
Selectins—The Two Dr. Jekyll and Mr. Hyde Faces of Adhesion Molecules—A Review



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
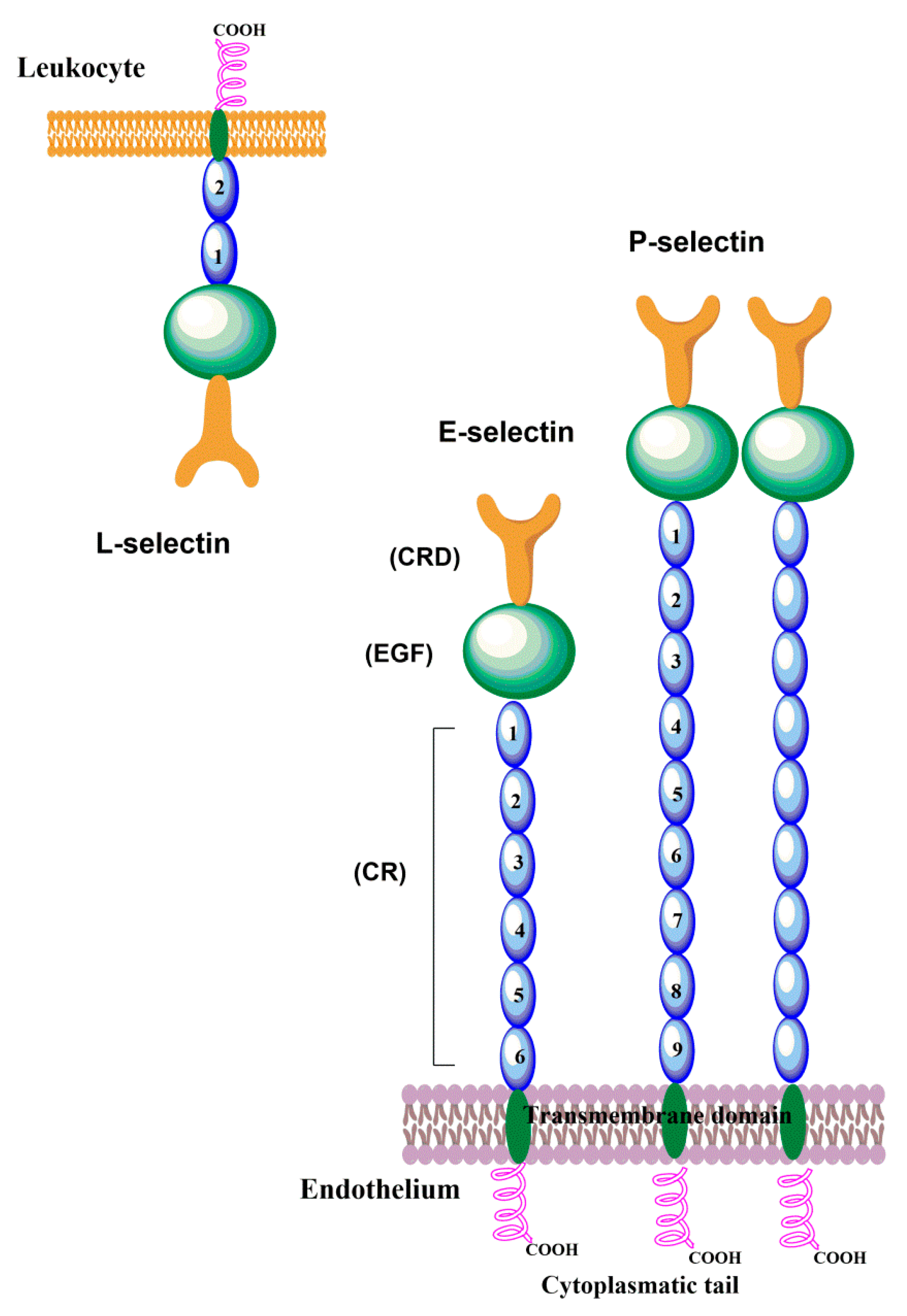
2. The Structure of Selectins
3. Selectin Ligands
3.1. Glycans as Minimal Recognition Determinants for Selectins
3.2. P-Selectin Ligands
3.3. E-Selectin Ligands
3.4. L-Selectin Ligands
4. Glycosyltransferases Involved in the Biosythesis of Glycan Determinants
4.1. The Glycosyltransferase Polypeptide UDP-GalNAc Transferase
4.2. Glycosyltransferases Core-1 β-1,3-Galactosyltransferase and Core-2 β-1,6-GlcNAc-Transferase
4.3. Glycosyltransferase β-1,4-Galactosyltransferase-1
4.4. Glycosyltransferases α-1,3-Fucosyltransferase and α-2,3-Sialyltransferase
4.5. Sulfotransferases GlcNAc-6-Sulfotransferase and Tyrosylprotein Sulfotransferase
5. The Biological Role of Selectins
5.1. Selectins in Inflammatory Processes
5.2. Mechanism of Selectin-Ligand Interaction
5.3. Selectins in Hemostasis and Thrombosis
5.4. Selectins in Cancer
5.5. Signaling Functions of Selectins and Selectin Ligands
6. The inhibition of Selectin-Ligand Interactions
6.1. Inhibition of the Expression of Selectins
6.2. Glycomimetic Inhibitors
6.3. Macromolecular Inhibitors
6.4. Non-Carbohydrate Inhibitors
6.5. Compounds in Clinical Trials
7. Carbohydrate Processing Inhibitors
7.1. Substrate Analog Inhibitors
7.2. Non-Substrate Inhibitors
7.3. Transition State Analog Inhibitors
8. Summary and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Golias, C.; Tsoutsi, E.; Matziridis, A.; Makridis, P.; Batistatou, A.; Charalabopoulos, K. Leukocyte and endothelial cell adhesion molecules in inflammation focusing on inflammatory heart disease. In Vivo 2007, 21, 757–769. [Google Scholar] [PubMed]
- Samanta, D.; Almo, S.C. Nectin family of cell-adhesion molecules: Structural and molecular aspects of function and specificity. Cell Mol. Life Sci. 2015, 72, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Harjunpaa, H.; Llort Asens, M.; Guenther, C.; Fagerholm, S.C. Cell Adhesion Molecules and Their Roles and Regulation in the Immune and Tumor Microenvironment. Front. Immunol. 2019, 10, 1078. [Google Scholar] [CrossRef] [PubMed]
- Ley, K. Adhesion Molecules: Function and Inhibition. Birkhäuser 2007. [Google Scholar] [CrossRef]
- Feizi, T. Carbohydrate ligands for the leukocyte-endothelium adhesion molecules, selectins. Results Probl. Cell Differ. 2001, 33, 201–223. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.E.; Drickamer, K. Paradigms for glycan-binding receptors in cell adhesion. Curr. Opin. Cell Biol. 2007, 19, 572–577. [Google Scholar] [CrossRef]
- Ley, K. Functions of selectins. Results Probl. Cell Differ. 2001, 33, 177–200. [Google Scholar] [CrossRef]
- Zarbock, A.; Ley, K.; McEver, R.P.; Hidalgo, A. Leukocyte ligands for endothelial selectins: Specialized glycoconjugates that mediate rolling and signaling under flow. Blood 2011, 118, 6743–6751. [Google Scholar] [CrossRef]
- Lasky, L.A. Selectin-carbohydrate interactions and the initiation of the inflammatory response. Annu. Rev. Biochem. 1995, 64, 113–139. [Google Scholar] [CrossRef]
- Silva, M.; Videira, P.A.; Sackstein, R. E-Selectin Ligands in the Human Mononuclear Phagocyte System: Implications for Infection, Inflammation, and Immunotherapy. Front. Immunol. 2018, 8, 1878. [Google Scholar] [CrossRef]
- Ludwig, R.J.; Schon, M.P.; Boehncke, W.H. P-selectin: A common therapeutic target for cardiovascular disorders, inflammation and tumour metastasis. Expert Opin. Ther. Targets 2007, 11, 1103–1117. [Google Scholar] [CrossRef] [PubMed]
- Ley, K. The role of selectins in inflammation and disease. Trends Mol. Med. 2003, 9, 263–268. [Google Scholar] [CrossRef]
- St Hill, C.A. Interactions between endothelial selectins and cancer cells regulate metastasis. Front. Biosci. 2011, 16, 3233–3251. [Google Scholar] [CrossRef]
- Laubli, H.; Borsig, L. Selectins promote tumor metastasis. Semin. Cancer Biol. 2010, 20, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Kansas, G.S. Selectins and their ligands: Current concepts and controversies. Blood 1996, 88, 3259–3287. [Google Scholar] [CrossRef] [PubMed]
- McEver, R.P. Selectins: Initiators of leucocyte adhesion and signalling at the vascular wall. Cardiovasc. Res. 2015, 107, 331–339. [Google Scholar] [CrossRef]
- Witz, I.P. The selectin-selectin ligand axis in tumor progression. Cancer Metastasis Rev. 2008, 27, 19–30. [Google Scholar] [CrossRef]
- Borsig, L. Selectins in cancer immunity. Glycobiology 2018, 28, 648–655. [Google Scholar] [CrossRef]
- Sperandio, M.; Gleissner, C.A.; Ley, K. Glycosylation in immune cell trafficking. Immunol. Rev. 2009, 230, 97–113. [Google Scholar] [CrossRef]
- Bedard, P.W.; Kaila, N. Selectin inhibitors: A patent review. Expert Opin. Ther. Pat. 2010, 20, 781–793. [Google Scholar] [CrossRef]
- Natoni, A.; Macauley, M.S.; O’Dwyer, M.E. Targeting Selectins and Their Ligands in Cancer. Front. Oncol. 2016, 6, 93. [Google Scholar] [CrossRef] [PubMed]
- Lowe, J.B. Glycosylation in the control of selectin counter-receptor structure and function. Immunol. Rev. 2002, 186, 19–36. [Google Scholar] [CrossRef]
- Lorant, D.E.; Patel, K.D.; McIntyre, T.M.; McEver, R.P.; Prescott, S.M.; Zimmerman, G.A. Coexpression of GMP-140 and PAF by endothelium stimulated by histamine or thrombin: A juxtacrine system for adhesion and activation of neutrophils. J. Cell Biol. 1991, 115, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Johnston, G.I.; Cook, R.G.; McEver, R.P. Cloning of GMP-140, a granule membrane protein of platelets and endothelium: Sequence similarity to proteins involved in cell adhesion and inflammation. Cell 1989, 56, 1033–1044. [Google Scholar] [CrossRef]
