150.7 ppm, CNO2 could not be detected; MS m/z (Irel, %): 512 [M+] (2), 449 [M-Cl-N2]+ (1), 401 [M-PhCl]+ (2), 138 [PhClNCH]+ (100); HRMS (ESI+) m/z calcd for C20H17N6O2Cl4 [M + H]+: 512.0078; found: 512.0075.
Synthesis of 4-(4-Chlorophenyl)-1-[5-(dichloromethyl)-1-methyl-4-nitro-1H-pyrazol-3-yl]piperidin-4-ol (29a) (General method). To a suspension of azole 25g (0.529 g, 1.0 mmol) in MeOH (10 mL) at −10 °C, methylhydrazine (0.921 g, 2.0 mmol) was added dropwise. After 2 h, the solution was allowed reach r.t. and stirred for 1 d. The solution was then concentrated and 10% HCl (5 mL) was added. The resulting precipitate was filtered off, washed with water, and purified by column chromatography using chloroform. Yield 0.311 g (74%), yellow solid, m.p. 95–96 °C. IR (ATR) νmax = 2951, 1553, 1484, 1348, 1024, 746 cm−1. 1H NMR (400 MHz, CDCl3) δ = 1.65 (1H, s, OH), 1.78–1.85 (2H, m, CCH2), 2.28 (2H, ddd, J = 13.1 Hz, J = 4.2 Hz, J = 13.1 Hz, CCH2), 3.33 (2H, ddd, J = 12.5 Hz, J = 2.5 Hz, J = 12.5 Hz, NCH2), 3.51–3.59 (2H, m, NCH2), 4.12 (3H, s, NCH3), 7.34 (2H, d, J = 8.7 Hz, H Ar), 7.47 (2H, d, J = 8.7 Hz, H Ar), 7.88 (1H, s, CHCl2) ppm; 13C NMR (100 MHz, CDCl3) δ = 38.0 (2 × CH2), 39.7 (NCH3), 46.0 (2 × NCH2), 57.8 (CHCl2), 71.1 (HO-C), 120.4 (CNO2), 126.1 (CH), 129.2 (CH), 133.0 (CCl), 137.9, 146.5, 152.8 (NC-pyr) ppm; MS m/z (Irel, %): 418 [M+] (3), 401 [M-OH]+ (6), 383 [M − Cl]+ (5), 367 [M-Cl-O]+ (4), 347 [M-Cl-HCl]+ (4), 100 (100); HRMS (ESI+) m/z calcd for C16H18N4O3Cl3 [M + H]+: 419.0439; found: 419.0437.
{4-[5-(Dichloromethyl)-1-methyl-4-nitro-1H-pyrazol-3-yl]piperazin-1-yl}(tetrahydrofuran-2-yl)methanone (29b). Same procedure as for 29a, using diene 25h (0.502 g, 1.0 mmol) and DCM as the eluent. Yield 0.310 g (79%), yellowish oil. IR (ATR) νmax = 2954, 1647, 1551, 1341, 1198, 745 cm−1. 1H NMR (400 MHz, CDCl3) δ = 1.86–2.12 (3H, m, CH2 Fur), 2.25–2.35 (1H, m, CH2 Fur), 3.17–3.31 (4H, m, NCH2), 3.63–3.73 (2H, m, NCH2), 3.79–3.98 (4H, m, NCH2, OCH2), 4.10 (3H, s, NCH3), 4.64 (1H, dd, J = 5.6 Hz, J = 7.5 Hz, COCHO), 7.84 (1H, s, CHCl2) ppm; 13C NMR (100 MHz, CDCl3) δ = 25.7 (CH2), 28.4 (CH2), 39.7 (NCH3), 41.6 (NCH2), 45.0 (NCH2), 49.6 (NCH2), 50.0 (NCH2), 57.6 (CH), 69.1 (OCH2), 75.9 (OCH), 120.5 (CNO2), 138.1, 152.1, 170.1 (C=O) ppm; MS m/z (Irel, %): 391 [M+] (100), 374 [M − OH]+ (20), 356 [M − Cl]+ (65), 320 [M − furan]+ (20), 292 [M-CO-furan]+ (35); HRMS (ESI+) m/z calcd for C14H20N5O4Cl2 [M + H]+: 392.0887; found: 392.0889.
1-(3-Chlorophenyl)-4-[5-(dichloromethyl)-1-methyl-4-nitro-1H-pyrazol-3-yl]piperazine (29c). Same proce-dure as for 29a, using diene 25i (0.514 g, 1.0 mmol) and a mixture of petroleum ether and ethyl acetate (5: 1) as the eluent. Yield 0.299 g (74%), yellow solid, m.p. 162–163 °C. IR (ATR) νmax = 2832, 1552, 1475, 1338, 936, 737 cm−1. 1H NMR (400 MHz, CDCl3) δ = 3.31–3.36 (4H, m, NCH2), 3.38–3.43 (4H, m, NCH2), 4.13 (3H, s, NCH3), 6.81–6.86 (2H, m, H Ar), 6.93 (1H, t, J = 2.1 Hz, H Ar), 7.18 (1H, t, J = 8.0 Hz, H Ar), 7.87 (1H, s, CHCl2) ppm; 13C NMR (100 MHz, CDCl3) δ = 39.7 (CH3), 48.5 (NCH2), 49.5 (NCH2), 57.7 (CHCl2), 114.1 (CH), 116.1 (CH), 119.7 (CH), 120.5 (CNO2), 130.1 (CH), 135.0 (CCl), 138.0, 152.2 (CN), 152.3 (CN) ppm; MS m/z (Irel, %): 403 [M+] (45), 357 [M − NO2]+ (6), 322 [M-NO2-Cl]+ (22), 138 [PhClNCH]+ (100); HRMS (ESI+) m/z calcd for C15H17N5O2Cl3 [M + H]+: 404.0442; found: 404.0444.
Synthesis of 3-[4-(3-Chlorophenyl)piperazin-1-yl]-1-methyl-4-nitro-1H-pyrazole-5-carbaldehyde (30). Pyrazole 29c (0.405 g, 1.0 mmol) was suspended in 10 mL sulfuric acid (25%) and heated to 95–100 °C for 10 h. After cooling to r.t., the solution was extracted with chloroform (3 × 10 mL) and purified by column chromatography using petroleum ether - ethyl acetate (5: 1). The product was dried in vacuo. Yield 0.199 g (57%), yellow solid, m.p. 159–160 °C. IR (ATR) νmax = 2827, 1683, 1546, 1474, 1236, 775 cm−1. 1H NMR (400 MHz, CDCl3) δ = 3.32–3.38 (4H, m, NCH2), 3.45–3.5 (4H, m, NCH2), 6.81–6.87 (2H, m, H Ph), 6.93 (1H, t, J = 2.0 Hz, H Ph), 7.19 (1H, t, J = 8.1 Hz, H Ph), 10.43 (1H, s, CHO) ppm; 13C NMR (100 MHz, CDCl3) δ = 40.9 (NCH3), 48.5 (2 × NCH2), 49.4 (2 × NCH2), 114.2 (CH), 116.1 (CH), 119.8 (CH), 125.7 (CNO2), 130.1 (CH), 135.0 (CCl), 135.2 (CCHO), 151.9, 152.1, 181.8 (CHO) ppm; MS m/z (Irel, %): 349 [M+] (100), 332 [M − OH]+ (5), 314 [M − Cl]+ (4), 304 [M-NO2+H]+ (20), 138 [ClPhNCH]+ (75); HRMS (ESI+) m/z calcd for C15H17N5O3Cl [M + H]+: 350.1014; found: 350.1016.
Synthesis of 2-(1H-Benzotriazol-1-yl)-4-(dichloromethylidene)-3-nitro-4H-pyrido[1,2-a]pyrimidine (31a) (General method). To a solution of azole 24 (0.437 g, 1.0 mmol) in THF (10 m), 2-aminopyridine (0.282 g, 3.0 mmol) was added at r.t. The solution was stirred for 8 h, then concentrated, and the residue treated with dilute HCl for 1 h, filtered off and washed with water (2 × 10 mL) and cold MeOH (5 mL). The product was dried in vacuo. Yield 0.304 g (81%), red solid, m.p. 158–160 °C. IR (KBr) νmax = 3082, 1628, 1552, 1434, 1307, 1215 cm−1. 1H NMR (200 MHz, DMSO-d6) δ = 7.50–7.64 (2H, m, H Ar), 7.64–7.75 (1H, m, H Bzt), 7.87 (1H, d, J = 8.3 Hz, H Ar), 7.90–7.95 (1H, m, H Bzt), 8.21 (1H, d, J = 7.8 Hz, H Bzt), 8.22–8.32 (1H, m, H Bzt), 8.99 (1H, dd, J = 6.8 Hz, 0.9 Hz, H Ar) ppm; 13C NMR (50 MHz, DMSO-d6) δ = 102.3 (CCl2), 113.0 (CH), 119.2 (CH), 120.0 (CH), 121.3 (CNO2), 123.7 (CH), 125.5 (CH), 126.3, 129.6 (CH), 132.3, 138.8 (CH), 143.6 (CH), 145.6, 148.0, 151.3 ppm; MS m/z (Irel, %): m/z (%) = 374 [M+] (1), 256 [M–benzotriazole]+ (14), 219 (18), 119 [benzotriazole] (14), 92 (100); HRMS (ESI+) m/z calcd for C15H9N6O2Cl2 [M + H]+: 375.0164; found: 375.0164.
2-(1H-Benzotriazol-1-yl)-7-chloro-4-(dichloromethylidene)-3-nitro-4H-pyrido[1,2-a]pyrimidine (31b). Same procedure as for 31a, using 2-amino-5-chloropyridine (0.386 g, 3.0 mmol) and heating the reaction mixture to 45 °C. Yield 0.352 g (86%), orange solid, m.p. 182–184 °C. IR (KBr) νmax = 3066, 1505, 1437, 1318, 1238, 1222 cm−1. 1H NMR (200 MHz, DMSO-d6) δ = 7.55 (1H, t, J = 7.9 Hz, H Bzt), 7.71 (1H, t, J = 8.1 Hz, H Bzt), 7.86 (1H, d, J = 9.3 Hz, H pyr), 7.92 (1H, d, J = 8.2 Hz, H Bzt), 8.23 (1H, d, J = 8.2 Hz, H Bzt), 8.35 (1H, dd, J = 9.4 Hz, J = 2.3 Hz, H pyr), 9.35 (1H, d, J = 2.3 Hz, H pyr) ppm; 13C NMR (50 MHz, DMSO-d6) δ = 102.8 (CCl2), 112.9 (CH), 119.9 (CH), 122.2 (CNO2), 124.7 (CCl), 124.8 (CH), 125.6 (CH), 129.6 (CH), 132.3, 136.6 (CH), 143.1 (CH), 145.6, 147.7 (NC), 150.6 ppm; MS m/z (Irel, %): 408 [M+] (1), 380 [M − N2]+ (1), 373 [M − Cl]+ (2), 334 [M-N2-NO2]+ (7), 112 (100); HRMS (ESI+) m/z calcd for C15H8N6O2Cl3 [M + H]+: 408.9769; found: 408.9768.
Synthesis of 4-(dichloromethylidene)-2-[4-(4-fluorophenyl)piperazin-1-yl]-3-nitro-4H-pyrido[1,2-a]pyrimi-dine (32a) (General method). To a solution of pyrimidine 31a (0.375 g, 1 mmol) in MeOH (10 mL) 1-(4-fluorophenyl)piperazine (0.216 g, 1.2 mmol) was added and the mixture stirred at 40 °C for 5 h. Subsequently, the mixture was cooled to 0 °C and treated with dilute HCl for 1 h. The precipitate was filtered off and washed with water (2 × 5 mL) and cold MeOH (5 mL). The product was dried in vacuo. Yield 0.423 g (97%), yellow solid, m.p. 126–127 °C. IR (KBr) νmax = 1639, 1551, 1504, 1253, 1145, 933 cm−1. 1H NMR (200 MHz, DMSO-d6) δ = 3.15–3.50 (4H, m, NCH2), 3.78–4.31 (4H, m, NCH2), 6.78–7.18 (5H, m, H Pyr, 4H Ph-F), 7.31 (1H, d, J = 8.6 Hz, H pyr), 7.94 (1H, ddd, J = 8.6 Hz, J = 1.3 Hz, J = 7.3 Hz, H pyr), 8.60 (1H, dd, J = 7.2 Hz, J = 1.3 Hz, H pyr) ppm; 13C NMR (50 MHz, DMSO-d6) δ = 49.5 (4 × NCH2), 94.9 (CCl2), 114.7 (CH), 115.6 (JC,F = 21.6 Hz, 2 × CH Ph), 118.1 (JC,F = 7.9 Hz, 2 × CH Ph), 120.5 (CNO2), 122.7 (CH), 128.2 (CCCl2), 137.1 (CH), 142.0 (CH), 147.3 (NC Ph), 151.5, 156.6 (JC,F = 235.9 Hz, CF), 156.1 (NCN) ppm; MS m/z (Irel, %): 435 [M+] (7), 256 [M-morph-Ph-F]+ (10), 179 [Morph-Ph-F]+ (15), 95 (100); HRMS (ESI+) m/z calcd for C19H17N5O2Cl2F [M + H]+: 436.0738; found: 436.0736.
7-Chloro-4-(dichloromethylidene)-2-[4-(4-fluorophenyl)piperazin-1-yl]-3-nitro-4H-pyrido[1,2-a] pyrimidine (32b). Same procedure as for 32a, using compound 31b (0.410 g, 1.0 mmol) but without adding HCl. Yield 0.447 g (95%), yellow solid, m.p. 141–142 °C. IR (KBr) νmax = 1635, 1563, 1493, 1373, 1237, 827 cm−1. 1H NMR (200 MHz, DMSO-d6) δ = 3.14–3.29 (4H, m, NCH2), 3.50–4.33 (4H, m, NCH2), 6.95–7.14 (4H, m, H Ph), 7.31 (1H, d, J = 9.4 Hz, H pyr), 7.99 (1H, dd, J = 9.5 Hz, J = 2.3 Hz, H pyr); 8.92 (1H, d, J = 8.9 Hz, H pyr) ppm; 13C NMR (50 MHz, DMSO-d6) δ = 49.4 (4 × NCH2), 94.7 (CCl2), 115.3 (JC,F = 21.9 Hz, 2 × FCCH), 117.6 (JC,F = 7.7 Hz, 2 × CH Ph), 120.1 (CCl), 121.1 (CNO2), 123.9 (CH), 127.6, 134.7 (NCH), 141.8 (CH), 147.3 (JC,F = 1.8 Hz, NC Ph), 150.4, 156.2 (JC,F = 236.4 Hz, CF), 156.0 ppm; MS m/z (Irel, %): 469 [M+] (8), 290 [M-piperazine-Ph-F]+ (3), 179 [piperazine-Ph-F]+ (20), 112 (100); HRMS (ESI+) m/z calcd for C19H16N5O2Cl3F [M + H]+: 470.0348; found: 470.0350.
Synthesis of Ethyl {[4-(dichloromethylidene)-3-nitro-4H-pyrido[1,2-a]pyrimidin-2-yl]sulfanyl}acetate (33a) (General method). To a solution of pyrimidine 31a (0.375 g, 1.0 mmol) in EtOH (10 mL), ethyl 2-mercaptoacetate (0.240 g, 2.0 mmol) and sodium ethanolate (0.204 g, 3.0 mmol) were added. The solution was stirred at r.t. for 3 d. Subsequently, dilute HCl (5 mL) was added under stirring for 30 min. The resulting precipitate was filtered off, washed with water (2 × 5 mL) and cold MeOH (3 mL), and dried in vacuo. Yield 0.365 g (97%).
