2.1. Parent Molecule P(CH)3 and Its Molecular Electrostatic Potential (MEP)
The parent molecule P(CH)3 is pyramidal with C3v symmetry. Although the experimental structure of this molecule is not known, a derivative P(C-t-butyl)3 has been synthesized. An X-ray diffraction study of this molecule reported P-C distances of 1.837 to 1.860 Å, and C-C bond distances of 1.467 to 1.481 Å. The computed P-C distances of P(CH)3 are 1.857 Å and the C-C distances are 1.465 Å. In addition, the one-bond coupling constant 1J(P-C) has been measured experimentally and found to have an unsigned value of 37.9 Hz. The computed EOM-CCSD value of 1J(P-C) for P(CH)3 is –40.3 Hz. Thus, the computed structure and P-C coupling constant for P(CH)3 are consistent with the experimental values for the t-butyl derivative.
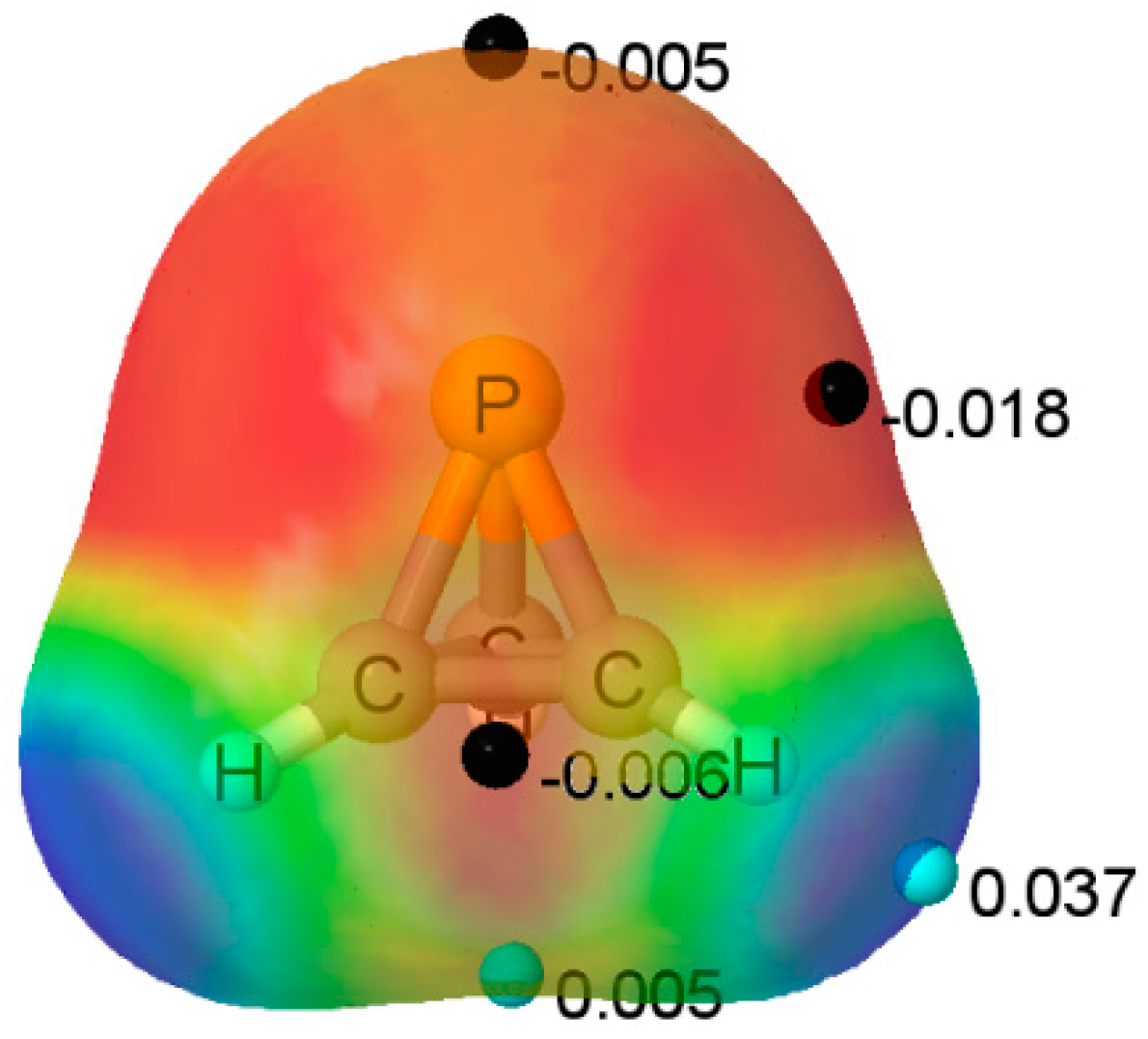
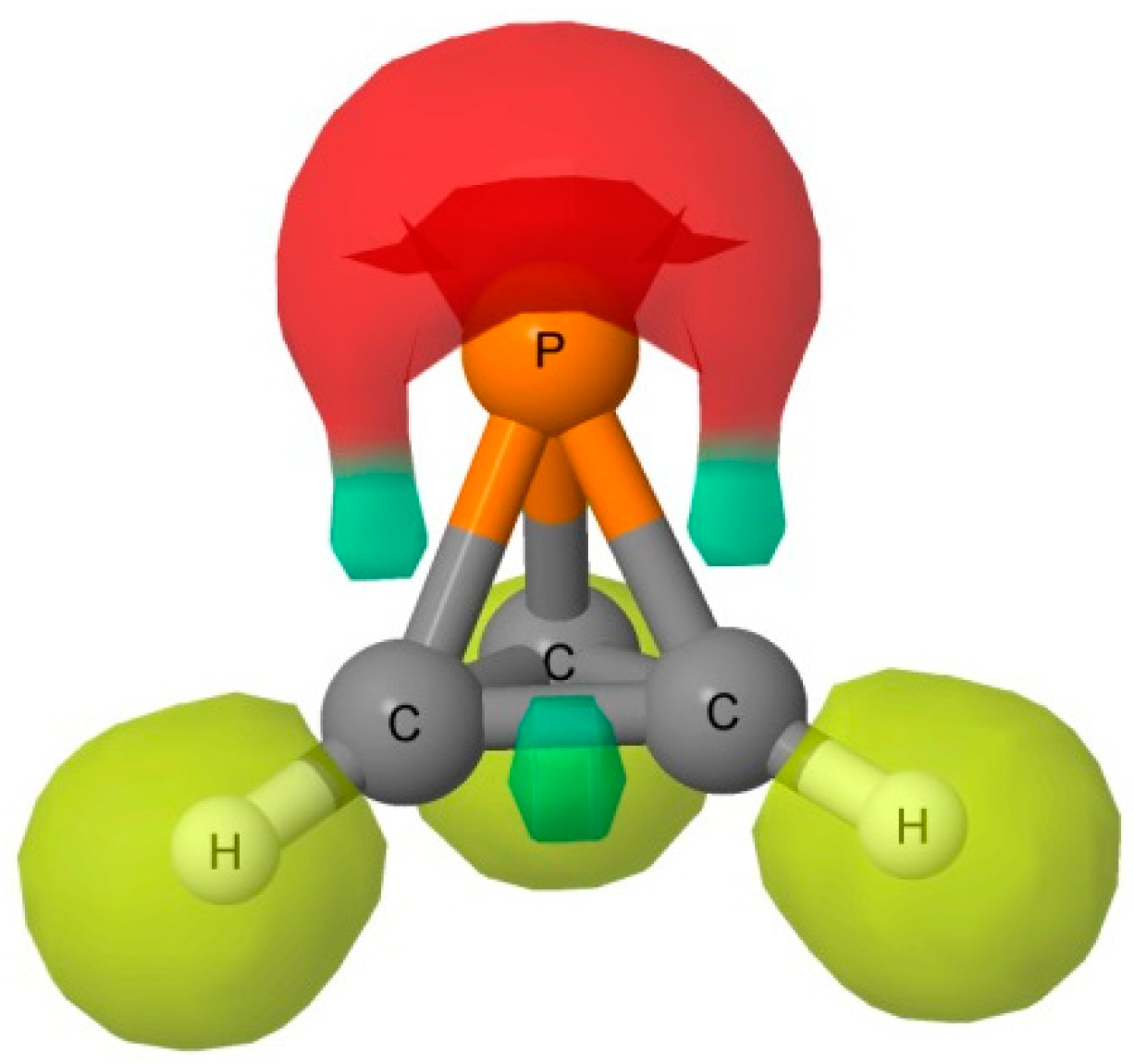
The MEP of the P(CH)
3 molecule is illustrated in
Figure 2. A triply degenerate MEP minimum with a value of −0.018 au is located at the P atom almost perpendicular to the C
3 symmetry axis. Three additional triply degenerate minima with values of −0.006 au are associated with the midpoint of each C-C bond in the ring. There is also a minimum of −0.005 au located on the C
3 axis at the P atom. At these minima, P(CH)
3 should act as an electron donor. There are also two unique maxima on this surface. One is a three-fold degenerate MEP maximum associated with the three C-H bonds. The second maximum has a value of 0.005 au, and is found below the centroid of the C-C-C ring on the C
3 symmetry axis. Bond formation in this region should have P(CH)
3 acting as the acid, that is, as an electron acceptor. The Electron Localization Function (ELF) analysis indicates that the deepest MEP minima on the 0.001 au electron density isosurface of −0.018 au arises from simultaneous contributions from the P-C bond basins and the P lone pair, as illustrated in
Figure 3.