- Johnston, G.I.; Kurosky, A.; McEver, R.P. Structural and biosynthetic studies of the granule membrane protein, GMP-140, from human platelets and endothelial cells. J. Biol. Chem. 1989, 264, 1816–1823. [Google Scholar]
- Bezouska, K.; Crichlow, G.V.; Rose, J.M.; Taylor, M.E.; Drickamer, K. Evolutionary conservation of intron position in a subfamily of genes encoding carbohydrate-recognition domains. J. Biol. Chem. 1991, 266, 11604–11609. [Google Scholar]
- Ramachandran, V.; Yago, T.; Epperson, T.K.; Kobzdej, M.M.; Nollert, M.U.; Cummings, R.D.; Zhu, C.; McEver, R.P. Dimerization of a selectin and its ligand stabilizes cell rolling and enhances tether strength in shear flow. Proc. Natl. Acad. Sci. USA 2001, 98, 10166–10171. [Google Scholar] [CrossRef]
- Geng, J.G.; Bevilacqua, M.P.; Moore, K.L.; McIntyre, T.M.; Prescott, S.M.; Kim, J.M.; Bliss, G.A.; Zimmerman, G.A.; McEver, R.P. Rapid neutrophil adhesion to activated endothelium mediated by GMP-140. Nature 1990, 343, 757–760. [Google Scholar] [CrossRef]
- Keelan, E.T.; Licence, S.T.; Peters, A.M.; Binns, R.M.; Haskard, D.O. Characterization of E-selectin expression in vivo with use of a radiolabeled monoclonal antibody. Am. J. Physiol. 1994, 266, H278–H290. [Google Scholar] [CrossRef]
- Collins, T.; Williams, A.; Johnston, G.I.; Kim, J.; Eddy, R.; Shows, T.; Gimbrone, M.A., Jr.; Bevilacqua, M.P. Structure and chromosomal location of the gene for endothelial-leukocyte adhesion molecule 1. J. Biol. Chem. 1991, 266, 2466–2473. [Google Scholar]
- Jutila, M.A.; Watts, G.; Walcheck, B.; Kansas, G.S. Characterization of a functionally important and evolutionarily well-conserved epitope mapped to the short consensus repeats of E-selectin and L-selectin. J. Exp. Med. 1992, 175, 1565–1573. [Google Scholar] [CrossRef] [PubMed]
- Von Andrian, U.H.; Hansell, P.; Chambers, J.D.; Berger, E.M.; Torres Filho, I.; Butcher, E.C.; Arfors, K.E. L-selectin function is required for beta 2-integrin-mediated neutrophil adhesion at physiological shear rates in vivo. Am. J. Physiol. 1992, 263, H1034–H1044. [Google Scholar] [CrossRef]
- Zimmerman, G.A.; Prescott, S.M.; McIntyre, T.M. Endothelial cell interactions with granulocytes: Tethering and signaling molecules. Immunol. Today 1992, 13, 93–100. [Google Scholar] [CrossRef]
- Graves, B.J.; Crowther, R.L.; Chandran, C.; Rumberger, J.M.; Li, S.; Huang, K.S.; Presky, D.H.; Familletti, P.C.; Wolitzky, B.A.; Burns, D.K. Insight into E-selectin/ligand interaction from the crystal structure and mutagenesis of the lec/EGF domains. Nature 1994, 367, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Preston, R.C.; Jakob, R.P.; Binder, F.P.; Sager, C.P.; Ernst, B.; Maier, T. E-selectin ligand complexes adopt an extended high-affinity conformation. J. Mol. Cell Biol. 2016, 8, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Somers, W.S.; Tang, J.; Shaw, G.D.; Camphausen, R.T. Insights into the molecular basis of leukocyte tethering and rolling revealed by structures of P- and E-selectin bound to SLe(X) and PSGL-1. Cell 2000, 103, 467–479. [Google Scholar] [CrossRef]
- Mehta-D’souza, P.; Klopocki, A.G.; Oganesyan, V.; Terzyan, S.; Mather, T.; Li, Z.; Panicker, S.R.; Zhu, C.; McEver, R.P. Glycan Bound to the Selectin Low Affinity State Engages Glu-88 to Stabilize the High Affinity State under Force. J. Biol. Chem. 2017, 292, 2510–2518. [Google Scholar] [CrossRef]
- Varki, A. Biological roles of glycans. Glycobiology 2017, 27, 3–49. [Google Scholar] [CrossRef]
- Roseman, S. Reflections on glycobiology. J. Biol. Chem. 2001, 276, 41527–41542. [Google Scholar] [CrossRef]
- Laine, R.A. A calculation of all possible oligosaccharide isomers both branched and linear yields 1.05 × 1012 structures for a reducing hexasaccharide: The Isomer Barrier to development of single-method saccharide sequencing or synthesis systems. Glycobiology 1994, 4, 759–767. [Google Scholar] [CrossRef]
- Varki, A. Selectin ligands: Will the real ones please stand up? J. Clin. Investig. 1997, 99, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Poppe, L.; Brown, G.S.; Philo, J.S.; Nokrad, P.V.; Shah, B.H. Conformation os sLex Tetrasaccharide, Free in Solution and Bound to E-, P-, and L-Selectin. J. Am. Chem. Soc. 1997, 119, 1727–1736. [Google Scholar] [CrossRef]
- Bizik, F.; Tvaroska, I. On the Flexibility of the Lewis x, Lewis a, Sialyl Lewis x, and Sialyl Lewis a Oligosaccharides. Conformational Analysis in Solution by Molecular Modelling. Chem. Pap. 1996, 50, 84–96. [Google Scholar]
- Haselhorst, T.; Weimar, T.; Peters, T. Molecular recognition of sialyl Lewis(x) and related saccharides by two lectins. J. Am. Chem. Soc. 2001, 123, 10705–10714. [Google Scholar] [CrossRef]
- Binder, F.P.; Lemme, K.; Preston, R.C.; Ernst, B. Sialyl Lewis(x): A “pre-organized water oligomer”? Angew. Chem. Int. Ed. Engl. 2012, 51, 7327–7331. [Google Scholar] [CrossRef]
- Bowman, K.G.; Cook, B.N.; de Graffenried, C.L.; Bertozzi, C.R. Biosynthesis of L-selectin ligands: Sulfation of sialyl Lewis x-related oligosaccharides by a family of GlcNAc-6-sulfotransferases. Biochemistry 2001, 40, 5382–5391. [Google Scholar] [CrossRef]
- Bowman, K.G.; Hemmerich, S.; Bhakta, S.; Singer, M.S.; Bistrup, A.; Rosen, S.D.; Bertozzi, C.R. Identification of an N-acetylglucosamine-6-0-sulfotransferase activity specific to lymphoid tissue: An enzyme with a possible role in lymphocyte homing. Chem. Biol. 1998, 5, 447–460. [Google Scholar] [CrossRef]