Alternative synthesis of pyrimidine33afrom nitrodiene18. To a solution of nitrobutadiene 18 (0.355 g, 1.0 mmol) in MeOH (10 mL) 2-aminopyridine (0.282 g, 3.0 mmol) was added. After stirring at r.t. for 1 d, the solution was concentrated and the resulting precipitate filtered off, washed with water (2 × 3 mL), cold MeOH (3 mL) and dried in vacuo. Yield 0.177 g (47%) of 33a, orange solid, m.p. 147–148 °C. IR (ATR) νmax = 1731, 1597, 1451, 1202, 1141, 764 cm−1. 1H NMR (400 MHz, CDCl3) δ = 1.27 (3H, t, J = 7.1 Hz, OCH2CH3); 3.85 (2H, s, SCH2), 4.20 (2H, q, J = 7.1 Hz, OCH2), 7.00 (1H, ddd, J = 1.3 Hz, J = 6.9 Hz, J = 6.9 Hz, CH), 7.23 (1H, ddd, J = 1.2 Hz, J = 0.6 Hz, J = 9.0 Hz, CH), 7.72 (1H, ddd, J = 1.7 Hz, J = 7.1 Hz, J = 8.8 Hz, CH), 8.04 (1H, ddd, J = 1.6 Hz, J = 0.7 Hz, J = 6.9 Hz, NCH) ppm; 13C NMR (100 MHz, CDCl3) δ = 14.2 (CH3), 33.7 (SCH2), 61.5 (OCH2), 114.5 (CH), 117.1 (CCl2), 123.0 (CH), 125.1 (CNO2), 135.6 (NCH), 140.0 (CH), 149.7 (NCN), 164.1 (SCN), 169.3 (C=O) ppm, NCCHCl2 could not be detected; MS m/z (Irel, %): 375 [M+] (10), 340 [M-Cl]+ (9), 288 [M-CH2CO2Et]+ (25), 209 [M-HNO2-SCH2CO2Et]+ (95), 149 (100); HRMS (ESI+) m/z calcd for C13H11N3O4Cl2SNa [M + Na]+: 397.9745; found: 397.9746.
Ethyl {[7-chloro-4-(dichloromethylidene)-3-nitro-4H-pyrido[1,2-a]pyrimidin-2-yl]sulfanyl}acetate (33b). Same procedure as for 33a,but starting from compounds 31b (Yield 0.382 g, 93%), or 18 (Yield 0.226 g, 55%), orange solid, m.p. 171–172 °C. IR (ATR) νmax = 3074, 1727, 1626, 1489, 1235, 741 cm−1. 1H NMR (200 MHz, CDCl3) δ = 1.27 (3H, t, J = 7.1 Hz, CH3), 3.83 (2H, s, SCH2), 4.19 (2H, q, J = 7.2 Hz, OCH2), 7.17 (1H, dd, J = 9.4 Hz, J = 0.5 Hz, CH), 7.63 (1H, dd, J = 9.5 Hz, J = 2.3 Hz, CH), 8.05 (1H, dd, J = 2.3 Hz, J = 0.6 Hz, NCH) ppm; 13C NMR (50 MHz, CDCl3) δ = 14.3 (CH3), 33.6 (SCH2), 61.6 (OCH2), 117.5 (CCl2), 121.4 (CNO2), 122.2 (CCl), 123.2 (CH), 124.4 (NC), 133.0 (CH), 140.5 (CH), 148.2, 163.7 (SC), 169.2 (C=O) ppm; MS m/z (Irel, %): 409 [M+] (10), 373 [M-HCl]+ (18), 363 [M-NO2]+ (15), 321 [M-CH3CO2Et]+ (45), 242 (100); HRMS (ESI+) m/z calcd for C13H10N3O4Cl3SNa [M + Na]+: 431.9355; found: 431.9357.
Synthesis of N-[1-(benzylsulfanyl)-3,4,4-trichloro-2-nitrobuta-1,3-dien-1-yl]naphthalen-1-amine (35a) (General method). To a solution of diene 34a (0.359 g, 1.0 mmol) in MeOH (10 mL) at −10 °C, naphthalen-1-amine (0.315 g, 2.2 mmol) was added. The solution was kept at −10 °C for 2 h and then allowed to warm up to r.t. for 8 h. Subsequently, the solution was concentrated and the resulting precipitate was filtered off, washed with water (2 × 5 mL) and cold MeOH (5 mL), and dried in vacuo. Yield 0.428 g (92%), green-yellow solid, m.p. 132–133 °C. IR (ATR) νmax = 1536, 1332, 1149, 939, 768, 702 cm−1. 1H NMR (400 MHz, CDCl3) δ = 3.47 (2H, q, J = 12.4 Hz, SCH2), 6.86 (2H, dd, J = 7.7 Hz, J = 1.7 Hz, H Ph), 7.14–7.25 (3H, m, H Ar), 7.57 (1H, dd, J = 7.8 Hz, J = 7.8 Hz, H Ph), 7.59–7.65 (2H, m, H Ph), 7.73 (1H, d, J = 7.4 Hz, H Ar), 7.93 (1H, d, J = 8.2 Hz, H Ar), 7.94–7.99 (2H, m, H Ar), 13.32 (1H, s, NH) ppm; 13C NMR (100 MHz, CDCl3) δ = 38.5 (CH2), 121.5 (CH), 122.0 (CNO2), 122.9 (CH), 123.9 (CCl2), 125.4 (CH), 127.2 (CH), 127.8 (CH), 128.0 (CCl), 128.2 (CH), 128.5, 128.6 (CH), 128.7 (2 × CH), 128.7 (CH), 128.9 (2 × CH), 133.4, 134.1, 134.2, 159.7 (NCS) ppm; MS m/z (Irel, %): 464 [M+] (1), 447 [M − OH]+ (1), 418 [M − NO2]+ (1), 127 [naphthalene]+ (11), 91 [PhCH2]+ (100); HRMS (ESI+) m/z calcd for C21H16N2O2Cl3S [M + H]+: 464.9993; found: 464.9991.
N-{3,4,4-Trichloro-1-[(4-chlorophenyl)sulfanyl]-2-nitrobuta-1,3-dien-1-yl}naphthalen-1-amine (35b). Same procedure as for 33a, but starting from 34b. Yield 0.423 g (87%), green-yellow solid, m.p. 185–186 °C. IR (ATR) νmax = 1538, 1472, 1345, 1161, 824, 767 cm−1. 1H NMR (400 MHz, CDCl3) δ = 6.68 (2H, d, J = 8.8 Hz, H Ar), 6.71 (2H, d, J = 8.8 Hz, H Ar), 7.33–7.43 (3H, m, H Ar), 7.48 (1H, ddd, J = 6.8 Hz, J = 1.2 Hz, J = 8.1 Hz, H Ar), 7.55 (1H, d, J = 8.5 Hz, H Ar), 7.71–7.80 (2H, m, H Ar), 11.96 (1H, s, NH) ppm; 13C NMR (100 MHz, CDCl3) δ = 121.4, 121.3 (CH), 123.8 (CNO2), 124.6 (CH), 124.9 (CH), 125.6, 126.8 (CH), 126.9 (CH), 128.2 (CH), 128.4, 128.5, 128.7 (CH), 129.1 (2 × CH), 133.2, 133.6, 134.3 (2 × CH), 135.4, 160.8 (NCS) ppm; MS m/z (Irel, %): 484 [M+] (5), 449 [M-Cl]+ (5), 438 [M-NO2]+ (3), 356 [M − naphthalene]+ (2), 143 [naphthylamine]+ (60), 127 [naphthalene]+ (100); HRMS (ESI+) m/z calcd for C20H13N2O2Cl4S [M + H]+: 484.9446; found: 484.9443.
Synthesis of 2-(Benzylsulfanyl)-4-(dichloromethyl)-3-nitrobenzo[h]quinoline (36a) (General method). To a solution of diene 35a (0.466 g, 1.0 mmol) in chloroform (10 mL) at 0 °C, triethylamine (0.202 g, 2.0 mmol) was added. The solution was kept at 0 °C for 2 h and then allowed to warm to r.t. for 3 h. Subsequently, the solution was concentrated and diluted with 5 mL MeOH. The resulting precipitate was filtered off, washed with 10% aq. HCl (2 × 5 mL), water (2 × 5 mL) and cold MeOH (5 mL), and dried in vacuo to give benzoquinoline 36a. Yield 0.326 g (76%), yellow solid, m.p. 173–174 °C. IR (ATR) νmax = 3033, 1541, 1336, 1198, 828, 711 cm−1. 1H NMR (400 MHz, CDCl3) δ = 4.78 (2H, s, SCH2), 7.12 (1H, s, CHCl2), 7.27–7.36 (3H, m, H Ph), 7.48–7.52 (2H, m, H Ph), 7.57 (1H, ddd, J = 7.2 Hz, J = 1.1 Hz, J = 7.9 Hz, H Ar), 7.79 (1H, ddd, J = 7.3 Hz, J = 1.4 Hz, J = 7.3 Hz, H Ar), 7.95 (2H, d, J = 9.0 Hz, H Ar), 8.64 (1H, d, J = 9.4 Hz, H Ar), 9.18 (1H, dd, J = 7.6 Hz, J = 0.8 Hz, H Ar) ppm; 13C NMR (100 MHz, CDCl3) δ = 35.8 (SCH2), 63.4 (CHCl2), 119.2, 122.3 (CH), 125.4 (CH), 127.7 (CH), 127.9 (CH), 128.0 (CH), 128.5 (CH), 128.7 (2 × CH), 129.0 (2 × CH), 130.1 (CH), 130.4, 134.2, 134.8, 136.1, 139.4, 147.7, 149.8 ppm; MS m/z (Irel, %): 428 [M+] (2), 344 [M-CH2Cl2]+ (2), 91 [PhCH2]+ (100); HRMS (ESI+) m/z calcd for C21H14N2O2Cl2SNa [M + Na]+: 451.0051; found: 451.0050.
2-[(4-Chlorophenyl)sulfanyl]-4-(dichloromethyl)-3-nitrobenzo[h]quinoline (36b). Same procedure as for 36a, but starting from 35b. Yield 0.382 g (85%), yellow solid, m.p. 162–163 °C. IR (ATR) νmax = 3073, 1572, 1541, 1388, 1028, 739 cm−1. 1H NMR (400 MHz, CDCl3) δ = 7.19 (1H, s, CHCl2), 7.53 (2H, d, J = 8.6 Hz, H Ph), 7.58 (1H, ddd, J = 7.1 Hz, J = 1.3 Hz, J = 8.3 Hz, H Ph), 7.63 (2H, d, J = 8.6 Hz, H Ph), 7.72 (1H, ddd, J = 7.0 Hz, J = 1.1 Hz, J = 8.1 Hz, H Ph), 7.88 (1H, d, J = 7.9 Hz, H Ph), 7.92 (1H, d, J = 9.3 Hz, H Ph), 8.32 (1H, d, J = 8.3 Hz, H Ph), 8.61 (1H, d, J = 9.3 Hz, H Ph) ppm; 13C NMR (100 MHz, CDCl3) δ = 63.4 (CHCl2), 119.8, 122.0 (CH), 125.3 (CH), 127.2, 127.8 (CH), 128.0 (CH), 129.1 (CH), 129.6 (2 × CH), 130.1 (CH), 130.4 (SC Ph), 134.0 (CCl), 135.4, 136.4 (Cl2CHC), 137.5 (2 × CH), 138.9 (CNO2), 147.8 (NC), 149.8 (NCS) ppm; MS m/z (Irel, %): 448 [M+] (28), 413 [M − Cl]+ (7), 364 [M-CHCl2-NO2+H]+ (15), 175 (100), 111 [PhCl]+ (55); HRMS (ESI+) m/z calcd for C20H11N2O2Cl3SNa [M + Na]+: 470.9505; found: 470.9504.
Synthesis of 2-(Benzylsulfinyl)-4-(dichloromethyl)-3-nitrobenzo[h]quinoline (37a) (General method). To a solution of quinoline 36a (0.429 g, 1.0 mmol) in chloroform (5 mL) and glacial acetic acid (2 mL) at 0 °C, hydrogen peroxide (1.13 g, 10.0 mmol, 30% aq.) was added dropwise. After 3 h at 0 °C, the solution was allowed to reach r.t. and stirred additionally for 2 d. The solution was then extracted with chloroform (3 × 30 mL), washed with water (2 × 50 mL), dried with CaCl2 and dried in vacuo. Column chromatography was carried out with a solvent ratio of 2: 1 (petroleum ether-ethyl acetate). Yield 0.396 g (89%), orange solid, m.p. 138–139 °C. IR (ATR) νmax = 1697, 1533, 1353, 1076, 830, 695 cm−1. 1H NMR (400 MHz, CDCl3) δ = 4.57 (2H, q, J = 12.7 Hz, SCH2), 7.25–7.31 (6H in all, m, CHCl2 and 5H Ph overlapped), 7.81–7.92 (2H, m, H Ar), 8.02 (1H, dd, J = 7.5 Hz, J = 1.5 Hz, H Ar), 8.15 (1H, d, J = 9.4 Hz, H Ar), 8.76 (1H, d, J = 9.4 Hz, H Ar), 9.31 (1H, dd, J = 7.7 Hz, J = 1.0 Hz, H Ar) ppm; 13C NMR (100 MHz, CDCl3) δ = 62.2 (CH2), 62.5 (CHCl2), 122.0 (CH), 123.3, 126.2 (CH), 128.0 (CH), 128.6 (CH), 128.7 (CH), 128.8 (2 × CH), 129.5, 130.4 (2 × CH), 130.5, 131.1 (CH), 131.9 (CH), 134.0, 135.9, 138.8 (CNO2), 148.3, 153.2 ppm; MS m/z (Irel, %): 444 [M+] (10), 428 [M − O]+ (5), 360 [M − CH2Cl2] (5), 96 (100); HRMS (ESI+) m/z calcd for C21H14N2O3Cl2SNa [M + Na]+: 467.0000; found: 466.9998.
2-[(4-Chlorophenyl)sulfinyl]-4-(dichloromethyl)-3-nitrobenzo[h]quinoline (37b). Same procedure as for 37a, but starting from 36b (0.450 g, 1.0 mmol). Yield 0.424 g (91%), yellow solid, m.p. 166–168 °C. IR (ATR) νmax = 3082, 1546, 1342, 1092, 828, 750 cm−1. 1H NMR (400 MHz, CDCl3) δ = 7.20 (1H, s, CHCl2), 7.50 (2H, d, J = 8.6 Hz, H Ar), 7.84–7.91 (2H, m, H Ar), 7.95–8.03 (3H, m, H Ar), 8.12 (1H, d, J = 9.4 Hz, H Ar), 8.71 (1H, d, J = 9.4 Hz, H Ar), 9.28–9.33 (1H, m, H Ar) ppm; 13C NMR (100 MHz, CDCl3) δ = 62.4 (CHCl2), 122.0 (CH), 123.3, 125.8 (CH), 127.0 (2 × CH), 128.2 (CH), 128.8 (CH), 129.7 (2 × CH), 130.5, 131.0 (CH), 132.0 (CH), 134.0, 136.0, 138.0 (CNO2), 138.3, 141.1, 148.2, 153.4 (NCS) ppm; MS m/z (Irel, %): 464 [M+] (2), 418 [M-NO2]+ (2), 365 [M-O-CHCl2]+ (38), 335 [M-CHCl2-NO2]+ (12), 111 [PhCl]+ (50), 100 (100); HRMS (ESI+) m/z calcd for C20H11N2O3Cl3SNa [M + Na]+: 486.9454; found: 486.9454.