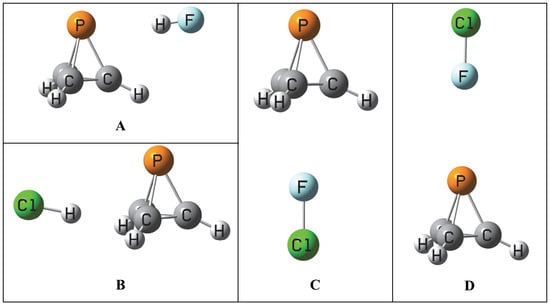
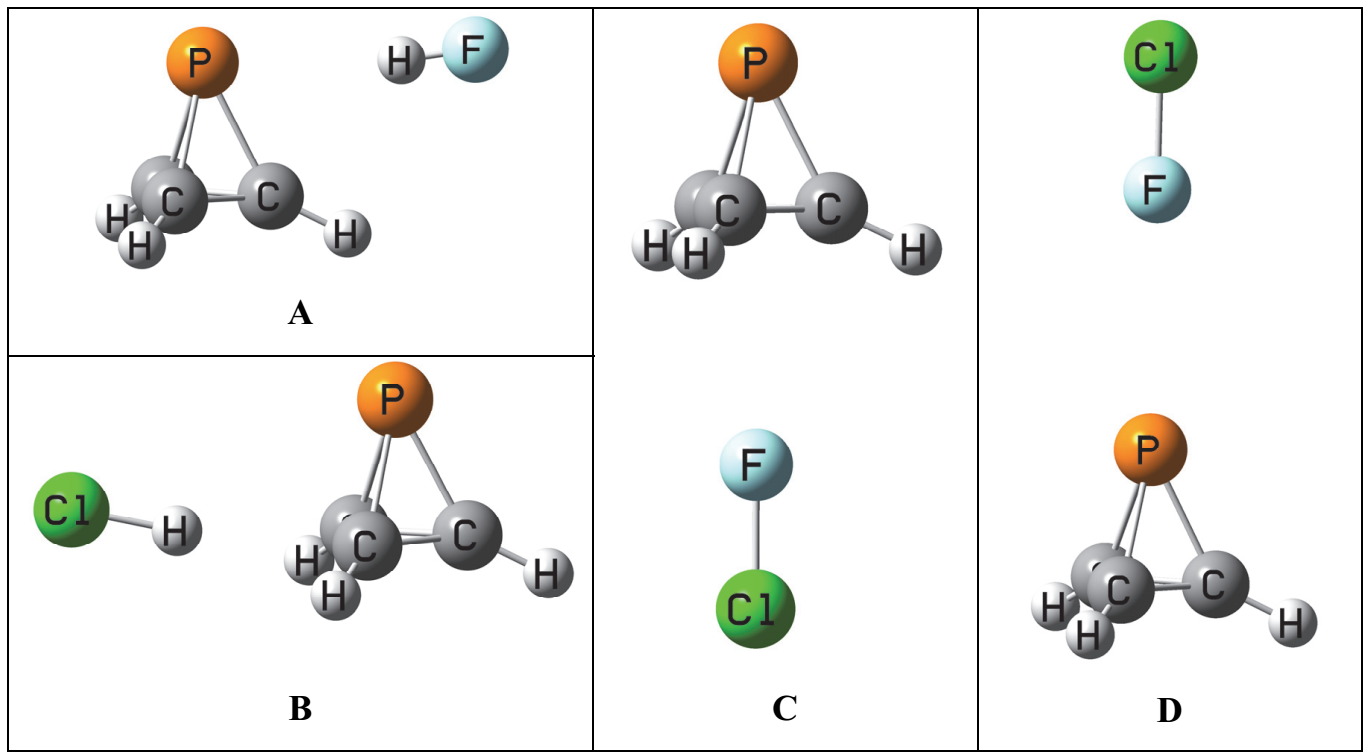
2.3. Binary Complexes A
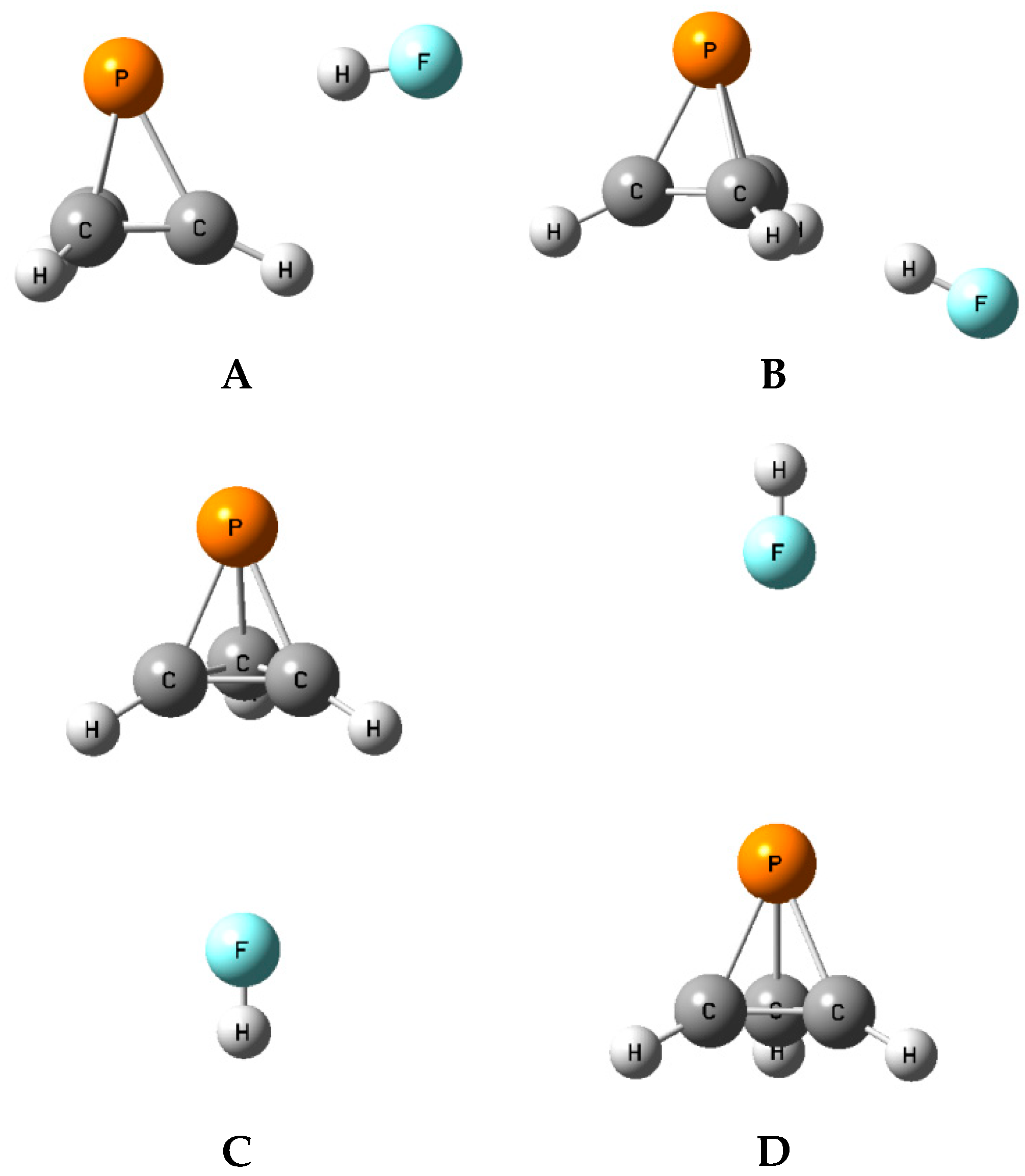
The binary complexes
A form at the local minimum identified in the MEP with a value of −0.018 au. The acid lies in the plane defined by the adjacent P-C and C-H bonds, so there are three such equilibrium structures on the potential energy surface. The structures, total energies, and molecular graphs of these complexes are reported in
Table S1. The binary complexes of P(CH)
3 with FH and ClH are stabilized by F-H⋯P and Cl-H⋯P hydrogen bonds, while the complex with ClF is stabilized by a P⋯Cl halogen bond.