- Brandley, B.K.; Kiso, M.; Abbas, S.; Nikrad, P.; Srivasatava, O.; Foxall, C.; Oda, Y.; Hasegawa, A. Structure-function studies on selectin carbohydrate ligands. Modifications to fucose, sialic acid and sulphate as a sialic acid replacement. Glycobiology 1993, 3, 633–641. [Google Scholar] [CrossRef]
- Jacob, G.S.; Kirmaier, C.; Abbas, S.Z.; Howard, S.C.; Steininger, C.N.; Welply, J.K.; Scudder, P. Binding of sialyl Lewis x to E-selectin as measured by fluorescence polarization. Biochemistry 1995, 34, 1210–1217. [Google Scholar] [CrossRef]
- Barra, P.A.; Jimenez, V.A.; Gavin, J.A.; Daranas, A.H.; Alderete, J.B. Discovery of New E-Selectin Inhibitors by Virtual Screening, Fluorescence Binding Assays, and STD NMR Experiments. ChemMedChem 2016, 11, 1008–1014. [Google Scholar] [CrossRef]
- Barra, P.A.; Ribeiro, A.J.; Ramos, M.J.; Jimenez, V.A.; Alderete, J.B.; Fernandes, P.A. Binding free energy calculations on E-selectin complexes with sLe(x) oligosaccharide analogs. Chem. Biol. Drug Des. 2017, 89, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Wild, M.K.; Huang, M.C.; Schulze-Horsel, U.; van der Merwe, P.A.; Vestweber, D. Affinity, kinetics, and thermodynamics of E-selectin binding to E-selectin ligand-1. J. Biol. Chem. 2001, 276, 31602–31612. [Google Scholar] [CrossRef] [PubMed]
- Tsukida, T.; Hiramatsu, Y.; Tsujishita, H.; Kiyoi, T.; Yoshida, M.; Kurokawa, K.; Moriyama, H.; Ohmoto, H.; Wada, Y.; Saito, T.; et al. Studies on selection blockers. 5. Design, synthesis, and biological profile of sialyl Lewis x mimetics based on modified serine-glutamic acid dipeptides. J. Med. Chem. 1997, 40, 3534–3541. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, K.; Ohmoto, H.; Kondo, N.; Tsujishita, H.; Hiramatsu, Y.; Inoue, Y.; Kondo, H. Studies on selectin blockers. 4. Structure-function relationships of sulfated sialyl Lewis X hexasaccharide ceramides toward E-, P-, and L-selectin binding. J. Med. Chem. 1997, 40, 455–462. [Google Scholar] [CrossRef]
- Croce, K.; Freedman, S.J.; Furie, B.C.; Furie, B. Interaction between soluble P-selectin and soluble P-selectin glycoprotein ligand 1: Equilibrium binding analysis. Biochemistry 1998, 37, 16472–16480. [Google Scholar] [CrossRef]
- Imai, Y.; Lasky, L.A.; Rosen, S.D. Sulphation requirement for GlyCAM-1, an endothelial ligand for L-selectin. Nature 1993, 361, 555–557. [Google Scholar] [CrossRef]
- Tsujishita, H.; Hiramatsu, Y.; Kondo, N.; Ohmoto, H.; Kondo, H.; Kiso, M.; Hasegawa, A. Selectin-ligand interactions revealed by molecular dynamics simulation in solution. J. Med. Chem. 1997, 40, 362–369. [Google Scholar] [CrossRef]
- Montreuil, J.; Vliegenthart, J.F.; Schachter, H. New Comprehensive Biochemistry of Glycoproteins; Elsevier: Amsterdam, The Netherlands, 1995; Volume 29, p. 644. [Google Scholar]
- Tvaroska, I. Atomistic insight into the catalytic mechanism of glycosyltransferases by combined quantum mechanics/molecular mechanics (QM/MM) methods. Carbohydr. Res. 2015, 403, 38–47. [Google Scholar] [CrossRef]
- Perez, S.; Tvaroska, I. Carbohydrate-protein interactions: Molecular modeling insights. Adv. Carbohydr. Chem. Biochem. 2014, 71, 9–136. [Google Scholar] [CrossRef]
- Lairson, L.L.; Henrissat, B.; Davies, G.J.; Withers, S.G. Glycosyltransferases: Structures, functions, and mechanisms. Annu. Rev. Biochem. 2008, 77, 521–555. [Google Scholar] [CrossRef]
- Fasting, C.; Schalley, C.A.; Weber, M.; Seitz, O.; Hecht, S.; Koksch, B.; Dernedde, J.; Graf, C.; Knapp, E.W.; Haag, R. Multivalency as a chemical organization and action principle. Angew. Chem. Int. Ed. Engl. 2012, 51, 10472–10498. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, M.; Pe’rez, S. Thermodynamics and chemical characterization of protein–carbohydrate interactions: The multivalency issue. Compt. Rend. Chim. 2011, 14, 74–95. [Google Scholar] [CrossRef]
- Cecioni, S.; Imberty, A.; Vidal, S. Glycomimetics versus multivalent glycoconjugates for the design of high affinity lectin ligands. Chem. Rev. 2015, 115, 525–561. [Google Scholar] [CrossRef] [PubMed]
- Ohtsubo, K.; Marth, J.D. Glycosylation in cellular mechanisms of health and disease. Cell 2006, 126, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.; Cummings, R.D.; McEver, R.P. Affinity and kinetic analysis of P-selectin binding to P-selectin glycoprotein ligand-1. J. Biol. Chem. 1998, 273, 32506–32513. [Google Scholar] [CrossRef] [PubMed]
- Leppanen, A.; Mehta, P.; Ouyang, Y.B.; Ju, T.; Helin, J.; Moore, K.L.; van Die, I.; Canfield, W.M.; McEver, R.P.; Cummings, R.D. A novel glycosulfopeptide binds to P-selectin and inhibits leukocyte adhesion to P-selectin. J. Biol. Chem. 1999, 274, 24838–24848. [Google Scholar] [CrossRef]
- Leppanen, A.; White, S.P.; Helin, J.; McEver, R.P.; Cummings, R.D. Binding of glycosulfopeptides to P-selectin requires stereospecific contributions of individual tyrosine sulfate and sugar residues. J. Biol. Chem. 2000, 275, 39569–39578. [Google Scholar] [CrossRef]
- Leppanen, A.; Yago, T.; Otto, V.I.; McEver, R.P.; Cummings, R.D. Model glycosulfopeptides from P-selectin glycoprotein ligand-1 require tyrosine sulfation and a core 2-branched O-glycan to bind to L-selectin. J. Biol. Chem. 2003, 278, 26391–26400. [Google Scholar] [CrossRef]
- Cummings, R.D. Structure and function of the selectin ligand PSGL-1. Braz. J. Med. Biol. Res. 1999, 32, 519–528. [Google Scholar] [CrossRef]
- Carlow, D.A.; Gold, M.R.; Ziltener, H.J. Lymphocytes in the peritoneum home to the omentum and are activated by resident dendritic cells. J. Immunol. 2009, 183, 1155–1165. [Google Scholar] [CrossRef]
- Tinoco, R.; Otero, D.C.; Takahashi, A.A.; Bradley, L.M. PSGL-1: A New Player in the Immune Checkpoint Landscape. Trends Immunol. 2017, 38, 323–335. [Google Scholar] [CrossRef]