Synthesis of 4-(Dichloromethyl)-3-nitro-2-(pyrrolidin-1-yl)benzo[h]quinoline (38). To a solution of quinoline 37a (0.445 g, 1.0 mmol) in toluene (10 mL), pyrrolidine (0.107 g, 1.5 mmol) was added and the mixture heated to 100 °C for 3 h. After cooling, the crude product was purified by column chromatography using petroleum ether - ethyl acetate (10: 1). Yield 0.290 g (77%), red solid, m.p. 169–170 °C. IR (ATR) νmax = 2884, 1584, 1506, 1359, 821, 734 cm−1. 1H NMR (400 MHz, CDCl3) δ = 1.99–2.09 (4H, m, NCCH2), 3.61–3.71 (4H, m, NCH2), 7.07 (1H, s, CHCl2), 7.61–7.71 (3H, m, H Ar), 7.85 (1H, dd, J = 7.6 Hz, J = 1.3 Hz, H Ar), 8.50 (1H, d, J = 9.3 Hz, H Ar), 9.03 (1H, d, J = 7.9 Hz, H Ar) ppm; 13C NMR (100 MHz, CDCl3) δ = 25.5 (2 × NCCH2), 48.1 (2 × NCH2), 64.0 (CHCl2), 113.9, 122.6 (CH), 124.1 (CH), 125.5 (CH), 126.6 (CH), 127.6 (CH), 129.2 (CH), 130.2, 130.4 (CNO2), 134.3, 135.6, 145.5, 147.5 ppm; MS m/z (Irel, %): 375 [M+] (100), 358 [M − OH]+ (15), 340 [M − Cl]+ (5), 329 [M − NO2]+ (12), 259 [M-NO2-Pyr]+ (30); HRMS (ESI+) m/z calcd for C18H15N3O2Cl2Na [M + Na]+: 398.0439; found: 398.0440.
Synthesis of 2-ethoxy-2-oxoethyl 4,5-dichloro-1,2-thiazole-3-carboxylate (41). To a solution of isothiazole 40 (0.198 g, 1.0 mmol) and ethyl bromoacetate (0.501 g, 3.0 mmol) in EtOH (10 mL), sodium ethanolate (0.204 g, 3.0 mmol) was added at r.t. The mixture was heated to reflux for 3 d. The product was extracted with chloroform (3 × 10 mL) and purified through a short column chromatography using petroleum ether – ethyl acetate (10:1). Yield 0.165 g (58%), yellowish solid, m.p. 31–33 °C. IR (KBr) νmax = 2983, 1743, 1427, 1193, 1038, 947 cm−1. 1H NMR (200 MHz, CDCl3) δ = 1.31 (3H, t, J = 7.1 Hz, OCH2CH3), 4.28 (2H, q, J = 7.1 Hz, OCH2CH3), 4.91 (2H, s, OCH2C=O) ppm; 13C NMR (50 MHz, CDCl3) δ = 14.0 (CH3), 61.7 (OCH2CH3, OCH2C=O), 126.1 (CCl), 150.8 (SCCl), 153.1 (C=N), 158.2, 166.7; MS m/z (Irel, %): 283 [M+] (18), 238 [M − OEt]+ (15), 210 [M − CO2Et]+ (12), 180 [M − OCH2CO2Et]+ (100); HRMS (ESI+) m/z calcd for C8H7NO4Cl2SNa [M + Na]+: 305.9371; found: 305.9370.
Synthesis of 2,2,2-Trifluoroethyl 4,5-dichloro-1,2-thiazole-3-carboxylate (43). A solution of isothiazole 42 (0.217 g, 1.0 mmol) and 2,2,2-trifluoroethanol (0.300 g, 4.0 mmol) in 20 mL dry THF was refluxed for 4 d. After cooling to r.t., the solution was concentrated, diluted with water (20 mL), extracted with hexane (3 × 20 mL), and washed with water (2 × 20 mL). Subsequently, the product was purified by column chromatography (hexane). Yield 0.179 g (64%), white solid, m.p. 51–52 °C. IR (KBr) νmax = cm−1: 1741, 1430, 1358, 1167, 1091, 745. 1H NMR (200 MHz, CDCl3) δ = 4.61 (2H, q, J = 8.2 Hz, OCH2); 13C NMR (50 MHz, CDCl3) δ = 61.3 (JC,F = 37.1 Hz, CH2), 122.6 (JC,F = 276.0 Hz, CF3), 126.3 (CCl), 151.3 (C=N), 152.3 (SCCl), 157.3 (C=O) ppm; MS m/z (Irel, %): 279 [M+] (15), 244 [M − Cl]+ (2), 197 [M-CH2CF3+H]+ (12), 180 [M-OCH2CF3]+ (100), 153 [M-CO2CH2CF3+H]+ (40); HRMS (ESI+) m/z calcd for C6H2NO2Cl2SF3Na [M + Na]+: 301.9033; found: 301.9035.
Synthesis of 4,5-Dichloro-N’-phenyl-1,2-thiazole-3-carbohydrazide (44a) (General method). To a suspension of phenylhydrazine (0.324 g, 3.0 mmol) in dry THF (10 mL), the isothiazole 42 (0.216 g, 1.0 mmol) was added at r.t. The mixture was stirred for 1 d. After removal of the solvent, the residue was treated with cold HCl (10%) and the resulting precipitate filtered off. Subsequently, it was washed with cold water (3 × 5 mL) and Et2O (2 × 3 mL). The product was dried in vacuo. Yield 0.216 g (75%), light brown solid, m.p. 145–147 °C. IR (KBr) νmax = 3254, 1678, 1605, 1351, 884, 746 cm−1. 1H NMR (200 MHz, CDCl3) δ = 6.88–6.96 (3H, m, H Ph), 7.20–7.26 (3H, m, NH, 2H Ph), 8.81 (1H, s, NH) ppm; 13C NMR (50 MHz, CDCl3) δ = 113.8 (2 × CH), 121.6 (CH), 125.3 (CCl), 129.2 (2 × CH), 147.3, 150.9 (SCCl), 155.3, 158.9 ppm; MS m/z (Irel, %): 287 [M+] (14), 180 [M-CONHNHPh]+ (5), 107 [PhNHNH]+ (100); HRMS (ESI+) m/z calcd for C10H7N3OCl2SNa [M + Na]+: 309.9585; found: 309.9585.
4,5-Dichloro-N’-[3-(trifluoromethyl)phenyl]-1,2-thiazole-3-carbohydrazide (44b). Same procedure as for 44a, but using 3-trifluoromethylphenylhydrazine (0.370 g, 2.1 mmol) and stirring the mixture at reflux for 4 h. Yield 0.338 g (95%), yellowish solid, m.p. 152–153 °C. IR (KBr) νmax = 3300, 1691, 1497, 1340, 1161, 798 cm−1. 1H NMR (200 MHz, DMSO-d6) δ = 7.03–7.09 (3H, m, H Ar), 7.34–7.49 (1H, m, H Ar), 8.57 (1H, s, NH), 10.79 (1H, s, NH) ppm; 13C NMR (50 MHz, DMSO-d6) δ = 108.2 (JC,F = 3.9 Hz, CH), 115.2 (JC,F = 4.0 Hz, CH), 116.1 (CH), 124.6 (JC,F = 271.5 Hz, CF3), 123.0 (CCl), 129.7 (JC,F = 31.3 Hz, CCF3), 130.2 (CH), 149.5 (CCl), 149.9, 157.8, 160.1 (C=O) ppm; MS m/z (Irel, %): 355 [M+] (25), 336 [M-F]+ (2), 180 [M-CF3-Ph-NHNH]+ (60), 175 [CF3-Ph-NHNH]+ (100), 152 [M-CONHNH-Ph-CF3]+ (10); HRMS (ESI+) m/z calcd for C11H6N3OCl2SF3Na [M + Na]+: 377.9458; found: 377.9459.
4,5-Dichloro-N-(4-cyano-2,5-difluorophenyl)-1,2-thiazole-3-carboxamide (44c). Same procedure as for 44a, but using 4-cyano-2,5-difluoro-aniline (0.354 g, 2.3 mmol) and stirring the mixture at reflux for 6 h. Yield 0.227 g (68%), white solid, m.p. 164–166 °C. IR (KBr) νmax = 3361, 2237 (CN), 1715, 1528, 1190, 673 cm−1. 1H NMR (200 MHz, CDCl3) δ = 7.41 (1H, dd, J = 9.8 Hz, J = 5.4 Hz, H Ar), 8.57 (1H, dd, J = 10.6 Hz, J = 6.2 Hz, H Ar), 9.49 (1H, s, NH) ppm; 13C NMR (50 MHz, CDCl3) δ = 95.6 (JC,F = 18.8 Hz, 9.6 Hz, C(CN)), 108.6 (JC,F = 28.7 Hz, 1.5 Hz, CH), 112.9 (JC,F = 2.3 Hz, C(CN)), 118.3 (JC,F = 24.5 Hz, 2.3 Hz, CH), 125.7 (CCl), 131.7 (JC,F = 11.9 Hz, NC Ph), 147.5 (JC,F = 244.0 Hz, 2.8 Hz, CF), 152.1 (CCl), 154.6, 156.3 (C=O), 160.1 (JC,F = 255.3 Hz, 2.3 Hz, CF) ppm; MS m/z (Irel, %): 333 [M+] (22), 180 [M-NH-Ph-F2-(CN)]+ (100), 152 [M-CONH-Ph-F2-(CN)]+ (15); HRMS (ESI+) m/z calcd for C11H3N3OCl2SF2Na [M + Na]+: 355.9240; found: 355.9241.
4,5-Dichloro-N-[2-(1H-indol-3-yl)ethyl]-1,2-thiazole-3-carboxamide (44d). Same procedure as for 44a, but using tryptamine (0.352 g, 2.2 mmol) and triethylamine (0.223 g, 2.1 mmol). Yield 0.265 g (78%), white solid, m.p. 183–185 °C. IR (KBr) νmax = 3302, 1650, 1537, 1349, 946, 738 cm−1. 1H NMR (200 MHz, CDCl3) δ = 3.08 (2H, t, J = 8.1 Hz, Ind-CH2), 3.72–3.82 (2H, m, NCH2), 7.07 (1H, s, NHC=O), 7.11–7.25 (3H, m, 2H Ar, NCH), 7.37 (1H, d, J = 7.7 Hz, H Ar), 7.63 (1H, d, J = 7.7 Hz, H Ar), 8.15 (1H, s, NH) ppm; 13C NMR (50 MHz, CDCl3) δ = 25.3 (CH2), 39.6 (CH2), 111.2 (CH), 112.6, 118.7 (CH), 119.5 (CH), 122.1 (CH, CCl), 122.2 (CH), 127.2, 136.4, 150.4 (CCl), 156.7, 159.0 (C=O) ppm; MS m/z (Irel, %): 339 [M+] (4), 180 [M-NHCH2-ind]+ (5), 143 [ind-CHCH2]+ (100), 130 [ind-CH2]+ (91); HRMS (ESI+) m/z calcd for C14H11N3OCl2SNa [M + Na]+: 361.9900; found: 361.9898.
4,5-Dichloro-N-[(6-chloropyridin-3-yl)methyl]-N-methyl-1,2-thiazole-3-carboxamide (44e). Same proce-dure as for 44a, but using isothiazole 42 and 1-(6-chloropyridin-3-yl)-N-methylmethanamine (0.313 g, 2.0 mmol) at 0 °C and then stirring at r.t. for 5 h. Yield 0.290 g (86%).
Alternative synthesis of thiazole44e: To a suspension of 4,5-dichloro-N’-phenyl-1,2-thiazole-3-carbohydrazide (44a) (0.288 g, 1.0 mmol) in DMSO (10 mL), 1-(6-chloropyridin-3-yl)-N-methylmethanamine (0.313 g, 2.0 mmol) was added. The mixture was heated to 100 °C for 8 h. After cooling to r.t., water (50 mL) was added and the mixture extracted with chloroform (3 × 15 mL). Subsequently, the product was purified by column chromatography using petroleum ether-ethyl acetate (3: 1). The product was dried in vacuo. Yield 0.232 g (69%), a mixture of two rotamers in relation 10: 6, yellowish solid, m.p. 43–45 °C. IR (KBr) νmax = 2934, 1651, 1460, 1349, 1106, 834 cm−1. 1H NMR (600 MHz, CDCl3) for major isomer δ = 2.90 (3H, s, NCH3), 4.73 (2H, s, NCH2), 7.34 (1H, d, J = 8.2 Hz, H pyr), 7.71 (1H, dd, J = 2.5 Hz, J = 8.3 Hz, H pyr), 8.37 (1H, d, J = 2.3 Hz, H pyr) ppm; minor isomer δ = 3.02 (1.8H, s, NCH3), 4.48 (1.2H, s, NCH2), 7.34 (0.6H, d, J = 8.2 Hz, H pyr), 7.72 (0.6H, dd, J = 2.5 Hz, J = 8.3 Hz, H pyr), 8.28 (0.6H, d, J = 2.3 Hz, H pyr) ppm; 13C NMR (150 MHz, CDCl3) for major isomer δ = 35.7 (NCH3), 47.8 (NCH2), 122.5 (CCl), 124.6 (CH), 130.7 (CH2C), 138.8 (CH), 149.2 (CH), 149.6 (CCl), 151.2 (NCCl), 159.6, 162.8 (C=O) ppm; minor isomer δ = 32.8 (NCH3), 51.2 (NCH2), 123.2 (CCl), 124.5 (CH), 130.3 (CH2C), 138.1 (CH), 148.9 (CH), 149.8 (CCl), 151.5 (NCCl), 159.5, 162.4 (C=O) ppm; 15N NMR (43.4 MHz, CDCl3, doped with nitromethane (0.0 ppm)) δ = -267.5 (NMe, minor isomer), -266.3 (NMe, major isomer), -75.7 (NCCl, major isomer), -75.5 (NCCl, minor isomer) ppm, NS not detected; MS m/z (Irel, %): 337 [M+] (3), 319 [M-CH3]+ (2), 300 [M-Cl]+ (6), 180 [M-CH3-N-CH2-isothiazole]+ (14), 155 [isothiazole-N-CH2-CH3]+ (100); HRMS (ESI+) m/z calcd for C11H8N3OCl3SNa [M + Na]+: 357.9351; found: 357.9352.
4-Chloro-5-(morpholin-4-yl)-N’-phenyl-1,2-thiazole-3-carbohydrazide (45). Following the alternative procedure for azole 44e using morpholine (0.348 g, 4.0 mmol). The mixture was stirred at 90–95 °C for 4 h. Yield 0.264 g (78%), yellowish solid, m.p. 163–164 °C. IR (ATR) νmax = 3294, 1695, 1494, 1403, 1115, 688 cm−1. 1H NMR (400 MHz, CDCl3) δ = 3.32–3.41 (4H, m, NCH2), 3.82–3.91 (4H, m, OCH2), 6.24 (1H, s, Ph-NH), 6.88–6.95 (3H, m, H Ph), 7.21–7.26 (2H, m, H Ph), 8.78 (1H, s, CONH) ppm; 13C NMR (100 MHz, CDCl3) δ = 50.8 (2 × NCH2), 66.0 (2 × OCH2), 108.9, 113.8 (2 × CH), 121.4 (CH), 129.2 (2 × CH), 147.7, 156.0 (NCC=O), 160.1 (C=O), 173.1 (morph-C) ppm; MS m/z (Irel, %): 338 [M+] (75), 303 [M-Cl]+ (8), 231 [M-PhNHNH]+ (35), 197 [M-PhNHNH-Cl+H]+ (100), 107 [PhNHNH]+ (60); HRMS (ESI+) m/z calcd for C14H15N4O2ClSNa [M + Na]+: 361.0502; found: 361.0502.