Table 1 presents the binding energies, intermolecular distances, selected angles, charge-transfer energies, and coupling constants across intermolecular bonds for these complexes. The most stable complex is the halogen-bonded complex FCl:P(CH)
3 with a binding energy of 32 kJ mol
−1, followed by the hydrogen-bonded complexes FH:P(CH)
3 and ClH:P(CH)
3 with binding energies of 20 and 17 kJ mol
−1, respectively. The corresponding intermolecular F-P and Cl-P distances are 3.41 and 3.87 Å in the hydrogen-bonded complexes, respectively, and 2.55 Å in the halogen-bonded complex. The C-P-X angles vary between 65 and 77°, consistent with the observation made from the MEP that complex formation should occur at P in a direction almost perpendicular to the local C
3 axis. The H-X-P angles of 5° indicate that the hydrogen bonds deviate only slightly from linearity, while the P-Cl-F angle of 169° aligns the Cl-F molecule to accept a pair of electrons from P through the σ-hole on Cl for the formation of the halogen bond. In the halogen-bonded complex, there is a significant elongation of the P-C bond that interacts with Cl, as evident from
Table 1. While this bond length is 1.881 and 1.875 Å in the complexes with FH and ClH, respectively, it increases dramatically to 1.946 Å in the complex with FCl. Further discussion of bond length changes and
1J(P-C) coupling constants for isomers
A,
B,
C, and
D will be given below.
Charge-transfer energies Plp―σ*X-H of 17.6 and 16.4 kJ mol−1 are found for the hydrogen-bonded complexes. This energy changes dramatically in the halogen-bonded FCl:P(CH)3 complex. The largest charge-transfer energy arises from electron donation by a σ(P-C) bonding orbital to an antibonding Cl-F orbital, with an energy of 83 kJ mol−1. There is a second Plp―σ*Cl-F charge-transfer with an energy of 36.5 kJ mol−1. The smallest charge-transfer energy of 17 kJ mol−1 is a reverse charge-transfer, from the chlorine lone pair to an antibonding P-C orbital.
Spin–spin coupling constants
2hJ(X-P) are also reported in
Table 1, and have values of 28 and 3 Hz for the hydrogen-bonded complexes with FH and ClH, respectively.
1x(Cl-P) is 232 Hz for the halogen-bonded FCl:P(CH)
3 complex. The components of these coupling constants are reported in
Table S5 and indicate that although these coupling constants are dominated by the Fermi contact (FC) term, the paramagnetic spin orbit (PSO) and spin dipole (SD) terms make non-negligible contributions.
Figure S1 of the Supplementary Materials provides a plot of
2hJ(F-P) versus the F-P distance along the intrinsic reaction path connecting F and P.
2hJ(F-P) varies from 9 Hz at an F-P distance of 3.71 Å to 64 Hz at a distance of 3.21 Å. The second-order trendline in
Figure S1 has a correlation coefficient of 0.998, and suggests that the F-H
…P hydrogen bond is a traditional hydrogen bond.
Figure S2 is a plot along the intrinsic Cl-P reaction path for FCl:P(CH)
3. The third-order trendline has a correlation coefficient of 0.999. The curvature of this trendline at short distances suggests that complexes with these distances would have chlorine-shared halogen bonds [
27,
28]. Thus, the Cl
…P halogen bond in FCl:P(CH)
3 isomer
A would appear to have increased chlorine-shared character.
The properties of the complexes with P(CH)3 may be compared to the properties of the same acids with PH3. The X-P distances in the complexes XY:P(CH)3 are longer than the corresponding distances in the XY:PH3 complexes, which are 3.270, 3.800, and 2.183 Å for the complexes with FH, ClH, and ClF, respectively. The binding energies of the complexes FH:PH3 and ClH:PH3 are 21.4 and 14.9 kJ mol−1, respectively, which are similar to those for the corresponding complexes with P(CH)3. However, the FCl:P(CH)3 binding energy of 31.6 kJ mol−1 is significantly less than that of FCl:PH3 which is 51.2 kJ mol−1. This large difference may be attributed primarily to the weakening of the interacting P-C bond of P(CH)3, as indicated by the significant lengthening of this bond.