- Snapp, K.R.; Craig, R.; Herron, M.; Nelson, R.D.; Stoolman, L.M.; Kansas, G.S. Dimerization of P-selectin glycoprotein ligand-1 (PSGL-1) required for optimal recognition of P-selectin. J. Cell Biol. 1998, 142, 263–270. [Google Scholar] [CrossRef]
- Epperson, T.K.; Patel, K.D.; McEver, R.P.; Cummings, R.D. Noncovalent association of P-selectin glycoprotein ligand-1 and minimal determinants for binding to P-selectin. J. Biol. Chem. 2000, 275, 7839–7853. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.L.; Eaton, S.F.; Lyons, D.E.; Lichenstein, H.S.; Cummings, R.D.; McEver, R.P. The P-selectin glycoprotein ligand from human neutrophils displays sialylated, fucosylated, O-linked poly-N-acetyllactosamine. J. Biol. Chem. 1994, 269, 23318–23327. [Google Scholar] [PubMed]
- Baisse, B.; Galisson, F.; Giraud, S.; Schapira, M.; Spertini, O. Evolutionary conservation of P-selectin glycoprotein ligand-1 primary structure and function. BMC Evol. Biol. 2007, 7, 166. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, A.; Kojima, N.; Uchimura, K.; Muramatsu, T.; Tamatani, T.; Berndt, M.C.; Kansas, G.S.; Kannagi, R. Distinct sulfation requirements of selectins disclosed using cells that support rolling mediated by all three selectins under shear flow. L-selectin prefers carbohydrate 6-sulfation totyrosine sulfation, whereas p-selectin does not. J. Biol. Chem. 2002, 277, 32578–32586. [Google Scholar] [CrossRef]
- Woelke, A.L.; Kuehne, C.; Meyer, T.; Galstyan, G.; Dernedde, J.; Knapp, E.W. Understanding selectin counter-receptor binding from electrostatic energy computations and experimental binding studies. J. Phys. Chem. B 2013, 117, 16443–16454. [Google Scholar] [CrossRef]
- Aigner, S.; Ruppert, M.; Hubbe, M.; Sammar, M.; Sthoeger, Z.; Butcher, E.C.; Vestweber, D.; Altevogt, P. Heat stable antigen (mouse CD24) supports myeloid cell binding to endothelial and platelet P-selectin. Int. Immunol. 1995, 7, 1557–1565. [Google Scholar] [CrossRef]
- Aigner, S.; Ramos, C.L.; Hafezi-Moghadam, A.; Lawrence, M.B.; Friederichs, J.; Altevogt, P.; Ley, K. CD24 mediates rolling of breast carcinoma cells on P-selectin. FASEB J. 1998, 12, 1241–1251. [Google Scholar] [CrossRef]
- Nelson, R.M.; Cecconi, O.; Roberts, W.G.; Aruffo, A.; Linhardt, R.J.; Bevilacqua, M.P. Heparin oligosaccharides bind L- and P-selectin and inhibit acute inflammation. Blood 1993, 82, 3253–3258. [Google Scholar] [CrossRef]
- Sackstein, R. Glycoengineering of HCELL, the human bone marrow homing receptor: Sweetly programming cell migration. Ann. Biomed. Eng. 2012, 40, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Pouyani, T.; Seed, B. PSGL-1 recognition of P-selectin is controlled by a tyrosine sulfation consensus at the PSGL-1 amino terminus. Cell 1995, 83, 333–343. [Google Scholar] [CrossRef]
- Sako, D.; Comess, K.M.; Barone, K.M.; Camphausen, R.T.; Cumming, D.A.; Shaw, G.D. A sulfated peptide segment at the amino terminus of PSGL-1 is critical for P-selectin binding. Cell 1995, 83, 323–331. [Google Scholar] [CrossRef]
- Asa, D.; Raycroft, L.; Ma, L.; Aeed, P.A.; Kaytes, P.S.; Elhammer, A.P.; Geng, J.G. The P-selectin glycoprotein ligand functions as a common human leukocyte ligand for P- and E-selectins. J. Biol. Chem. 1995, 270, 11662–11670. [Google Scholar] [CrossRef]
- Lenter, M.; Levinovitz, A.; Isenmann, S.; Vestweber, D. Monospecific and common glycoprotein ligands for E- and P-selectin on myeloid cells. J. Cell Biol. 1994, 125, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Bruehl, R.E.; Springer, T.A.; Bainton, D.F. Quantitation of L-selectin distribution on human leukocyte microvilli by immunogold labeling and electron microscopy. J. Histochem. Cytochem. 1996, 44, 835–844. [Google Scholar] [CrossRef]
- Gonatas, J.O.; Mourelatos, Z.; Stieber, A.; Lane, W.S.; Brosius, J.; Gonatas, N.K. MG-160, a membrane sialoglycoprotein of the medial cisternae of the rat Golgi apparatus, binds basic fibroblast growth factor and exhibits a high level of sequence identity to a chicken fibroblast growth factor receptor. J. Cell Sci. 1995, 108, 457–467. [Google Scholar]
- Steegmaier, M.; Levinovitz, A.; Isenmann, S.; Borges, E.; Lenter, M.; Kocher, H.P.; Kleuser, B.; Vestweber, D. The E-selectin-ligand ESL-1 is a variant of a receptor for fibroblast growth factor. Nature 1995, 373, 615–620. [Google Scholar] [CrossRef]
- Steegmaier, M.; Borges, E.; Berger, J.; Schwarz, H.; Vestweber, D. The E-selectin-ligand ESL-1 is located in the Golgi as well as on microvilli on the cell surface. J. Cell Sci. 1997, 110, 687–694. [Google Scholar]
- Levinovitz, A.; Muhlhoff, J.; Isenmann, S.; Vestweber, D. Identification of a glycoprotein ligand for E-selectin on mouse myeloid cells. J. Cell Biol. 1993, 121, 449–459. [Google Scholar] [CrossRef]
- 92 Huang, M.C.; Zollner, O.; Moll, T.; Maly, P.; Thall, A.D.; Lowe, J.B.; Vestweber, D. P-selectin glycoprotein ligand-1 and E-selectin ligand-1 are differentially modified by fucosyltransferases Fuc-TIV and Fuc-TVII in mouse neutrophils. J. Biol. Chem. 2000, 275, 31353–31360. [Google Scholar] [CrossRef]
- Hidalgo, A.; Peired, A.J.; Wild, M.; Vestweber, D.; Frenette, P.S. Complete identification of E-selectin ligands on neutrophils reveals distinct functions of PSGL-1, ESL-1, and CD44. Immunity 2007, 26, 477–489. [Google Scholar] [CrossRef]
- Jacob, G.S.; Welply, J.K.; Scudder, P.R.; Kirmaier, C.; Abbas, S.Z.; Howard, S.C.; Keene, J.L.; Schmuke, J.J.; Broschat, K.; Steininger, C. Studies on selectin-carbohydrate interactions. Adv. Exp. Med. Biol. 1995, 376, 283–290. [Google Scholar] [CrossRef]
- Naor, D.; Sionov, R.V.; Ish-Shalom, D. CD44: Structure, function, and association with the malignant process. Adv. Cancer Res. 1997, 71, 241–319. [Google Scholar] [CrossRef] [PubMed]