Synthesis of 4-chloro-5-[(4-chlorophenyl)sulfanyl]-N’-phenyl-1,2-thiazole-3-carbohydrazide (46a) (General method). To a solution of isothiazole 44a (0.288 g, 1.0 mmol) in DMSO (10 mL), 4-chlorothiophenol (0.173 g, 1.2 mmol) and sodium ethanolate (0.082 g, 1.2 mmol) were added. The mixture was stirred at 110 °C for 8 h. After concentration at 100 °C, it was poured into 50 mL diluted HCl (5%) and extracted with chloroform (3 × 20 mL). The combined organic phases were washed with water, dried over calcium chloride, and purified by column chromatography using petroleum ether-ethyl acetate (2:1). Subsequently, the dried product was washed with cold methanol (1 × 3 mL). Yield 0.266 g (67%), white solid, m.p. 169–170 °C. IR (ATR) νmax = 3250, 1655, 1493, 1094, 896, 695 cm−1. 1H NMR (400 MHz, CDCl3) δ = 5.11 (1H, s br, Ph-NH), 6.88–6.95 (3H, m, H Ph), 7.20–7.26 (2H, m, H Ph), 7.43 (2H, d, J = 8.5 Hz, H Ar), 7.53 (2H, d, J = 8.5 Hz, H Ar), 8.7 (1H, s, CONH) ppm; 13C NMR (100 MHz, CDCl3) δ = 113.8 (2 × CH), 121.6 (CH), 123.3, 128.2 (CCl), 129.2 (2 × CH), 130.5 (2 × CH), 134.7 (2 × CH), 136.7 (CS-Thiaz), 147.5, 155.5 (CC=O), 159.3, 160.9 (C=O) ppm; MS m/z (Irel, %): 395 [M+] (20), 288 [M-PhNHNH]+ (10), 261 [M-PhNHNHCO+H]+ (4), 225 [M-PhNHNHCO-Cl]+ (15), 107 [NHNHPh]+ (100); HRMS (ESI+) m/z calcd for C16H11N3OCl2S2Na [M + Na]+: 417.9618; found: 417.9617.
4-Chloro-5-[(4-chlorophenyl)sulfanyl]-N’-[3-(trifluoromethyl)phenyl]-1,2-thiazole-3-carbohydrazide (46b). Same procedure as for 46a, but using 44b (0.356 g, 1.0 mmol). The product was purified using petroleum ether-ethyl acetate (10: 1). Yield 0.325 g (70%), white solid, m.p. 144–145 °C. IR (ATR) νmax = 3246, 1662, 1476, 1338, 1123, 694 cm−1. 1H NMR (400 MHz, CDCl3) δ = 6.39 (1H, s br, NH), 7.05 (1H, dd, J = 8.0 Hz, J = 2.1 Hz, H Ar), 7.11 (1H, s, H Ar), 7.15 (1H, d, J = 7.9 Hz, H Ar), 7.32 (1H, t, J = 8.0 Hz, H Ar), 7.44 (2H, d, J = 8.6 Hz, H Ar), 7.54 (2H, d, J = 8.6 Hz, H Ar), 8.77 (1H, s, NH). 13C (100 MHz, CDCl3) δ = 110.2 (JC,F = 4.1 Hz, CH), 116.8 (CH), 118.1 (JC,F = 3.9 Hz, CH), 123.9 (JC,F = 272.5 Hz, CF3), 123.1 (CCl), 128.0 (CCl), 129.7 (CH), 130.9 (2 × CH), 131.6 (JC,F = 32.2 Hz, CCF3), 134.8 (2 × CH), 136.9 (CS), 148.1 (N2H2C), 155.1 (SCS), 159.5 (CCO), 161.5 (C=O) ppm; MS m/z (Irel, %): 463 [M+] (35), 288 [M-N2H2PhCF3]+ (93), 225 [M-CON2H2PhCF3]+ (80), 145 [PhCF3]+ (100); HRMS (ESI+) m/z calcd for C17H10N3OCl2S2F3Na [M + Na]+: 485.9492; found: 485.9494.
4-Chloro-5-[(4-chlorophenyl)sulfanyl]-N-(4-cyano-2,5-difluorophenyl)-1,2-thiazole-3-carboxamide (46c). Same procedure as for 46a, but using 44c (0.334 g, 1.0 mmol). The product was purified by using petroleum ether-ethyl acetate (10: 1). Yield 0.367 g (83%), white solid, m.p. 152–153 °C. IR (ATR) νmax = 3384, 2241 (CN), 1703, 1473, 1196, 617 cm−1. 1H NMR (400 MHz, CDCl3) δ = 7.24–7.66 (5H, m, H Ar), 8.57 (1H, s, H Ar), 9.47 (1H, s, NH) ppm; 13C NMR (101 MHz, CDCl3) δ = 95.4 (JC,F = 19.8 Hz, 11.0 Hz, Ar CCN), 108.6 (JC,F = 28.6 Hz, CH), 112.9 (Ph-CN), 118.3 (JC,F = 24.2 Hz, CH), 123.0 (CCl), 127.6 (CCl), 130.7 (2 × CH), 131.9 (JC,F = 11.7 Hz, CNH), 135.0 (2 × CH), 137.1 (SC), 147.5 (JC,F = 244.3 Hz, CF), 154.9 (SCS), 156.9 (SNC), 160.2 (JC,F = 253.8 Hz, CF), 163.0 (C=O) ppm; MS m/z (Irel, %): 441 [M+] (40), 288 [M-NHPhF2CN]+ (100), 225 [M-ClCOPhF2CN]+ (80), 143 [SPhCl]+ (53); HRMS (ESI+) m/z calcd for C17H7N3OCl2S2F2Na [M + Na]+: 463.9273; found: 463.9271.
4-Chloro-5-[(4-chlorophenyl)sulfanyl]-N-[2-(1H-indol-3-yl)ethyl]-1,2-thiazole-3-carboxamide (46d). Same procedure as for 46a, but using 44d (0.340 g, 1.0 mmol). After addition of HCl the crude product precipitated and was filtered off, washed with diluted HCl (3 mL), water (2 × 5 mL), cold MeOH (3 mL) and dried in vacuo. Yield 0.399 g (89%), white solid, m.p. 176–177 °C. IR (ATR) νmax = 3280, 1667, 1525, 1338, 821, 740 cm−1. 1H NMR (400 MHz, DMSO-d6) δ = 2.88–2.99 (2H, m, Ind-CH2), 3.47–3.58 (2H, m, NCH2), 6.98 (1H, t, J = 7.0 Hz, H ind), 7.06 (1H, t, J = 7.0 Hz, H ind), 7.19 (1H, s, H ind), 7.34 (1H, d, J = 7.7 Hz, H ind), 7.51–7.73 (5H, m, H ind, 4H Ar), 8.84 (1H, s, CONH), 10.82 (1H, s, NH ind); 13C NMR (101 MHz, DMSO-d6) δ = 25.1 (ind-CH2), 39.7 (NCH2), 111.5, 111.6 (CH), 118.4 (CH), 118.5 (CH), 121.1 (CH), 121.5, 122.8 (NCH), 127.4, 128.8, 130.6 (2 × CH), 134.5 (2 × CH), 135.3 (SC), 136.4, 158.6 (CCO), 159.4 (SCS), 159.8 (C=O) ppm; MS m/z (Irel, %): 447 [M+] (3), 288 [M-NHC2H4-ind]+ (1), 143 [SPhCl]+ (100), 130 [I ndCH2]+ (53); HRMS (ESI+) m/z calcd for C20H15N3OCl2S2Na [M + Na]+: 469.9931; found: 469.9931.
2-Ethoxy-2-oxoethyl 4-chloro-5-[(4-chlorophenyl)sulfanyl]-1,2-thiazole-3-carboxylate (46e). Same proce-dure as for 46a, but using 41 (0.284 g, 1.0 mmol). The product was purified by using petroleum ether-ethyl acetate (10: 1). Yield 0.267 g (68%), yellowish solid, m.p. 72–73 °C. IR (ATR) νmax = 1755, 1722, 1344, 1198, 1084, 505 cm−1. 1H NMR (400 MHz, CDCl3) δ = 1.28 (3H, t, J = 7.0 Hz, OCH2CH3), 4.25 (2H, q, J = 7.0 Hz, OCH2), 4.87 (2H, s, OCH2), 7.42 (2H, d, J = 8.7 Hz, H Ar), 7.51 (2H, d, J = 8.6 Hz, H Ar) ppm; 13C NMR (101 MHz, CDCl3) δ = 14.1 (CH3), 61.6 (OCH2CH3), 61.7 (COOCH2), 124.0 (CCl), 128.1 (CCl), 130.5 (2 × CH), 134.6 (2 × CH), 136.7 (SC Ar), 153.5 (NC), 158.6, 160.9 (C=O), 166.8 (COOEt); MS m/z (Irel, %): 391 [M+] (48), 288 [M-OCH2CO2Et]+ (50), 225 [M-CO2CH2CO2Et-Cl]+ (70), 144 [ClPhSH]+ (100); HRMS (ESI+) m/z calcd for C14H11NO4Cl2S2Na [M + Na]+: 413.9404; found: 413.9405.
Synthesis of 5-methyl-5-phenyl-3-(trichloroethenyl)-4,5-dihydro-1,2-oxazole (47) (General method). To a solution of nitrodiene 1 (0.271 g, 1.0 mmol) in dry toluene (10 mL) prop-1-en-2-ylbenzene (1.182 g, 10.0 mmol), 18-crown-6 (0.066 g, 0.25 mmol), activated molar sieves 4 Å (0.250 g), and powdered NaOH (0.120 g, 3.0 mmol) were added under nitrogen atmosphere. The mixture was then stirred at 60 °C for 16 h, and at 80 °C for another 16 h. After completion of the reaction, the solvent was evaporated, 10% aq. HCl (10 mL) was added, and the mixture was extracted with DCM (3 × 10 mL). The crude product was purified by column chromatography using petroleum ether-ethyl acetate (10: 1). Yield 0.221 g (76%), yellow viscous oil. IR (ATR) νmax = 2977, 1684, 1446, 1265, 859, 697 cm−1. 1H NMR (400 MHz, CDCl3) δ = 1.77 (3H, s, CH3), 3.43 (1H, d, J = 16.9 Hz, ONCCH2), 3.50 (1H, d, J = 16.9 Hz, ONCH2), 7.37–7.48 (5H, m, H Ph) ppm; 13C NMR (100 MHz, CDCl3) δ = 27.8 (CH3), 50.1 (CH2), 90.3 (OCCH3), 121.7, 124.5 (2 × CH), 127.7 (CH), 128.6 (2 × CH), 137.1 (CCl2), 144.2, 152.5 (NCCCl) ppm; MS m/z (Irel, %): 289 [M+] (4), 274 [M-CH3]+ (4), 117 [PhC2H5]+ (60), 105 (100); HRMS (ESI+) m/z calcd for C12H10NOCl3Na [M + Na]+: 311.9726; found: 311.9725.
tert-Butyl 3-(trichloroethenyl)-4,5-dihydro-1,2-oxazole-5-carboxylate (48). Same procedure as for 47, but using tert-butyl prop-2-enoate (1.282 g, 10.0 mmol). Yield 0.195 g (65%), yellowish oil. IR (ATR) νmax = 2981, 1732, 1368, 1232, 1150, 838 cm−1. 1H NMR (400 MHz, CDCl3) δ = 1.47 (9H, s, CCH3), 3.53 (2H, d, J = 9.4 Hz, ONCCH2), 5.03 (1H, t, J = 9.4 Hz, OCH) ppm; 13C NMR (100 MHz, CDCl3) δ = 27.8 (3 × CH3), 40.0 (CH2), 80.2 (OCH), 83.1 (OCMe3), 120.5 (CCl), 125.2 (CCl2), 152.3 (ONC), 168.0 (C=O) ppm; 15N NMR (43.4 MHz, CDCl3, doped with nitromethane (0.0 ppm)) δ = 7.8 ppm; MS m/z (Irel, %): 299 [M+] (12), 200 [M-CO2CMe3]+ (100), 170 [M-C2Cl3]+ (12), 129 [C2Cl3]+ (52), 100 [CO2CMe3-H]+ (95); HRMS (ESI+) m/z calcd for C10H12NO3Cl3Na [M + Na]+: 321.9781; found: 321.9781.
2-Ethylhexyl 3-(trichloroethenyl)-4,5-dihydro-1,2-oxazole-5-carboxylate (49). Same procedure as for 47, but using 2-ethylhexyl prop-2-enoate (1.843 g, 10.0 mmol). Yield 0.216 g (60%), yellowish oil. IR (ATR) νmax = 2958, 2860, 1732, 1461, 1162, 851 cm−1. 1H NMR (400 MHz, CDCl3) δ = 0.86–0.88 (6H, m, 2 × CH3), 1.19–1.42 (8H, m, 4 × CH2), 1.60–1.64 (1H, m, CH), 3.60 (1H, d, J = 10.7 Hz, ONCCH2), 3.61 (1H, d, J = 7.8 Hz, ONCCH2), 4.06–4.18 (2H, m, OCH2), 5.17 (1H, dd, J = 10.7 Hz, J = 7.7 Hz, OCH) ppm; 13C NMR (100 MHz, CDCl3) δ = 10.9 (CH3), 14.0 (CH3), 22.9 (CH2), 23.6 (CH2), 28.8 (CH2), 30.2 (CH2), 28.6 (CH), 40.2 (ONCCH2), 68.5 (OCH2), 79.6 (OCH), 120.4 (CCl), 125.5 (CCl2), 152.4 (ONC), 169.2 (C=O) ppm; MS m/z (Irel, %): 355 [M+] (10), 320 [M-Cl]+ (22), 198 [M-CO2C8H17]+ (100); HRMS (ESI+) m/z calcd for C14H20NO3Cl3Na [M + Na]+: 378.0407; found: 378.0406.
3-(Trichloroethenyl)-3a,4,5,6,7,7a-hexahydro-1,2-benzoxazole (50). Same procedure as for 47, but using cyclohexene (0.821 g, 10.0 mmol). Yield 0.104 g (41%), yellowish oil. IR (ATR) νmax = 2935, 2863, 1448, 912, 838, 739 cm−1. 1H NMR (400 MHz, CDCl3) δ = 1.23–1.43 (2H, m, CH2), 1.48–1.64 (3H, m, CH2), 1.72–1.82 (1H, m, CH2), 1.83–1.93 (1H, m, CH2), 2.00–2.09 (1H, m, CH2), 3.32–3.41 (1H, m, CH), 4.58 (1H, ddd, J = 8.2 Hz, J = 4.2 Hz, J = 4.2 Hz, CH) ppm; 13C (100 MHz, CDCl3) δ = 19.9 (CH2), 21.7 (CH2), 24.8 (CH2), 25.3 (CH2), 46.1 (CH), 81.4 (CH), 121.0 (CCl), 124.4 (CCl2), 159.1 (ONC) ppm; MS m/z (Irel, %): 253 [M+] (75), 236 [M-OH]+ (5), 218 [M-Cl]+ (7), 182 [M-Cl-HCl]+ (16), 91 (100); HRMS (ESI+) m/z calcd for C9H10NOCl3Na [M + Na]+: 275.9726; found: 275.9725.
Synthesis of 1-[1,1-dichloro-3-(1,3-dithiolan-2-ylidene)-3-nitroprop-1-en-2-yl]-4-(4-fluorophenyl)pipera- zine (52). A solution of dithiolane 51 (0.292 g, 1.0 mmol) in MeOH (10 mL) and 1-(4-fluorophenyl)piperazine (0.433 g, 2.4 mmol) was stirred at reflux for 7 d. After cooling, the solution was concentrated and treated with water for 30 min. The resulting precipitate was filtered off and washed with water (2 × 5 mL). Yield 0.393 g (90%), orange solid, m.p. 47–50 °C. IR (KBr) νmax = 2825, 1509, 1272, 1226, 954, 816 cm−1. 1H NMR (200 MHz, CDCl3) δ = 3.08–3.39 (8H, m, NCH2), 3.48–3.61 (4H, m, SCH2), 6.80–7.11 (4H, m, H Ar) ppm; 13C NMR (50 MHz, CDCl3) δ = 37.6 (SCH2), 40.1 (SCH2), 49.2 (2 × NCH2), 50.6 (2 × NCH2), 113.3 (CCl2), 115.5 (JC,F = 21.9 Hz, 2 × CH), 118.2 (JC,F = 7.7 Hz, 2 × CH), 130.9 (CNO2), 139.0 (CN), 147.9 (JC,F = 2.2 Hz, NC Ar), 157.3 (JC,F = 238.9 Hz, CF), 170.4 (SCS) ppm; MS m/z (Irel, %): 439 [M+] (25), 418 [M − OH]+ (4), 389 [M − NO2]+ (7), 179 [F-Ph-piperazine]+ (15), 122 (100); HRMS m/z calcd for C16H16N3O2FCl2S2 [M]+: 435.0045; found: 435.0047.