2.4. Binary Complexes B
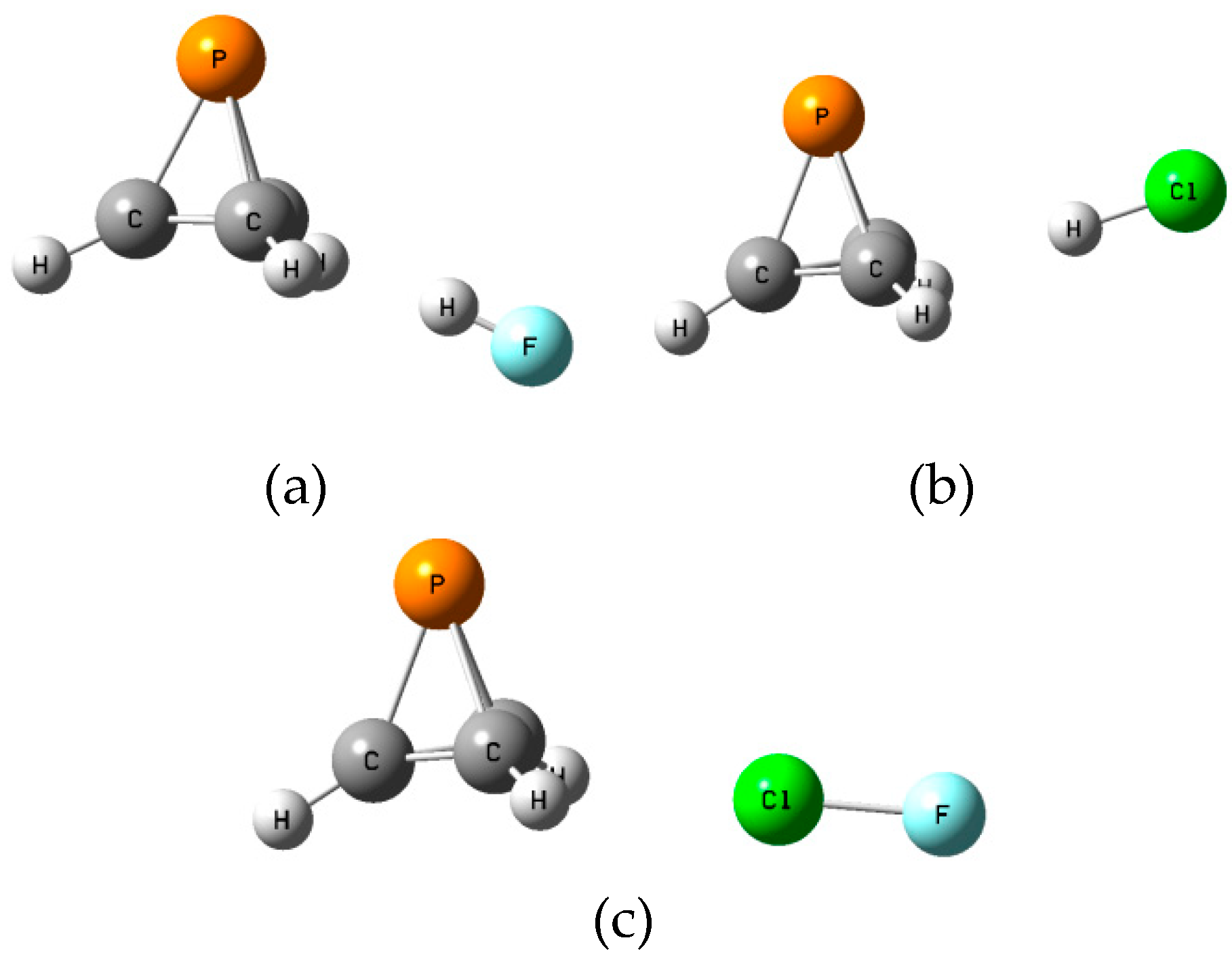
The
B isomers of the binary complexes XY:P(CH)
3 arise as a carbon-carbon bond of P(CH)
3 donates a pair of electrons to XY through its σ-hole. There are three such equilibrium complexes on the XY:P(CH)
3 surface, one at each C-C bond.
Table S2 of the Supplementary Materials provides the structures, total energies, and molecular graphs of these equilibrium complexes.
Table 2 reports their binding energies, the X-C distances, and the distances from X to the midpoint of the C-C bond (γ), the P-γ-X angles, charge-transfer energies, and coupling constants across the intermolecular bonds.
Figure 5 illustrates the structures of the
B isomers.
The binding energies of the
B isomers are less than those of the
A isomers, and extend over a smaller range, from 14.1 kJ mol
−1 at a distance of 2.19 Å from Cl to the midpoint of the C-C bond when ClH is the acid, to 17.3 kJ mol
−1 at a distance of 2.83 Å from the midpoint of the C-C bond to the Cl atom when ClF is the acid. The angles defined by P, the midpoint of the C-C bond, and X are similar when FH and FCl are the acids, but smaller when ClH is the acid, as evident from
Figure 5 and
Table 2. The distances from X to one of the two C atoms range from 2.92 Å in the complex with ClF to 3.54 Å in the complex with ClH. The charge-transfer energies of these complexes are approximately 2 kJ mol
−1, and are, therefore, much less than the charge-transfer energies of the
A isomers. They arise from electron donation by the C-C bond to a σ antibonding X-Y orbital. The order of decreasing charge-transfer energies is the same as the order of decreasing binding energies.