- Ouhtit, A.; Rizeq, B.; Saleh, H.A.; Rahman, M.; Zayed, H. Novel CD44-downstream signaling pathways mediating breast tumor invasion. Int. J. Biol. Sci. 2018, 14, 1782–1790. [Google Scholar] [CrossRef] [PubMed]
- Dimitroff, C.J.; Lee, J.Y.; Fuhlbrigge, R.C.; Sackstein, R. A distinct glycoform of CD44 is an L-selectin ligand on human hematopoietic cells. Proc. Natl. Acad. Sci. USA 2000, 97, 13841–13846. [Google Scholar] [CrossRef] [PubMed]
- Dimitroff, C.J.; Lee, J.Y.; Schor, K.S.; Sandmaier, B.M.; Sackstein, R. differential L-selectin binding activities of human hematopoietic cell L-selectin ligands, HCELL and PSGL-1. J. Biol. Chem. 2001, 276, 47623–47631. [Google Scholar] [CrossRef]
- Sackstein, R. The biology of CD44 and HCELL in hematopoiesis: The ‘step 2-bypass pathway’ and other emerging perspectives. Curr. Opin. Hematol. 2011, 18, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Sackstein, R. The bone marrow is akin to skin: HCELL and the biology of hematopoietic stem cell homing. J. Investig. Dermatol. Sympos. Proc. 2004, 9, 215–223. [Google Scholar] [CrossRef]
- Dimitroff, C.J.; Lee, J.Y.; Rafii, S.; Fuhlbrigge, R.C.; Sackstein, R. CD44 is a major E-selectin ligand on human hematopoietic progenitor cells. J. Cell Biol. 2001, 153, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Sackstein, R.; Dimitroff, C.J. A hematopoietic cell L-selectin ligand that is distinct from PSGL-1 and displays N-glycan-dependent binding activity. Blood 2000, 96, 2765–2774. [Google Scholar] [CrossRef] [PubMed]
- Hanley, W.D.; Burdick, M.M.; Konstantopoulos, K.; Sackstein, R. CD44 on LS174T colon carcinoma cells possesses E-selectin ligand activity. Cancer Res. 2005, 65, 5812–5817. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.; Fung, R.K.F.; Donnelly, C.B.; Videira, P.A.; Sackstein, R. Cell-Specific Variation in E-Selectin Ligand Expression among Human Peripheral Blood Mononuclear Cells: Implications for Immunosurveillance and Pathobiology. J. Immunol. 2017, 198, 3576–3587. [Google Scholar] [CrossRef]
- Sackstein, R.; Merzaban, J.S.; Cain, D.W.; Dagia, N.M.; Spencer, J.A.; Lin, C.P.; Wohlgemuth, R. Ex vivo glycan engineering of CD44 programs human multipotent mesenchymal stromal cell trafficking to bone. Nat. Med. 2008, 14, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Sackstein, R. Glycosyltransferase-programmed stereosubstitution (GPS) to create HCELL: Engineering a roadmap for cell migration. Immunol. Rev. 2009, 230, 51–74. [Google Scholar] [CrossRef] [PubMed]
- McEver, R.P.; Moore, K.L.; Cummings, R.D. Leukocyte trafficking mediated by selectin-carbohydrate interactions. J. Biol. Chem. 1995, 270, 11025–11028. [Google Scholar] [CrossRef]
- Laszik, Z.; Jansen, P.J.; Cummings, R.D.; Tedder, T.F.; McEver, R.P.; Moore, K.L. P-selectin glycoprotein ligand-1 is broadly expressed in cells of myeloid, lymphoid, and dendritic lineage and in some nonhematopoietic cells. Blood 1996, 88, 3010–3021. [Google Scholar] [CrossRef]
- Merzaban, J.S.; Burdick, M.M.; Gadhoum, S.Z.; Dagia, N.M.; Chu, J.T.; Fuhlbrigge, R.C.; Sackstein, R. Analysis of glycoprotein E-selectin ligands on human and mouse marrow cells enriched for hematopoietic stem/progenitor cells. Blood 2011, 118, 1774–1783. [Google Scholar] [CrossRef]
- Rosen, S.D. Ligands for L-selectin: Homing, inflammation, and beyond. Ann. Rev. Iimmunol. 2004, 22, 129–156. [Google Scholar] [CrossRef]
- Rosen, S.D. Endothelial ligands for L-selectin: From lymphocyte recirculation to allograft rejection. Am. J. Pathol. 1999, 155, 1013–1020. [Google Scholar] [CrossRef]
- Rosen, S.D.; Tsay, D.; Singer, M.S.; Hemmerich, S.; Abraham, W.M. Therapeutic targeting of endothelial ligands for L-selectin (PNAd) in a sheep model of asthma. Am. J. Pathol. 2005, 166, 935–944. [Google Scholar] [CrossRef]
- Hemmerich, S.; Rosen, S.D. Carbohydrate sulfotransferases in lymphocyte homing. Glycobiology 2000, 10, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, M.W.; Barclay, A.N.; Singer, M.S.; Rosen, S.D.; van der Merwe, P.A. Affinity and kinetic analysis of L-selectin (CD62L) binding to glycosylation-dependent cell-adhesion molecule-1. J. Biol. Chem. 1998, 273, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Rosenbloom, C.L.; Anderson, D.C.; Manning, A.M. Selective inhibition of E-selectin, vascular cell adhesion molecule-1, and intercellular adhesion molecule-1 expression by inhibitors of I kappa B-alpha phosphorylation. J. Immunol. 1995, 155, 3538–3545. [Google Scholar]
- Satomaa, T.; Renkonen, O.; Helin, J.; Kirveskari, J.; Makitie, A.; Renkonen, R. O-glycans on human high endothelial CD34 putatively participating in L-selectin recognition. Blood 2002, 99, 2609–2611. [Google Scholar] [CrossRef]
- Hernandez Mir, G.; Helin, J.; Skarp, K.P.; Cummings, R.D.; Makitie, A.; Renkonen, R.; Leppanen, A. Glycoforms of human endothelial CD34 that bind L-selectin carry sulfated sialyl Lewis x capped O- and N-glycans. Blood 2009, 114, 733–741. [Google Scholar]
- Nielsen, J.S.; McNagny, K.M. Novel functions of the CD34 family. J. Cell Sci. 2008, 121, 3683–3692. [Google Scholar] [CrossRef]
- Hoke, D.; Mebius, R.E.; Dybdal, N.; Dowbenko, D.; Gribling, P.; Kyle, C.; Baumhueter, S.; Watson, S.R. Selective modulation of the expression of L-selectin ligands by an immune response. Curr. Biol. 1995, 5, 670–678. [Google Scholar] [CrossRef]
- Berg, E.L.; McEvoy, L.M.; Berlin, C.; Bargatze, R.F.; Butcher, E.C. L-selectin-mediated lymphocyte rolling on MAdCAM-1. Nature 1993, 366, 695–698. [Google Scholar] [CrossRef]