Synthesis of 1-[2-Chloro-5-(ethenylsulfanyl)-4-nitrothiophen-3-yl]-4-(4-fluorophenyl)piperazine (53). An aqueous solution of NaOH (3.0 mmol, 40%) was added dropwise to a solution of dithiolane 52 (0.436 g, 1.0 mmol) in DMSO (10 mL) at 0 °C. After 1 h at 0 °C, the mixture was stirred at r.t. for an additional 3 h. Subsequently, cold water was added and the mixture was treated dropwise with diluted HCl until a precipitate formed. The precipitate was filtered off and washed with water (2 × 5 mL) and cold MeOH (3 mL). Yield 0.340 g (85%), yellowish solid, m.p. 83–86 °C. IR (KBr) νmax = 2829, 1543, 1511, 1322, 1233, 831 cm−1. 1H NMR (200 MHz, CDCl3) δ = 3.16–3.26 (4H, m, NCH2), 3.31–3.47 (4H, m, NCH2), 5.83 (1H, d, J = 9.1 Hz, SCHCH2), 8.55 (1H, d, J = 5.9 Hz, SCHCH2), 6.55 (1H, dd, J = 16.4 Hz, J = 9.2 Hz, SCH), 6.86–7.09 (4H, m, H Ar) ppm; 13C NMR (50 MHz, CDCl3) δ = 50.0 (2 × NCH2), 51.5 (2 × NCH2), 115.6 (JC,F = 21,9 Hz, 2 × CH), 118.6 (JC,F = 8.1 Hz, 2 × CH), 119.1 (CCl), 126.2 (CH2 vin), 126.8 (CH vin), 139.4 (CNO2), 141.0 (CN), 143.3 (SCS), 148.5 (JC,F = 3.7 Hz, NC Ar), 157.7 (JC,F = 234.5 Hz, CF) ppm; MS m/z (Irel, %): 399 [M+] (85), 352 [M-HNO2]+ (17), 317 [M-HNO2-Cl]+ (4), 122 (100); HRMS (ESI+) m/z calcd for C16H15N3O2ClFS2 [M + H]+: 399.0278; found: 399.0278.
Synthesis of 5-(Ethenylsulfanyl)-3-[4-(4-fluorophenyl)piperazin-1-yl]-4-nitrothiophene-2-carbaldehyde (54). A solution of thiophene 53 (0.400 g, 1.0 mmol) and POCl3 (0.307 g, 2.0 mmol) in dry DMF was stirred at r.t. for 1 d and then at 55 °C for 2 d. After cooling down to 0 °C, cold water (15 mL) was added and the mixture extracted with chloroform (3 × 10 mL). After drying over CaCl2, the crude product was purified by column chromatography using petroleum ether-ethyl acetate (2: 1). Yield 0.252 g (64%), orange solid, m.p. 104–105 °C. IR (ATR) νmax = 2833, 1621, 1531, 1505, 1223, 997 cm−1. 1H NMR (400 MHz, CDCl3) δ = 3.25–3.33 (4H, m, NCH2), 3.50–3.59 (4H, m, OCH2), 5.99 (2H, dd, J = 16.7 Hz, J = 9.4 Hz, CH2 vin), 6.65 (1H, dd, J = 16.6 Hz, J = 9.3 Hz, SCH vin), 6.88–7.03 (4H, m, H Ar), 10.03 (1H, s, CHO) ppm; 13C NMR (100 MHz, CDCl3) δ = 50.7 (2 × NCH2), 53.5 (2 × OCH2), 115.6, 115.7 (JC,F = 22.2 Hz, 2 × CH Ar), 118.6 (JC,F = 7.7 Hz, 2 × CH Ar), 124.7 (CN), 125.6 (SCH), 128.7 (CH2 vin), 136.2 (CNO2), 147.6 (JC,F = 2.2 Hz, NC Ar), 150.5 (SCS), 157.6 (JC,F = 239.9 Hz, CF), 159.8 (SC), 180.2 (CHO) ppm; MS m/z (Irel, %): 393 [M+] (15), 376 [M − OH]+ (5), 346 [M − HNO2]+ (3), 179 [piperazine-Ph-F]+ (7), 122 (100); HRMS (ESI+) m/z calcd for C17H16N3FO3S2Na [M + Na]+: 416.0515 ; found: 416.0514.
Synthesis of {[5-(Ethenylsulfonyl)-3-(morpholin-4-yl)-4-nitrothiophen-2-yl]methylidene}propanedinitrile (57). A solution of thiophene 56 (0.348 g, 1.0 mmol) and hydrogen peroxide (3.0 mmol, 35%) in acetic acid was stirred at 50–55 °C for 3 h. After cooling, ice was poured into the mixture and the resulting precipitate was filtered off and washed with water (3 × 5 mL). Yield 0.312 g (82%), red solid, m.p. 121–123 °C. IR (KBr) νmax = 2857, 2223 (CN), 1541, 1340, 1111, 857 cm−1. 1H NMR (200 MHz, CDCl3) δ = 3.23–3.30 (4H, m, NCH2), 3.84–3.90 (4H, m, OCH2), 6.15 (1H, dd, J = 9.3, J = 1.4, CH2, cis vin), 6.45 (1H, dd, J = 16.2, J = 1.3, CH2, trans vin), 7.09 (1H, dd, J = 16.3, J = 9.3, CH vin), 8.02 (1H, s, SCCH). 13C NMR (50 MHz, CDCl3) δ = 52.0 (2 × NCH2), 67.0 (2 × OCH2), 80.3 (C(CN)2), 112.2 (2 × CN), 113.3, 124.1 (CH2 vin), 127.4 (SC), 138.5 (CH vin), 146.8 (SCCH), 151.4 (C-morph), 166.1 (SO2C), CNO2 could not be detected. Mass spectrum, m/z (Irel, %): 380 [M+] (3), 363 [M-OH]+ (20), 346 [M-2(OH)]+ (100), 289 [M-SO2C2H3]+ (2); HRMS (ESI+) m/z calcd for C14H12N4O5S2Na [M + Na]+: 403.0147; found: 403.0146.
Synthesis of 2,2’-{[5-(Ethenylsulfanyl)-3-(morpholin-4-yl)-4-nitrothiophen-2-yl]methanediyl}bis(5,5- di-methylcyclohexane-1,3-dione) (58). A solution of thiophene 55 (0.300 g, 1.0 mmol), dimedone (0.308 g, 2.2 mmol) and pyridine (8.0 mg, 0.1 mmol) in MeOH (15 mL) was stirred at r.t. for 1 d and then at 35–40 °C for 2 d. After cooling to r.t., the mixture was treated with diluted HCl and the resulting precipitate was filtered off and washed with water (2 × 5 mL) and cold Et2O (3 mL). Yield 0.411 g (73%), yellow solid, m.p. 159–160 °C. IR (KBr) νmax = 3243, 2961, 1717, 1626, 1542, 1068 cm−1. 1H NMR (200 MHz, CDCl3) δ = 1.03 (12H, s, 4CH3), 2.34 (8H, s, 4COCH2), 2.60–3.80 (10H, m, 2NCH2, 2OCH2, 2COCH), 5.77 (2H, dd, J = 16.4 Hz, J = 9.3 Hz, CH2 vin), 6.41–6.51 (1H, m, SCH), 6.67 (1H, dd, J = 16.4 Hz, J = 9.3 Hz, CH vin) ppm; 13C NMR (50 MHz, CDCl3) δ = 26.7 (SCCH), 28.1 (2 × CH3C), 31.6 (4 × CH3), 46.7 (2 × NCH2), 49.6 (4 × COCH2), 67.4 (2 × OCH2), 77.2 (2 × CC=O), 116.1 (SC), 124.4 (CH2 vin), 127.6 (SCH), 137.2 (CNO2), 141.1 (C-morph), 143.2 (SCS), 189.9 (4 × C=O) ppm; MS m/z (Irel, %): 562 [M+] (12), 543 [M-H2O-H]+ (12), 526 [M-2(H2O)]+ (42), 458 [M-SC2H3-NO2+H]+ (10); HRMS (ESI+) m/z calcd for C27H34N2O7S2Na [M + Na]+: 585.1705; found: 585.1705.
Synthesis of 4-[5-(Ethenylsulfanyl)-4-nitro-2-{[2-(2,4,6-trichlorophenyl)hydrazinylidene]methyl} thiophen-3-yl]morpholine (59). A solution of thiophene 55 (0.300 g, 1.0 mmol) and (2,4,6-trichlorophenyl)-hydrazine (0.634 g, 3.0 mmol) in MeOH (15 mL) was refluxed for 3 h. After cooling to r.t., the mixture was treated with diluted HCl and the resulting precipitate was filtered off and washed with water (2 × 5 mL) and cold MeOH (3 mL). Yield 0.425 g (86%), orange solid, m.p. 192–194 °C. IR (KBr) νmax = 3228, 1520, 1445, 1332, 1107, 851 cm−1. 1H NMR (200 MHz, CDCl3) δ = 3.05–3.19 (4H, m, NCH2), 3.75–3.82 (4H, m, OCH2), 5.87 (2H, dd, J = 16.7 Hz, J = 9.4 Hz, CH2 vin), 6.65 (1H, dd, J = 16.7 Hz, J = 9.4 Hz, SCH vin), 7.36 (2H, s, H Ar), 7.64 (1H, br s, NH), 8.01 (1H, s, NCH) ppm; 13C (50 MHz, CDCl3) δ = 51.0 (2 × NCH2), 67.5 (2 × OCH2), 126.4 (CH2 vin), 126.8 (SCH vin), 127.6 (2 × CCl), 129.0 (2 × CH), 129.1 (CCl), 130.1 (SC), 133.3 (NCH), 135.8 (NHC), 139.5 (CNO2), 142.3 (SCS), 148.5 (C-morph) ppm; MS m/z (Irel, %): 492 [M+] (12), 433 [M-SC2H3]+ (2), 298 [M-NHPhCl3]+ (55), 195 [H2NPhCl3]+ (100); HRMS (ESI+) m/z calcd for C17H15N4O3Cl3S2Na [M + Na]+: 514.9549; found: 514.9547.
Synthesis of 5-{[5-(Ethenylsulfanyl)-3-(morpholin-4-yl)-4-nitrothiophen-2-yl]methylidene}-2,2-dimethyl- 1,3-dioxane-4,6-dione (60). A solution of thiophene 55 (0.300 g, 1.0 mmol), 2,2-dimethyl-1,3-dioxane-4,6-dione (0.288 g, 2.0 mmol) and piperidine (8.5 mg, 0.1 mmol) in MeOH (15 mL) was stirred at r.t. for 1 d. Then another 0.288 g of 2,2-dimethyl-1,3-dioxane-4,6-dione were added and the mixture was stirred for an additional 2 d. Subsequently, the mixture was treated with diluted HCl and the resulting precipitate was filtered off and washed with water (2 × 5 mL) and cold MeOH (3 mL). Yield 0.333 g (73%), red solid, m.p. 173–174 °C. IR (ATR) νmax = 1683, 1516, 1332, 1197, 1113, 787 cm−1. 1H NMR (200 MHz, CDCl3) δ = 1.76 (6H, s, CH3), 3.33–3.39 (4H, m, NCH2), 3.93–3.96 (4H, m, OCH2), 6.00 (2H, dd, J = 16.8 Hz, J = 9.5 Hz, CH2 vin), 6.75 (1H, dd, J = 16.8 Hz, J = 9.5 Hz, SCH vin), 8.68 (1H, s, SCCH) ppm; 13C (200 MHz, CDCl3) δ = 27.5 (2 × CH3), 53.2 (2 × NCH2), 67.1 (2 × OCH2), 103.6 (CMe2), 104.7 (CC=O), 119.5 (SC), 124.9 (SCH vin), 128.7 (CH2 vin), 137.2 (CNO2), 144.1 (SCCH), 156.9 (SCS), 161.4 (C-morph), 162.3 (C=O), 163.4 (C=O) ppm; MS m/z (Irel, %): 426 [M+] (100), 409 [M-OH]+ (10), 351 [M-Me2CO2H]+ (24), 295 [M-NO2-morph+H]+ (27); HRMS (ESI+) m/z calcd for C17H18N2O7S2Na [M + Na]+: 449.0453; found: 449.0453.