The DFT-SAPT data for the
A and
B complexes in
Table 3 allow for further insight into the binding energies of these isomers. There is only one destabilizing interaction, and that is the first-order exchange energy which varies from 27 kJ mol
−1 for ClH:P(CH)
3 isomer
B to 270 kJ mol
−1 for FCl:P(CH)
3 isomer
A. The remaining SAPT terms are stabilizing and are sufficient to overcome the exchange term, leading to SAPT binding energies [−ΔE(SAPT)] which range from 10 kJ mol
−1 for the
B isomers of ClH:P(CH)
3 and FCl:P(CH)
3 to 35 kJ mol
−1 for the
A isomer of FCl:P(CH)
3. The electrostatic interaction term is the most stabilizing term for all of these isomers except ClH:P(CH)
3 isomer
B, in which case the dispersion term is the leading stabilizing term. For FCl:P(CH)
3 isomer
B, the dispersion term is about 1 kJ mol
−1 less stabilizing than the electrostatic term. The SAPT binding energies are less than the MP2 binding energies except for FCl:P(CH)
3 isomer
A which has a SAPT binding energy that is 3 kJ mol
−1 greater than the MP2 energy.
Spin–spin coupling constants across intermolecular bonds for the
B isomers are also reported in
Table 2. The coupling constants are negligible at 0.1 and 0.2 Hz for the complexes with HCl and ClF, respectively. The coupling constant with the largest absolute value is
2hJ(F-C) for FH:P(CH)
3 with a value of only −1.8 Hz. It is interesting to note from
Table S5 that the dominant term for
2hJ(F-Cl) is the PSO term followed by the SD term. That the FC term is negligible is quite unusual for a hydrogen-bonded complex, but the hydrogen bonds in these
B complexes are not typical hydrogen bonds.
2.5. Binary Complexes C and D
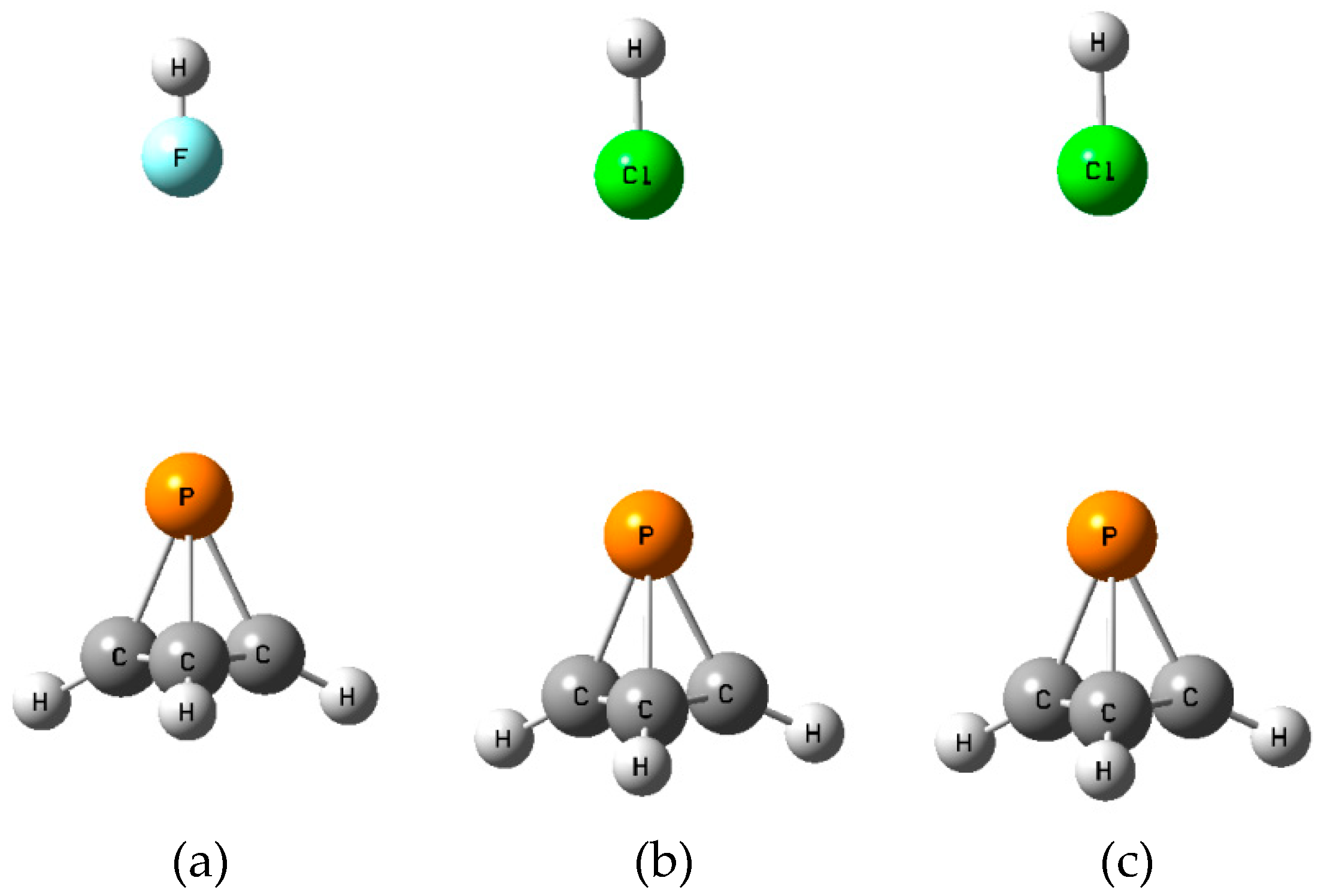
There are two additional equilibrium
C and
D isomers on the XY:P(CH)
3 surfaces. In
C the dipole moment vectors of XY and P(CH)
3 are favorably aligned head-to-tail, while in
D there is an unfavorable alignment, as illustrated in
Figure S3 of the Supplementary Materials. One might question why the reverse alignments of FH, ClH, and ClF in
D which would lead to favorable dipole alignments are not the equilibrium structures. This and related questions will be addressed below.