- Patel, K.D.; Cuvelier, S.L.; Wiehler, S. Selectins: Critical mediators of leukocyte recruitment. Semin. Immunol. 2002, 14, 73–81. [Google Scholar] [CrossRef]
- Berlin, C.; Bargatze, R.F.; Campbell, J.J.; von Andrian, U.H.; Szabo, M.C.; Hasslen, S.R.; Nelson, R.D.; Berg, E.L.; Erlandsen, S.L.; Butcher, E.C. alpha 4 integrins mediate lymphocyte attachment and rolling under physiologic flow. Cell 1995, 80, 413–422. [Google Scholar] [CrossRef]
- Kleene, R.; Berger, E.G. The molecular and cell biology of glycosyltransferases. Biochim. Biophys. Acta 1993, 1154, 283–325. [Google Scholar] [CrossRef]
- Beyer, T.A.; Sadler, J.E.; Rearick, J.I.; Paulson, J.C.; Hill, R.L. Glycosyltransferases and their use in assessing oligosaccharide structure and structure-function relationships. Adv. Enzymol. Relat. Areas Mol. Biol. 1981, 52, 23–175. [Google Scholar] [PubMed]
- Campbell, J.A.; Davies, G.J.; Bulone, V.; Henrissat, B. A classification of nucleotide-diphospho-sugar glycosyltransferases based on amino acid sequence similarities. Biochem. J. 1997, 32, 929–939. [Google Scholar] [CrossRef]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucl. Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef]
- Tenno, M.; Ohtsubo, K.; Hagen, F.K.; Ditto, D.; Zarbock, A.; Schaerli, P.; von Andrian, U.H.; Ley, K.; Le, D.; Tabak, L.A.; et al. Initiation of protein O glycosylation by the polypeptide GalNAcT-1 in vascular biology and humoral immunity. Mol. Cell Biol. 2007, 27, 8783–8796. [Google Scholar] [CrossRef]
- Ten Hagen, K.G.; Fritz, T.A.; Tabak, L.A. All in the family: The UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferases. Glycobiology 2003, 13, 1R–6R. [Google Scholar] [CrossRef]
- Fritz, T.A.; Raman, J.; Tabak, L.A. Dynamic association between the catalytic and lectin domains of human UDP-GalNAc:polypeptide alpha-N-acetylgalactosaminyltransferase-2. J. Biol. Chem. 2006, 281, 8613–8619. [Google Scholar] [CrossRef]
- Kubota, T.; Shiba, T.; Sugioka, S.; Furukawa, S.; Sawaki, H.; Kato, R.; Wakatsuki, S.; Narimatsu, H. Structural basis of carbohydrate transfer activity by human UDP-GalNAc: Polypeptide alpha-N-acetylgalactosaminyltransferase (pp-GalNAc-T10). J. Mol. Biol. 2006, 359, 708–727. [Google Scholar] [CrossRef]
- Tenno, M.; Kezdy, F.J.; Elhammer, A.P.; Kurosaka, A. Function of the lectin domain of polypeptide N-acetylgalactosaminyltransferase 1. Biochem. Biophys. Res. Commun. 2002, 298, 755–759. [Google Scholar] [CrossRef]
- Trnka, T.; Kozmon, S.; Tvaroska, I.; Koca, J. Stepwise catalytic mechanism via short-lived intermediate inferred from combined QM/MM MERP and PES calculations on retaining glycosyltransferase ppGalNAcT2. PLoS Comput. Biol. 2015, 11, e1004061. [Google Scholar] [CrossRef]
- Janos, P.; Trnka, T.; Kozmon, S.; Tvaroska, I.; Koca, J. Different QM/MM Approaches To Elucidate Enzymatic Reactions: Case Study on ppGalNAcT2. J. Chem. Theory Comput. 2016, 12, 6062–6076. [Google Scholar] [CrossRef] [PubMed]
- Pak, J.E.; Arnoux, P.; Zhou, S.; Sivarajah, P.; Satkunarajah, M.; Xing, X.; Rini, J.M. X-ray crystal structure of leukocyte type core 2 beta1,6-N-acetylglucosaminyltransferase. Evidence for a convergence of metal ion-independent glycosyltransferase mechanism. J. Biol. Chem. 2006, 281, 26693–26701. [Google Scholar] [CrossRef]
- Tvaroska, I.; Kozmon, S.; Wimmerova, M.; Koca, J. A QM/MM investigation of the catalytic mechanism of metal-ion-independent core 2 beta1,6-N-acetylglucosaminyltransferase. Chem. Eur. J. 2013, 19, 8153–8162. [Google Scholar] [CrossRef] [PubMed]
- Angata, K.; Lee, W.; Mitoma, J.; Marth, J.D.; Fukuda, M. Cellular and molecular analysis of neural development of glycosyltransferase gene knockout mice. Methods Enzymol. 2006, 417, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Ellies, L.G.; Tsuboi, S.; Petryniak, B.; Lowe, J.B.; Fukuda, M.; Marth, J.D. Core 2 oligosaccharide biosynthesis distinguishes between selectin ligands essential for leukocyte homing and inflammation. Immunity 1998, 9, 881–890. [Google Scholar] [CrossRef]
- Snapp, K.R.; Heitzig, C.E.; Ellies, L.G.; Marth, J.D.; Kansas, G.S. Differential requirements for the O-linked branching enzyme core 2 beta1-6-N-glucosaminyltransferase in biosynthesis of ligands for E-selectin and P-selectin. Blood 2001, 97, 3806–3811. [Google Scholar] [CrossRef]
- Sperandio, M.; Thatte, A.; Foy, D.; Ellies, L.G.; Marth, J.D.; Ley, K. Severe impairment of leukocyte rolling in venules of core 2 glucosaminyltransferase-deficient mice. Blood 2001, 97, 3812–3819. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.Y.; Antonopoulos, A.; Gupta, R.; Qu, J.; Dell, A.; Haslam, S.M.; Neelamegham, S. Competition between core-2 GlcNAc-transferase and ST6GalNAc-transferase regulates the synthesis of the leukocyte selectin ligand on human P-selectin glycoprotein ligand-1. J. Biol. Chem. 2013, 288, 13974–13987. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, B.; Qasba, P.K. Crystal structure of lactose synthase reveals a large conformational change in its catalytic component, the beta-1,4-galactosyltransferase-I. J. Mol. Biol. 2001, 310, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, B.; Balaji, P.V.; Qasba, P.K. Crystal structure of beta-1,4-galactosyltransferase complex with UDP-Gal reveals an oligosaccharide acceptor binding site. J. Mol. Biol. 2002, 318, 491–502. [Google Scholar] [CrossRef]
- Krupicka, M.; Tvaroska, I. Hybrid quantum mechanical/molecular mechanical investigation of the beta-1,4-galactosyltransferase-I mechanism. J. Phys. Chem. B 2009, 113, 11314–11319. [Google Scholar] [CrossRef]