 ,
, 
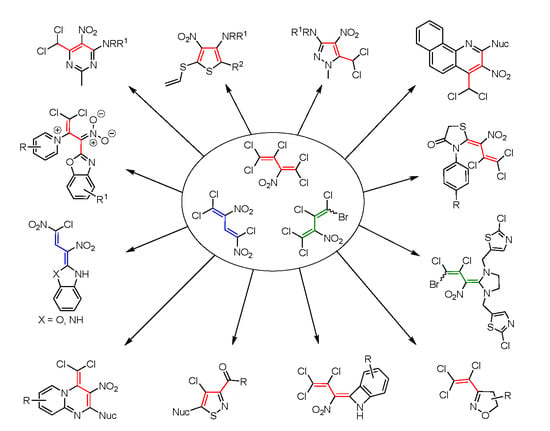
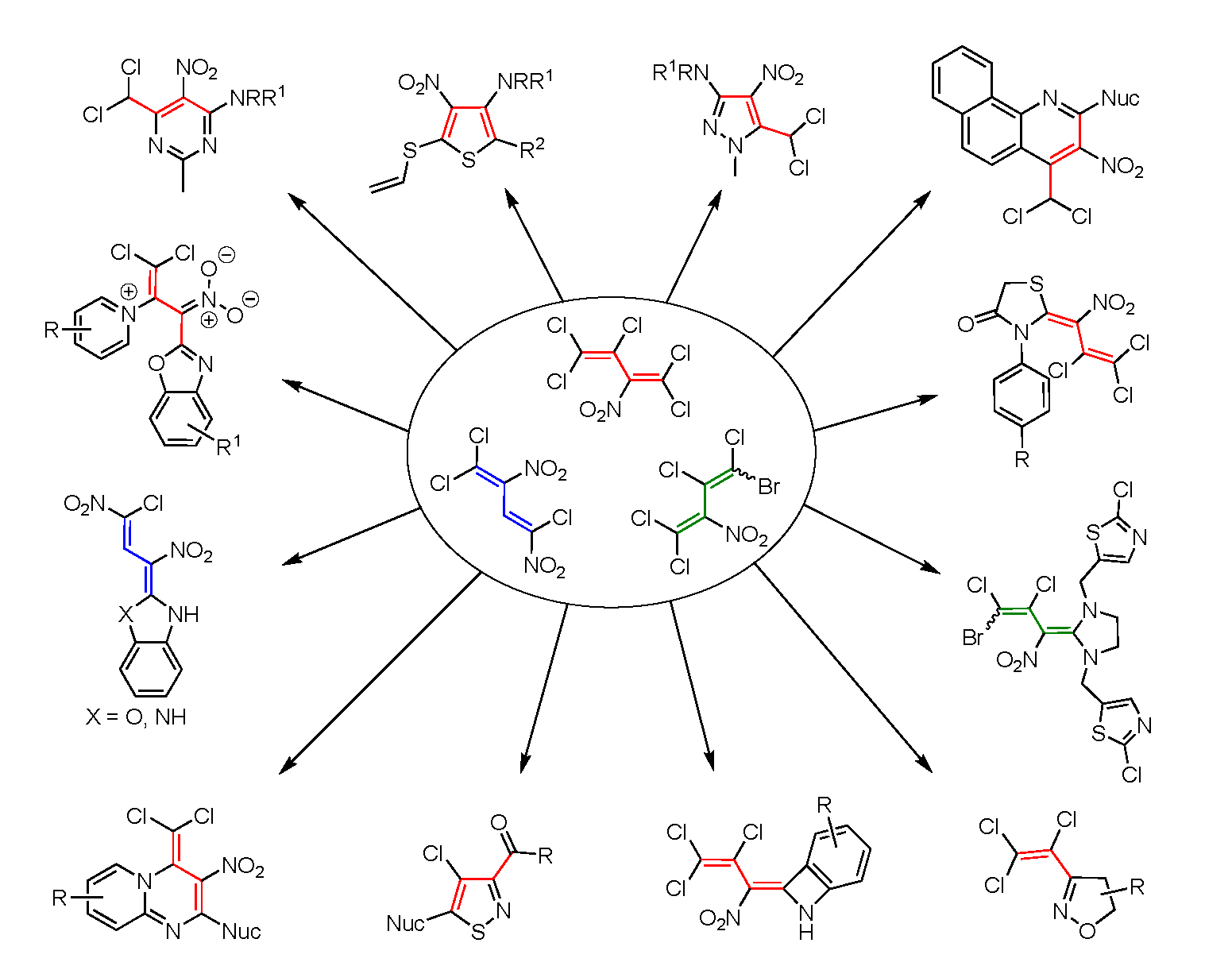{kind=link}
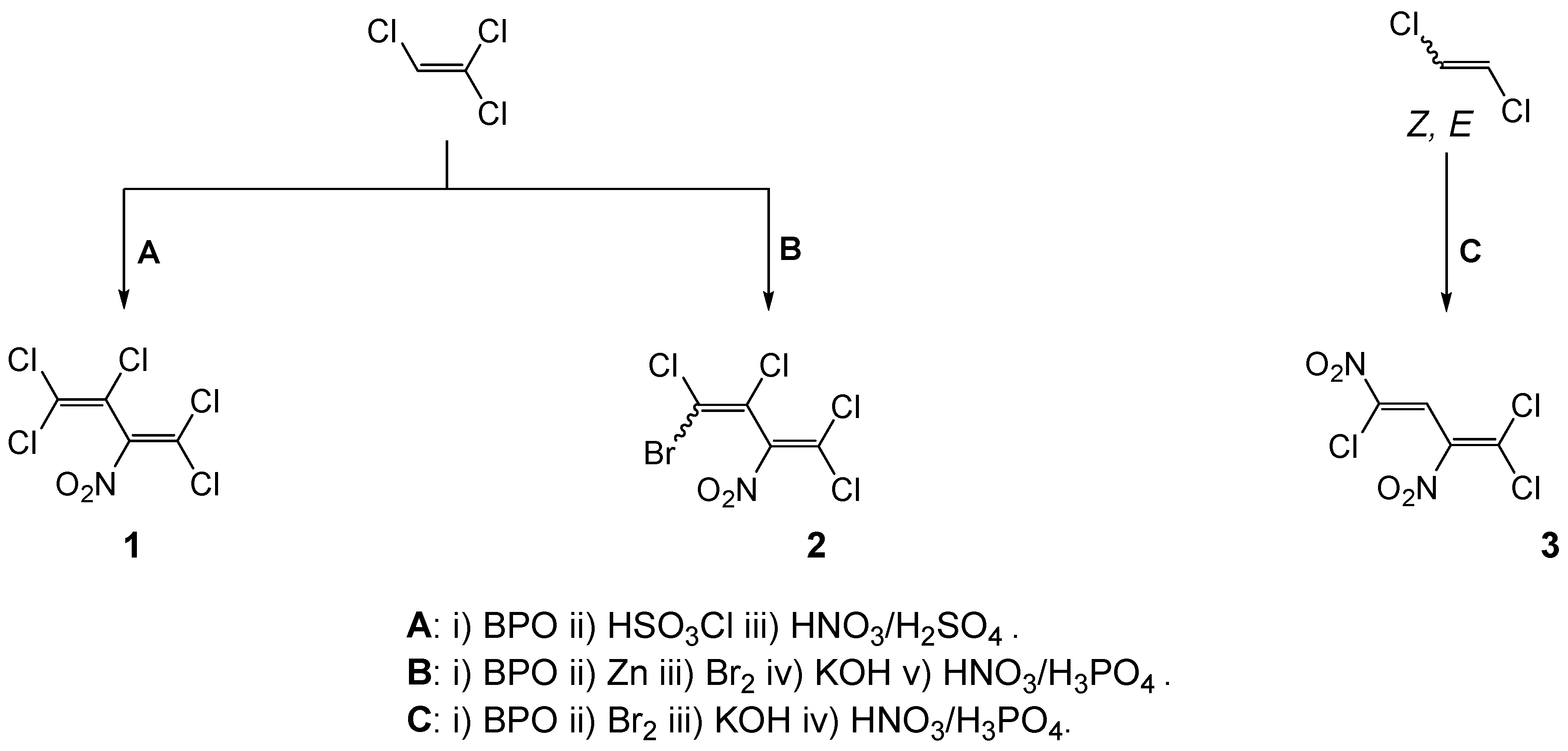{kind=link}
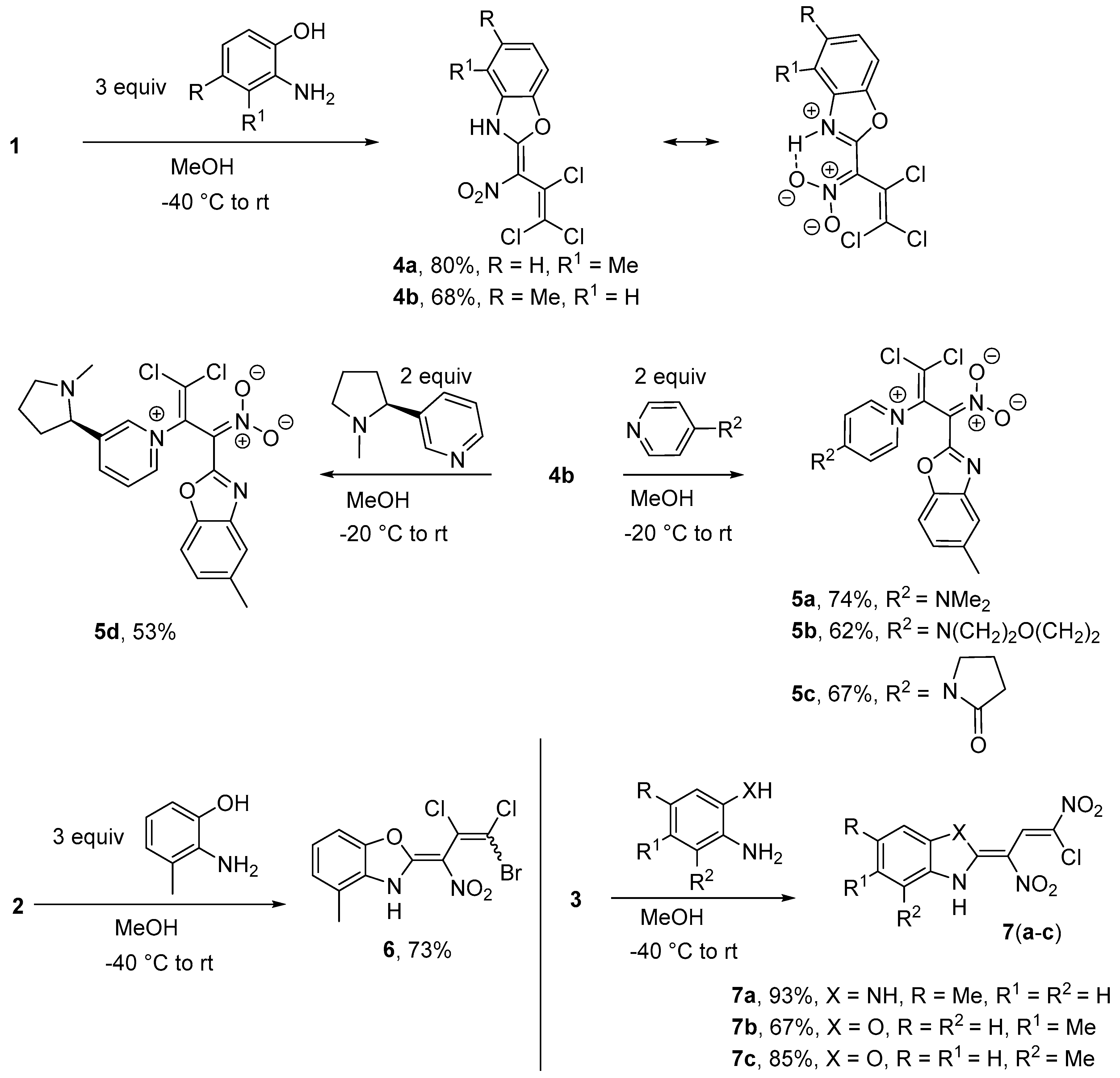{kind=link}
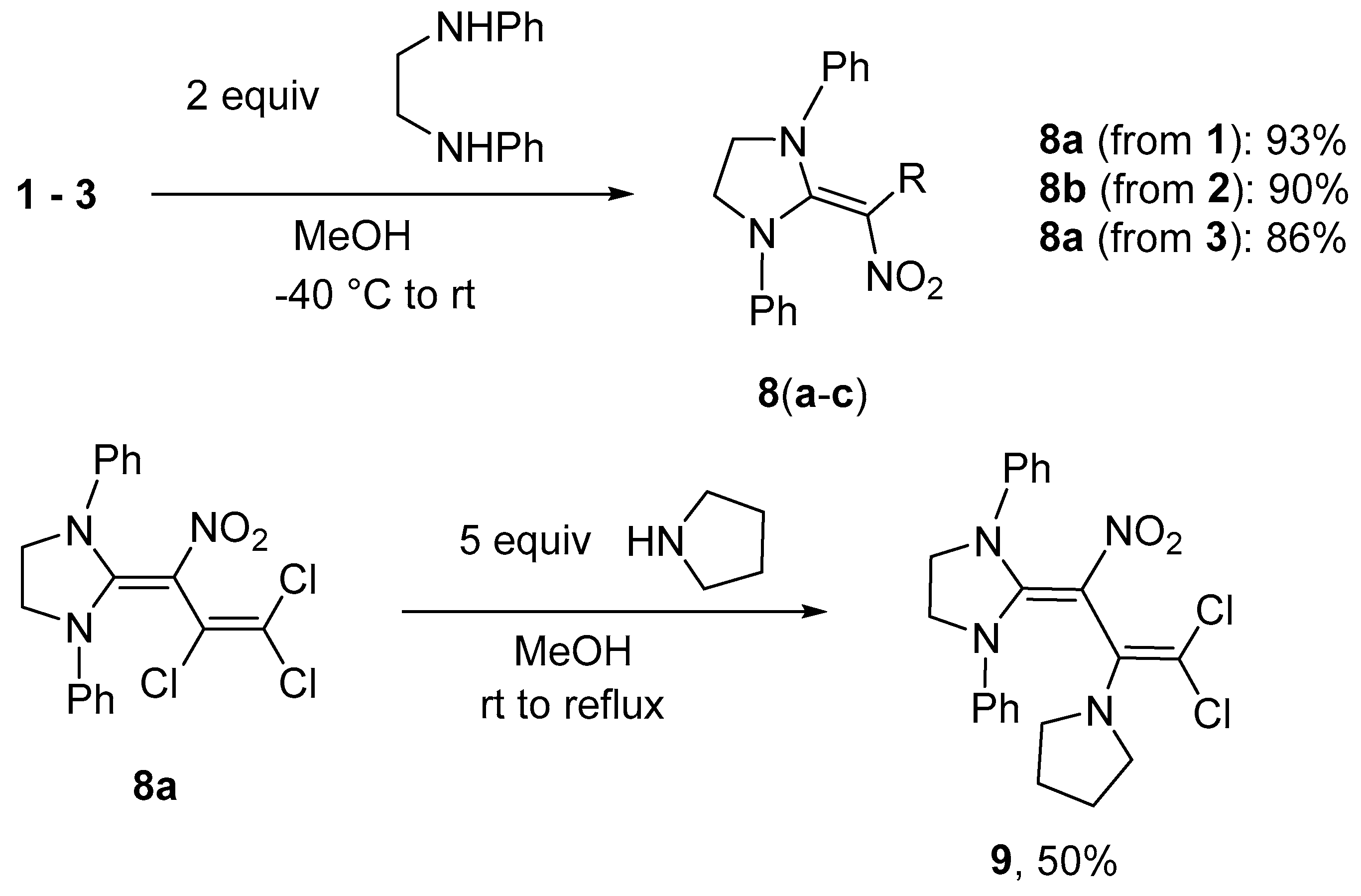{kind=link}