The structures, total energies, and molecular graphs of the
C isomers are reported in
Table S3 of the Supplementary Materials, and
Figure 6 illustrates these isomers. It is noteworthy that it is the F atom and not the Cl that interacts with P(CH)
3 in the FCl:P(CH)
3 complex. The structure in which Cl interacts with P(CH)
3 is not an equilibrium structure.
Table 4 provides the binding energies, intermolecular distances, and spin–spin coupling constants of the
C isomers. It is not surprising that the binding energies no longer follow the order of binding energies found for the hydrogen-bonded
A and
B isomers, but decrease instead in the order ClH > ClF > HF. The binding energies range from 4.5 kJ mol
−1 for FH:P(CH)
3 to 9.2 kJ mol
−1 for ClH:P(CH)
3. The X-C and X-P distances decrease in the order ClF > ClH > FH. That the mostly weakly bound complex with FH has the shortest X-C and X-P distances may be attributed at least in part to the smaller van der Waals radius of F.
Table 4 also reports coupling constants J(X-C) and J(X-P). The coupling constants J(X-C) are small, ranging from 0.0 Hz in the complex with ClF to 3.2 Hz in the complex with FH. Coupling with P is most interesting, since the interaction is through the tetrahedron of P(CH)
3 at very long distances. J(F-P) is −15.8 Hz at a distance of 6.22 Å in the complex with ClF and −12.1 Hz in the complex with FH. When the acid is ClH, J(Cl-P) has a value of −3.2 Hz.
The structures, total energies, and molecular graphs of the
D isomers are reported in
Table S4 of the Supplementary Materials, and these isomers are illustrated in
Figure 7 which shows that the F atom of FH and ClF, and the Cl atom of ClH are adjacent to P. From
Figure S3 it can be seen that these complexes have an unfavorable dipole alignment. The binding energies of the
D isomers are reported in
Table 5, and are noticeably less than those of the
C isomers. The
D isomers have binding energies that range from 0.8 kJ mol
−1 for HF:P(CH)
3 at a F-P distance of 3.49 Å, to 3.9 kJ mol
−1 for HCl:P(CH)
3 at a Cl-P distance of 3.63 Å. The binding energy of the FCl:P(CH)
3 complex is slightly less at 3.6 kJ mol
−1, but the Cl-P distance of 3.22 Å is much shorter.
The
D isomers have large spin–spin coupling constants J(X-P), and
Table S5 shows that total J is essentially equal to the FC term. The smallest coupling constant with a value of 45 Hz is J(Cl-P) in HCl:P(CH)
3. HF:P(CH)
3 has a value of J(F-P) equal to 118 Hz. The largest coupling constant among the
D isomers is J(F-P) for FCl:P(CH)
3 with a value of 299 Hz.
The SAPT energies for the
C and
D isomers are given in
Table 6. The SAPT binding energies are less than the MP2 binding energies. In fact, SAPT predicts that the HF:P(CH)
3 isomer
D is unbound with an unfavorable electrostatic interaction energy of 0.5 kJ mol
−1. As usual, the first-order SAPT exchange term is the destabilizing term. However, the electrostatic term is not the primary stabilizing term in these isomers as it is in the A and B isomers, but rather, it is the second-order dispersion term. In the FH:P(CH)
3 isomer
C, the dispersion term cancels the exchange term. In the remaining complexes, it has an absolute value that is no more than 2 kJ mol
−1 less than the exchange term. All of the other terms are stabilizing, except for the induction term for the
C and
D isomers of ClF:P(CH)
3. The sum of the stabilizing terms leads to binding energies between 3 and 5 kJ mol
−1 for the
C isomers, and between 1 and 2 kJ mol
−1 for the bound
D isomers.
Up to this point, the discussion of coupling constants has focused on coupling across intermolecular bonds. However, it is also of interest to examine how
1J(P-C) for P(CH)
3 changes, depending on whether the P-C bond interacts or does not interact with FH, ClH, or ClF. The data required for this analysis are given in
Table S6. For the
B,
C, and
D isomers,
1J(P-C) varies by about ±1 Hz relative to isolated P(CH)
3. For these,
1J(P-C) is essentially independent of bond formation by P(CH)
3. Bond formation in these isomers occurs at relatively long distances, and does not produce a large enough change in the electron distributions of P and C to produce changes in
1J(P-C). This is also evident from the small values of the charge-transfer energies in these isomers.
For the A isomers, 1J(P-C) coupling constants in complexes with FH and ClH span a slightly greater range compared to the isomers B, C, and D. However, it is the A isomer with ClF that exhibits dramatic changes. This is consistent with the stronger halogen bonding interaction between P(CH)3 and ClF which leads to a significant lengthening of the P-C bond to 1.946 Å, and changes in the electron density of that bond. The electron density changes are evident from the charge-transfer energies, and from the coupling constant for the interacting P-C bond, which decreases in absolute value from −40 Hz in isolated P(CH)3 to −33 Hz in this complex. Although the noninteracting P-C bonds decrease in length by only 0.007 Å, 1J(P-C) for these bonds increases in absolute value to −45 Hz in response to the changes in the electron density of this isomer.