- Asano, M.; Nakae, S.; Kotani, N.; Shirafuji, N.; Nambu, A.; Hashimoto, N.; Kawashima, H.; Hirose, M.; Miyasaka, M.; Takasaki, S.; et al. Impaired selectin-ligand biosynthesis and reduced inflammatory responses in beta-1,4-galactosyltransferase-I-deficient mice. Blood 2003, 102, 1678–1685. [Google Scholar] [CrossRef] [PubMed]
- Asano, M.; Furukawa, K.; Kido, M.; Matsumoto, S.; Umesaki, Y.; Kochibe, N.; Iwakura, Y. Growth retardation and early death of beta-1,4-galactosyltransferase knockout mice with augmented proliferation and abnormal differentiation of epithelial cells. EMBO J. 1997, 16, 1850–1857. [Google Scholar] [CrossRef] [PubMed]
- Oriol, R.; Mollicone, R.; Cailleau, A.; Balanzino, L.; Breton, C. Divergent evolution of fucosyltransferase genes from vertebrates, invertebrates, and bacteria. Glycobiology 1999, 9, 323–334. [Google Scholar] [CrossRef]
- de Vries, T.; Knegtel, R.M.; Holmes, E.H.; Macher, B.A. Fucosyltransferases: Structure/function studies. Glycobiology 2001, 11, 119R–128R. [Google Scholar] [CrossRef]
- Ma, B.; Simala-Grant, J.L.; Taylor, D.E. Fucosylation in prokaryotes and eukaryotes. Glycobiology 2006, 16, 158R–184R. [Google Scholar] [CrossRef]
- Huang, X.; Wei, C.; Li, F.; Jia, L.; Zeng, P.; Li, J.; Tan, J.; Sun, T.; Jiang, S.; Wang, J.; et al. PCGF6 regulates stem cell pluripotency as a transcription activator via super-enhancer dependent chromatin interactions. Protein Cell 2019, 10, 709–725. [Google Scholar] [CrossRef]
- Sun, H.Y.; Lin, S.W.; Ko, T.P.; Pan, J.F.; Liu, C.L.; Lin, C.N.; Wang, A.H.; Lin, C.H. Structure and mechanism of Helicobacter pylori fucosyltransferase. A basis for lipopolysaccharide variation and inhibitor design. J. Biol. Chem. 2007, 282, 9973–9982. [Google Scholar] [CrossRef]
- Murray, B.W.; Wittmann, V.; Burkart, M.D.; Hung, S.C.; Wong, C.H. Mechanism of human alpha-1,3-fucosyltransferase V: Glycosidic cleavage occurs prior to nucleophilic attack. Biochemistry 1997, 36, 823–831. [Google Scholar] [CrossRef]
- Maly, P.; Thall, A.; Petryniak, B.; Rogers, C.E.; Smith, P.L.; Marks, R.M.; Kelly, R.J.; Gersten, K.M.; Cheng, G.; Saunders, T.L.; et al. The alpha(1,3)fucosyltransferase Fuc-TVII controls leukocyte trafficking through an essential role in L-, E-, and P-selectin ligand biosynthesis. Cell 1996, 86, 643–653. [Google Scholar] [CrossRef]
- Weninger, W.; Ulfman, L.H.; Cheng, G.; Souchkova, N.; Quackenbush, E.J.; Lowe, J.B.; von Andrian, U.H. Specialized contributions by alpha(1,3)-fucosyltransferase-IV and FucT-VII during leukocyte rolling in dermal microvessels. Immunity 2000, 12, 665–676. [Google Scholar] [CrossRef]
- Harduin-Lepers, A.; Vallejo-Ruiz, V.; Krzewinski-Recchi, M.A.; Samyn-Petit, B.; Julien, S.; Delannoy, P. The human sialyltransferase family. Biochimie 2001, 83, 727–737. [Google Scholar] [CrossRef]
- Li, F.; Ding, J. Sialylation is involved in cell fate decision during development, reprogramming and cancer progression. Protein Cell 2019, 10, 550–565. [Google Scholar] [CrossRef] [PubMed]
- Audry, M.; Jeanneau, C.; Imberty, A.; Harduin-Lepers, A.; Delannoy, P.; Breton, C. Current trends in the structure-activity relationships of sialyltransferases. Glycobiology 2011, 21, 716–726. [Google Scholar] [CrossRef] [PubMed]
- Rao, F.V.; Rich, J.R.; Rakic, B.; Buddai, S.; Schwartz, M.F.; Johnson, K.; Bowe, C.; Wakarchuk, W.W.; Defrees, S.; Withers, S.G.; et al. Structural insight into mammalian sialyltransferases. Nat. Struct. Mol. Biol. 2009, 16, 1186–1188. [Google Scholar] [CrossRef]
- Meng, L.; Forouhar, F.; Thieker, D.; Gao, Z.; Ramiah, A.; Moniz, H.; Xiang, Y.; Seetharaman, J.; Milaninia, S.; Su, M.; et al. Enzymatic basis for N-glycan sialylation: Structure of rat alpha2,6-sialyltransferase (ST6GAL1) reveals conserved and unique features for glycan sialylation. J. Biol. Chem. 2013, 288, 34680–34698. [Google Scholar] [CrossRef]
- Kuhn, B.; Benz, J.; Greif, M.; Engel, A.M.; Sobek, H.; Rudolph, M.G. The structure of human alpha-2,6-sialyltransferase reveals the binding mode of complex glycans. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1826–1838. [Google Scholar] [CrossRef]
- Ellies, L.G.; Sperandio, M.; Underhill, G.H.; Yousif, J.; Smith, M.; Priatel, J.J.; Kansas, G.S.; Ley, K.; Marth, J.D. Sialyltransferase specificity in selectin ligand formation. Blood 2002, 100, 3618–3625. [Google Scholar] [CrossRef]
- Priatel, J.J.; Chui, D.; Hiraoka, N.; Simmons, C.J.; Richardson, K.B.; Page, D.M.; Fukuda, M.; Varki, N.M.; Marth, J.D. The ST3Gal-I sialyltransferase controls CD8+ T lymphocyte homeostasis by modulating O-glycan biosynthesis. Immunity 2000, 12, 273–283. [Google Scholar] [CrossRef]
- Chapman, E.; Best, M.D.; Hanson, S.R.; Wong, C.H. Sulfotransferases: Structure, mechanism, biological activity, inhibition, and synthetic utility. Angew. Chem. Int. Ed. Engl. 2004, 43, 3526–3548. [Google Scholar] [CrossRef]
- Bowman, K.G.; Bertozzi, C.R. Carbohydrate sulfotransferases: Mediators of extracellular communication. Chem. Biol. 1999, 6, R9–R22. [Google Scholar] [CrossRef]
- Brockhausen, I. Sulphotransferases acting on mucin-type oligosaccharides. Biochem. Soc. Trans. 2003, 31, 318–325. [Google Scholar] [CrossRef]
- Rath, V.L.; Verdugo, D.; Hemmerich, S. Sulfotransferase structural biology and inhibitor discovery. Drug Discov. Today 2004, 9, 1003–1011. [Google Scholar] [CrossRef]
- Negishi, M.; Pedersen, L.G.; Petrotchenko, E.; Shevtsov, S.; Gorokhov, A.; Kakuta, Y.; Pedersen, L.C. Structure and function of sulfotransferases. Arch. Biochem. Biophys. 2001, 390, 149–157. [Google Scholar] [CrossRef]
- Uchimura, K.; Gauguet, J.M.; Singer, M.S.; Tsay, D.; Kannagi, R.; Muramatsu, T.; von Andrian, U.H.; Rosen, S.D. A major class of L-selectin ligands is eliminated in mice deficient in two sulfotransferases expressed in high endothelial venules. Nat. Immunol. 2005, 6, 1105–1113. [Google Scholar] [CrossRef]