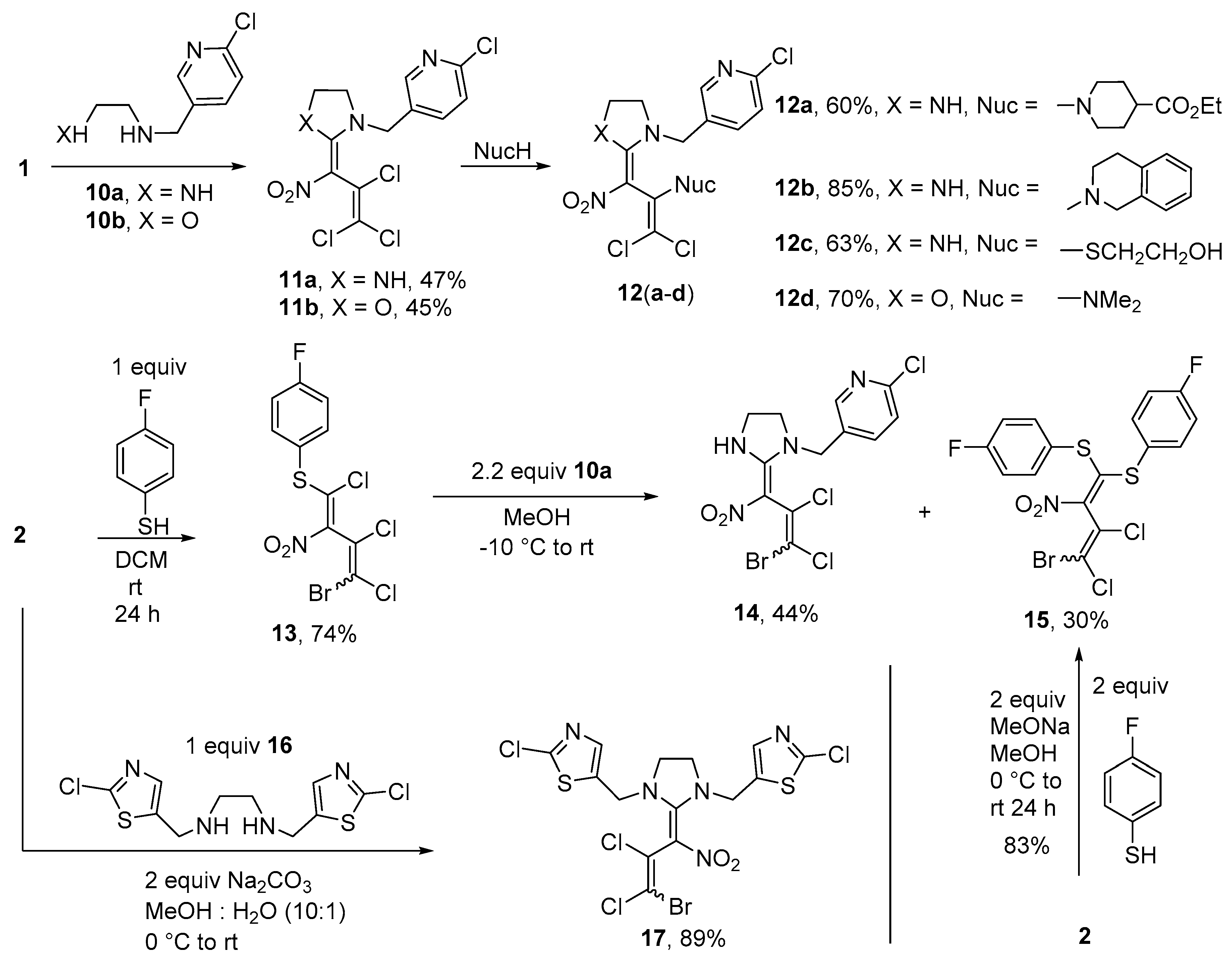{kind=link}
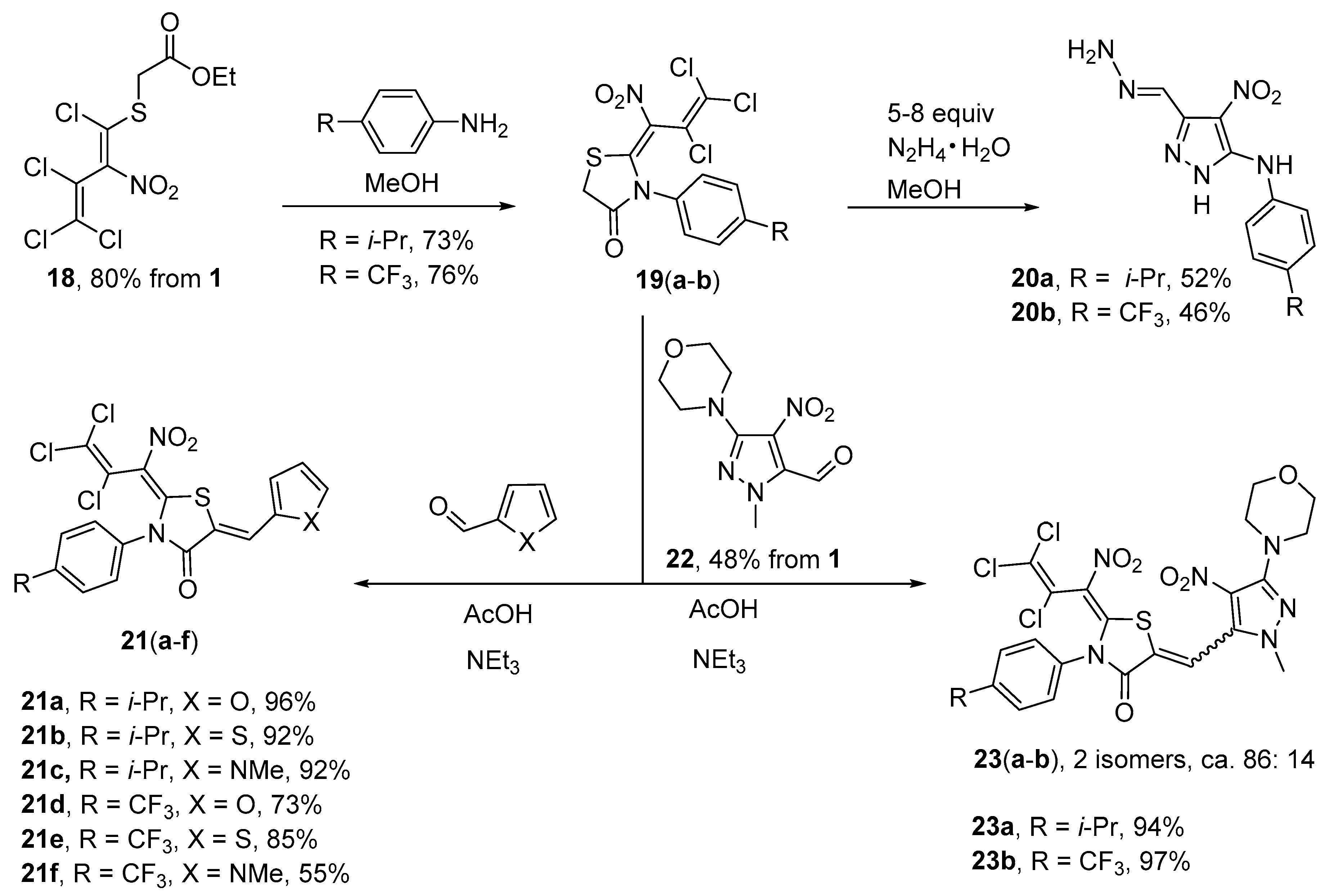{kind=link}
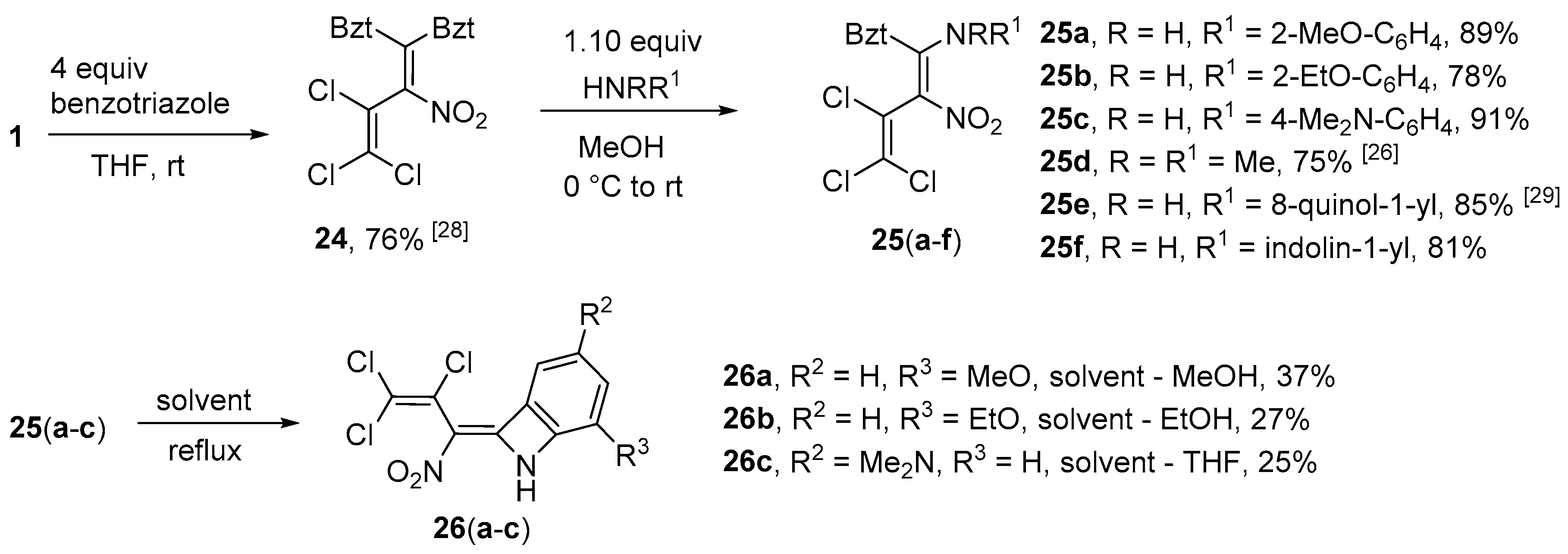{kind=link}
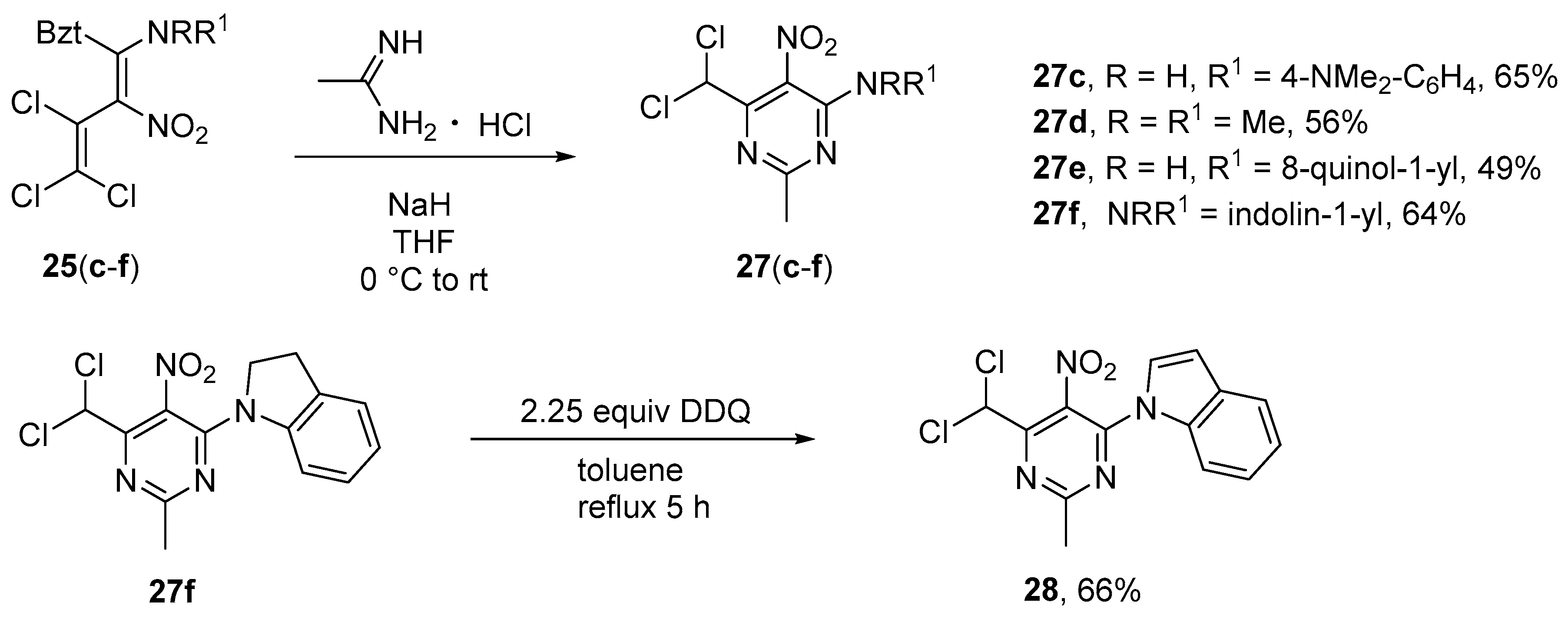{kind=link}
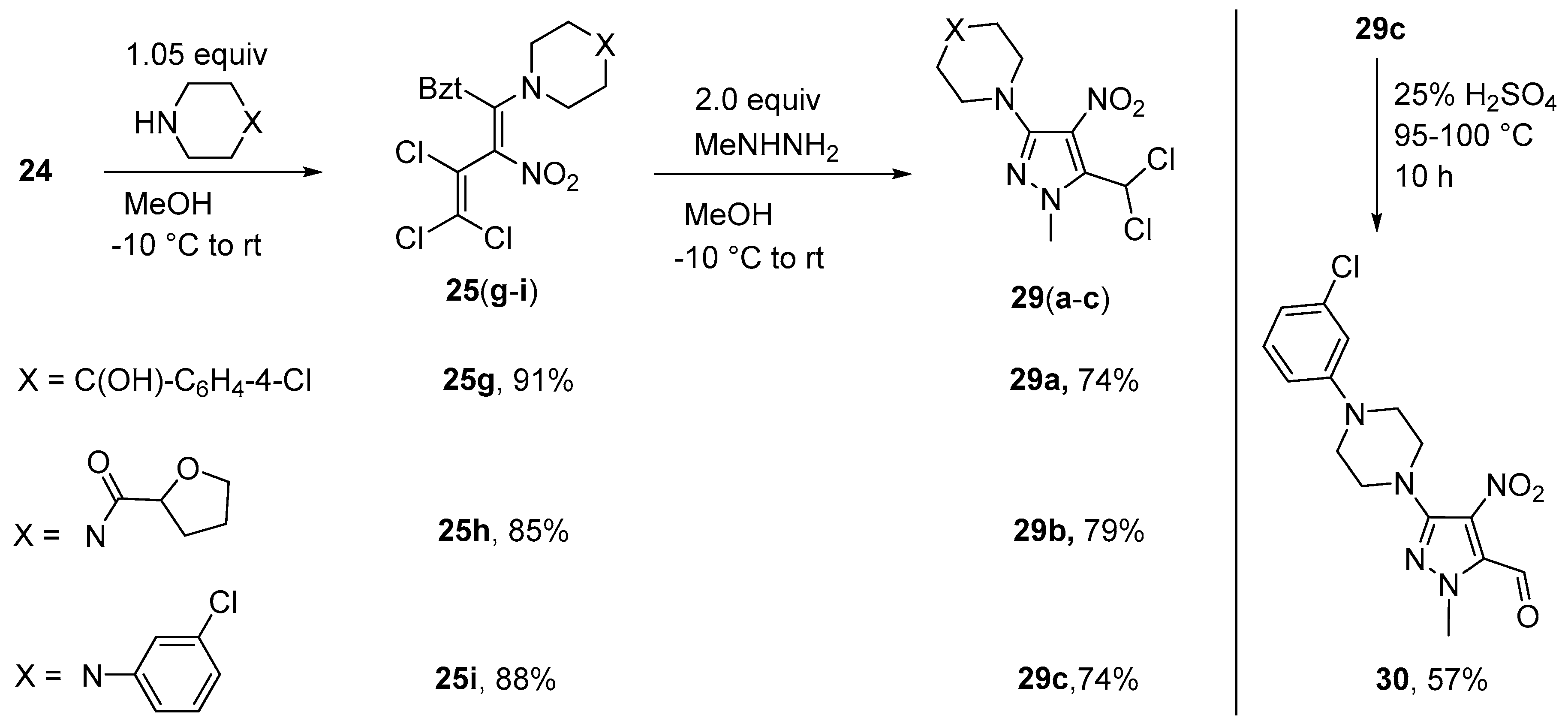{kind=link}