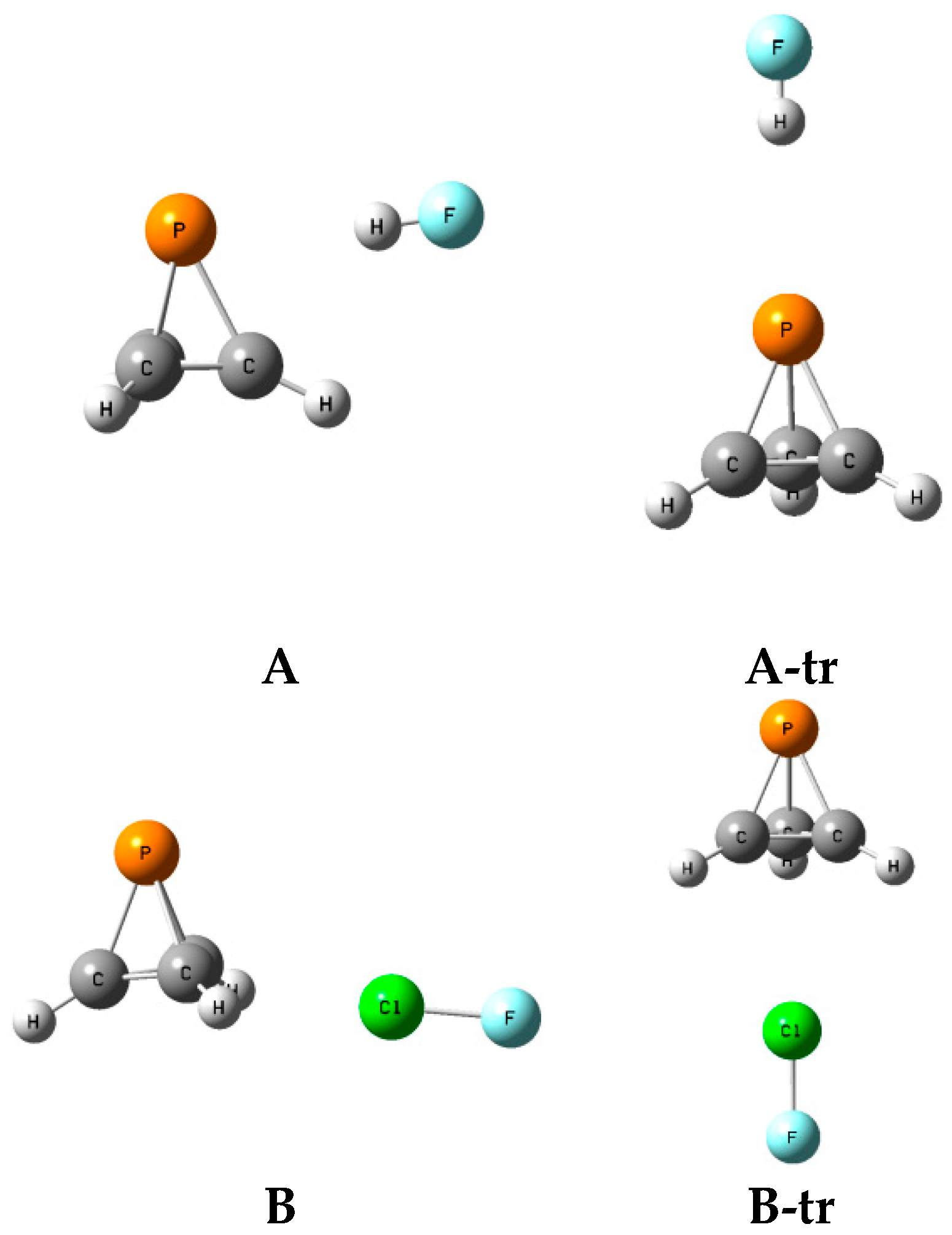
2.6. Structures A-tr and B-tr
The structures, total energies, and molecular graphs of the complexes
A-tr and
B-tr are given in
Table S7 of the Supplementary Materials, and the structures FH:P(CH)
3 and FH:P(CH)
3-
A-tr, and ClF:P(CH)
3 and ClF:P(CH)
3B-tr, are illustrated in
Figure 8. These complexes have C
3v symmetry and are not equilibrium structures on their potential surfaces, since they have two degenerate imaginary frequencies. Rather, complexes labeled
A-tr are the transition structures for converting one isomer
A to another one of the
A isomers, and
B-tr are the transition structures for converting one isomer
B to another one of the isomers
B.
Table 7 presents the binding energies, barriers, and intermolecular distances for the transition structures XY:P(CH)
3 A-tr and
B-tr. The intermolecular distances in these transition structures are longer than the X-P and X-C distances in the corresponding complexes
A and
B, respectively. The
A-tr structures are bound with stabilization energies ranging from 8 kJ mol
−1 for FH:P(CH)
3 A-tr and ClH:P(CH)
3 A-tr to 13 kJ mol
−1 for ClF:P(CH)
3 A-tr. The
B-tr isomers have binding energies that range from 10 kJ mol
−1 for FH:P(CH)
3 B-tr to 13 kJ mol
−1 for ClF:P(CH)
3 B-tr. From these energies of corresponding equilibrium complexes and transition structures, it is possible to evaluate the barrier for converting one
A or
B complex to another equivalent complex. These barriers are relatively high for the
A isomers, and vary from 9 kJ mol
−1 for ClH:P(CH)
3 to 19 kJ mol
−1 for ClF:P(CH)
3. Given the structure of the
B isomers, it is not surprising that the barriers to converting one isomer
B to another
B are much lower. These barriers vary from 2 kJ mol
−1 for ClH:P(CH)
3 to 4 kJ mol
−1 for FH:P(CH)
3 and FCl:P(CH)
3.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}