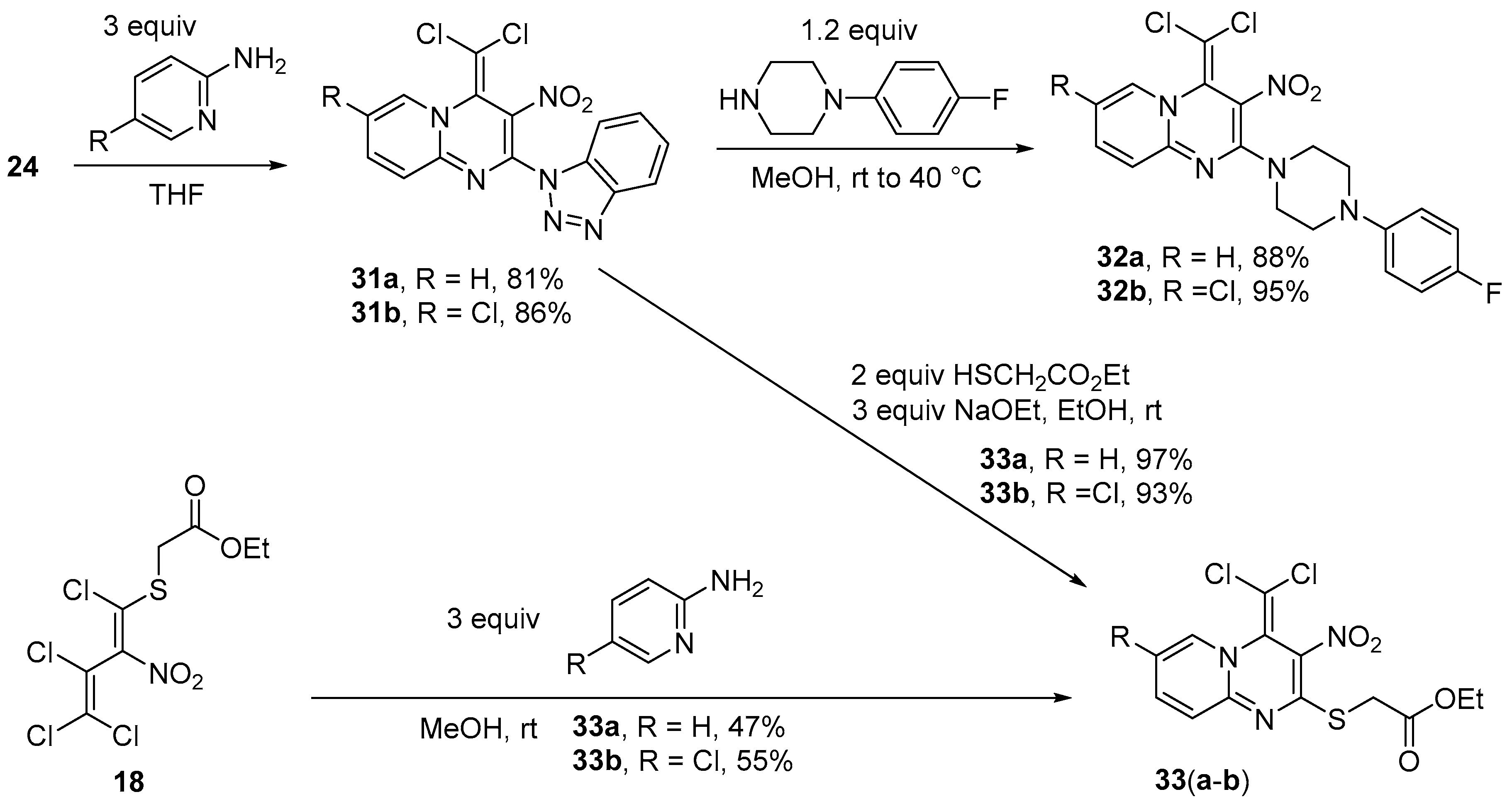{kind=link}
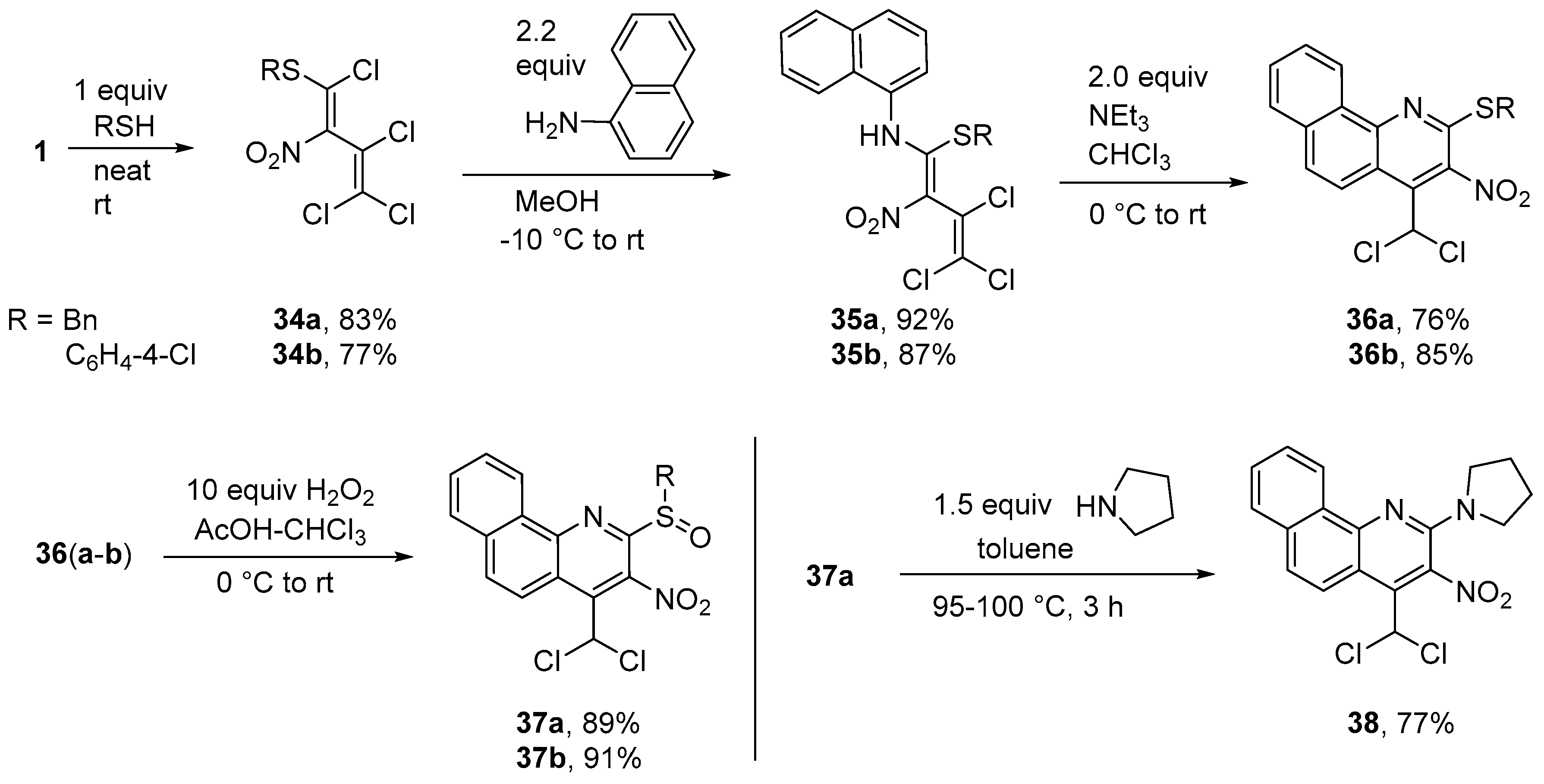{kind=link}
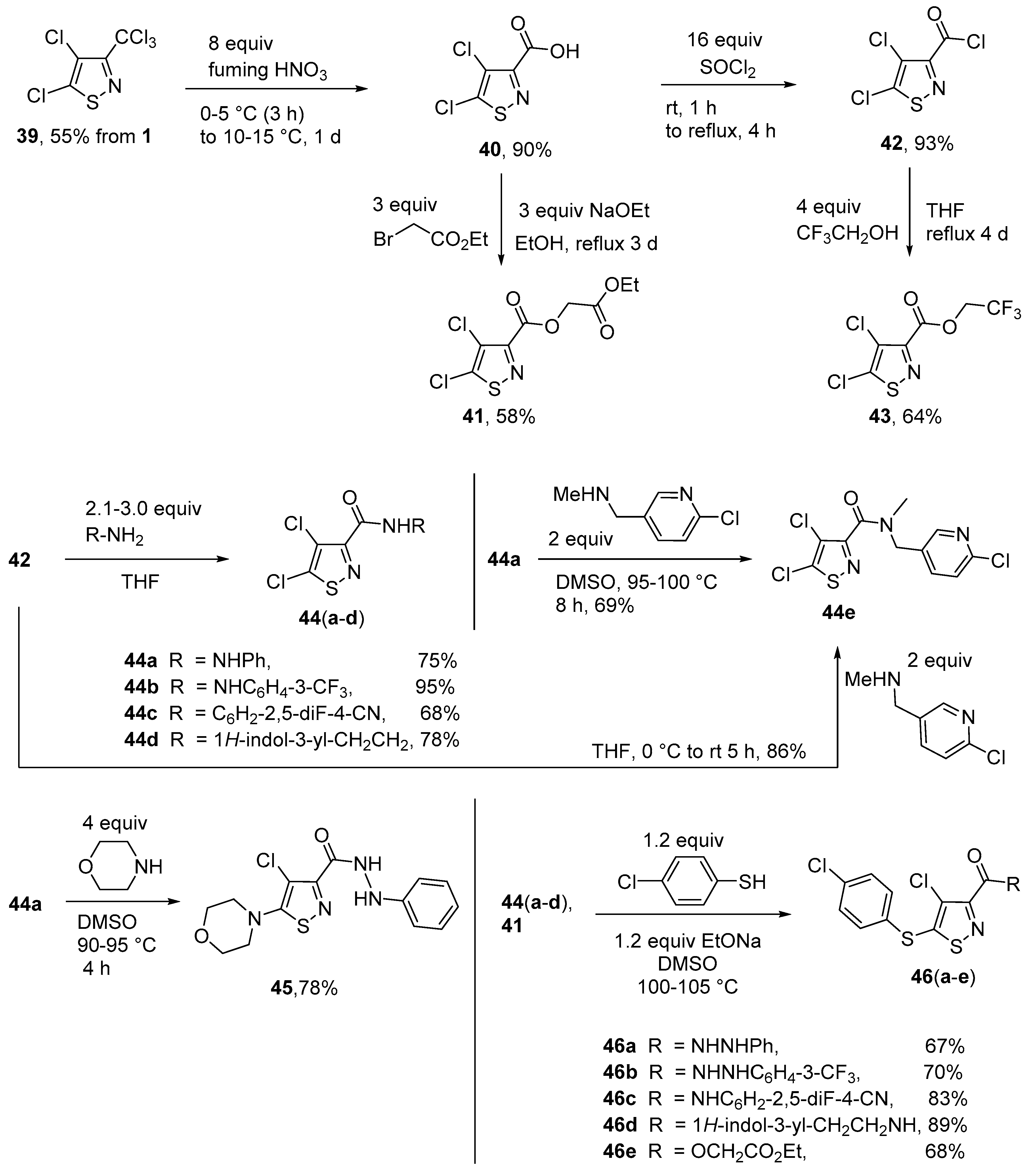{kind=link}
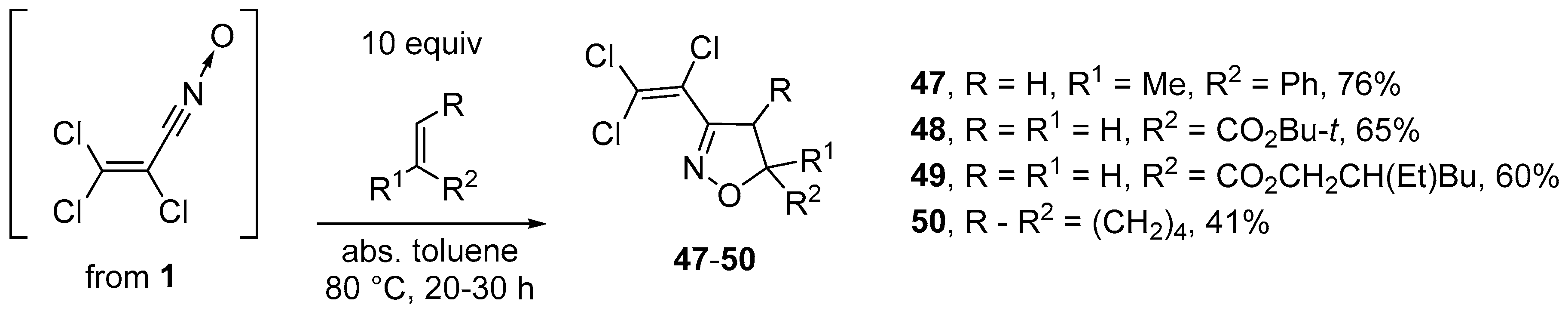{kind=link}
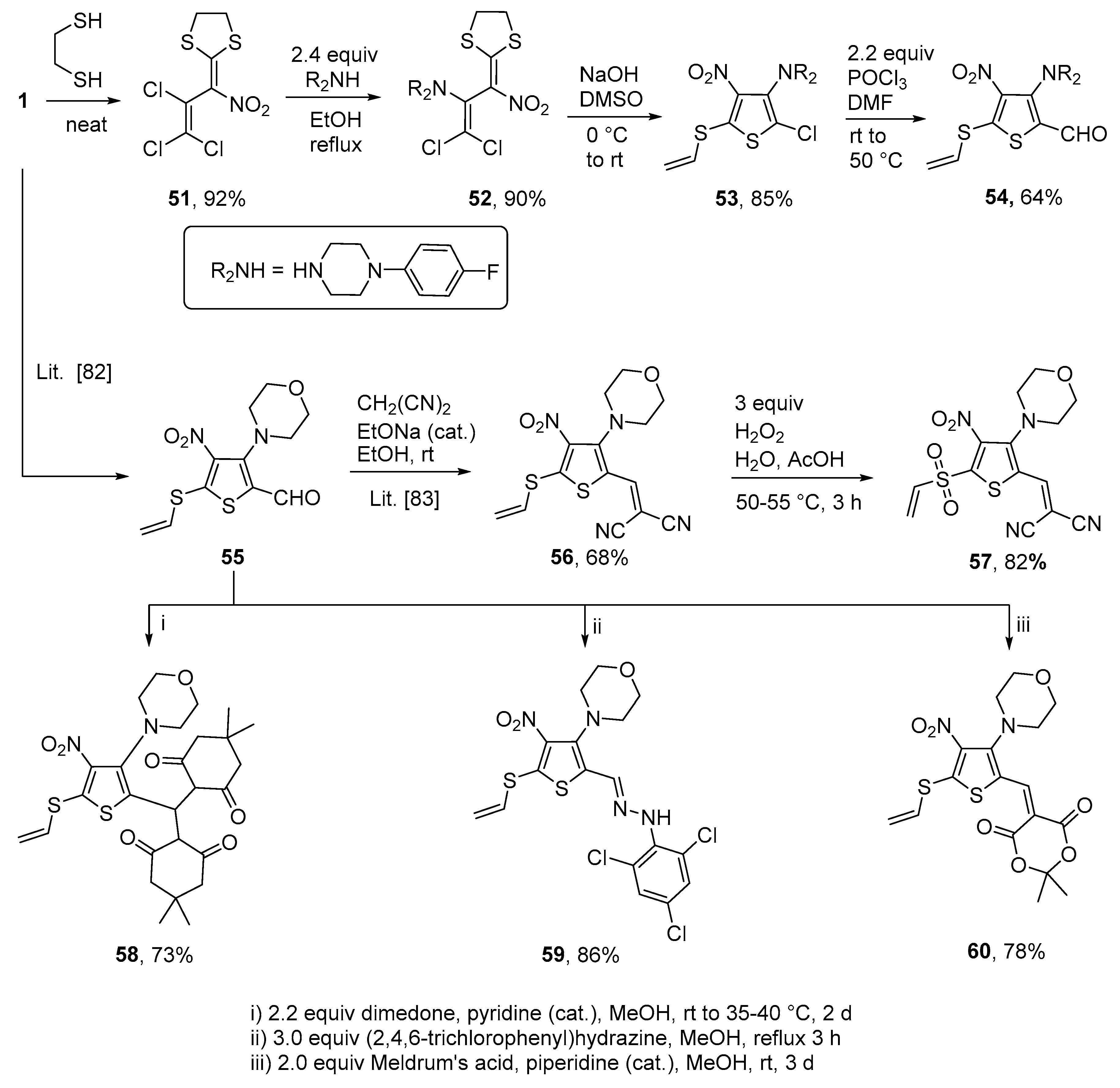{kind=link}