- Kawashima, H.; Petryniak, B.; Hiraoka, N.; Mitoma, J.; Huckaby, V.; Nakayama, J.; Uchimura, K.; Kadomatsu, K.; Muramatsu, T.; Lowe, J.B.; et al. N-acetylglucosamine-6-O-sulfotransferases 1 and 2 cooperatively control lymphocyte homing through L-selectin ligand biosynthesis in high endothelial venules. Nat. Immunol. 2005, 6, 1096–1104. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.L. The biology and enzymology of protein tyrosine O-sulfation. J. Biol. Chem. 2003, 278, 24243–24246. [Google Scholar] [CrossRef]
- Ramachandran, V.; Nollert, M.U.; Qiu, H.; Liu, W.J.; Cummings, R.D.; Zhu, C.; McEver, R.P. Tyrosine replacement in P-selectin glycoprotein ligand-1 affects distinct kinetic and mechanical properties of bonds with P- and L-selectin. Proc. Natl. Acad. Sci. USA 1999, 96, 13771–13776. [Google Scholar] [CrossRef] [PubMed]
- Teramoto, T.; Fujikawa, Y.; Kawaguchi, Y.; Kurogi, K.; Soejima, M.; Adachi, R.; Nakanishi, Y.; Mishiro-Sato, E.; Liu, M.C.; Sakakibara, Y.; et al. Crystal structure of human tyrosylprotein sulfotransferase-2 reveals the mechanism of protein tyrosine sulfation reaction. Nat. Commun. 2013, 4, 1572. [Google Scholar] [CrossRef]
- Marforio, T.D.; Giacinto, P.; Bottoni, A.; Calvaresi, M. Computational Evidence for the Catalytic Mechanism of Tyrosylprotein Sulfotransferases: A Density Functional Theory Investigation. Biochemistry 2015, 54, 4404–4410. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Y.B.; Moore, K.L. Molecular cloning and expression of human and mouse tyrosylprotein sulfotransferase-2 and a tyrosylprotein sulfotransferase homologue in Caenorhabditis elegans. J. Biol. Chem. 1998, 273, 24770–24774. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Hoffhines, A.J.; Moore, K.L.; Leary, J.A. Determination of the sites of tyrosine O-sulfation in peptides and proteins. Nat. Methods 2007, 4, 583–588. [Google Scholar] [CrossRef]
- Ley, K.; Kansas, G.S. Selectins in T-cell recruitment to non-lymphoid tissues and sites of inflammation. Nat. Rev. Immunol. 2004, 4, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Romano, S.J. Selectin antagonists: Therapeutic potential in asthma and COPD. Treat. Respir. Med. 2005, 4, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Romano, S.J.; Slee, D.H. Targeting selectins for the treatment of respiratory diseases. Curr. Opin. Investig. Drugs 2001, 2, 907–913. [Google Scholar]
- Czech, W.; Schopf, E.; Kapp, A. Soluble E-selectin in sera of patients with atopic dermatitis and psoriasis--correlation with disease activity. Br. J. Dermatol. 1996, 134, 17–21. [Google Scholar] [CrossRef]
- Schon, M.P.; Drewniok, C.; Boehncke, W.H. Targeting selectin functions in the therapy of psoriasis. Curr. Drug Targets Inflamm. Allergy 2004, 3, 163–168. [Google Scholar] [CrossRef]
- Bock, D.; Philipp, S.; Wolff, G. Therapeutic potential of selectin antagonists in psoriasis. Expert Opin. Investig. Drugs 2006, 15, 963–979. [Google Scholar] [CrossRef]
- Merten, M.; Thiagarajan, P. P-selectin in arterial thrombosis. Z. Kardiol. 2004, 93, 855–863. [Google Scholar] [CrossRef]
- Sfikakis, P.P.; Mavrikakis, M. Adhesion and lymphocyte costimulatory molecules in systemic rheumatic diseases. Clin. Rheumatol. 1999, 18, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J.; Gordon, J.L.; Gearing, A.J.; Pigott, R.; Woolf, N.; Katz, D.; Kyriakopoulos, A. The expression of the adhesion molecules ICAM-1, VCAM-1, PECAM, and E-selectin in human atherosclerosis. J. Pathol. 1993, 171, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.G.; Velji, R.; Guevara, N.V.; Hicks, M.J.; Chan, L.; Beaudet, A.L. P-Selectin or intercellular adhesion molecule (ICAM)-1 deficiency substantially protects against atherosclerosis in apolipoprotein E-deficient mice. J. Exp. Med. 2000, 191, 189–194. [Google Scholar] [CrossRef]
- Dong, Z.M.; Chapman, S.M.; Brown, A.A.; Frenette, P.S.; Hynes, R.O.; Wagner, D.D. The combined role of P- and E-selectins in atherosclerosis. J. Clin. Investig. 1998, 102, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Nishibayashi, Y.; Kita, K.; Ohno, O.; Imura, S.; Hirata, S. Adhesion molecules in the lymphoid cell distribution in rheumatoid synovial membrane. Bull. Hosp. Jt. Dis. 1993, 53, 23–28. [Google Scholar] [CrossRef]
- Chapman, P.T.; Jamar, F.; Keelan, E.T.; Peters, A.M.; Haskard, D.O. Use of a radiolabeled monoclonal antibody against E-selectin for imaging of endothelial activation in rheumatoid arthritis. Arthr. Rheum 1996, 39, 1371–1375. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Gao, Y.; Cheng, C.; Yan, M.; Wang, J. Upregulation of beta-1,4-galactosyltransferase I in rat spinal cord with experimental autoimmune encephalomyelitis. J. Mol. Neurosci. 2013, 49, 437–445. [Google Scholar] [CrossRef]
- Abdi, R.; Moore, R.; Sakai, S.; Donnelly, C.B.; Mounayar, M.; Sackstein, R. HCELL Expression on Murine MSC Licenses Pancreatotropism and Confers Durable Reversal of Autoimmune Diabetes in NOD Mice. Stem Cells 2015, 33, 1523–1531. [Google Scholar] [CrossRef] [PubMed]
- Konstantopoulos, K.; Thomas, S.N. Cancer cells in transit: The vascular interactions of tumor cells. Annu. Rev. Biomed. Eng. 2009, 11, 177–202. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Butcher, E.C. Leukocyte-endothelial cell recognition: Three (or more) steps to specificity and diversity. Cell 1991, 67, 1033–1036. [Google Scholar] [CrossRef]
- McEver, R.P.; Zhu, C. Rolling cell adhesion. Annu. Rev. Cell Dev. Biol. 2010, 26, 363–396. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, O.; Sanchez-Madrid, F. Molecular basis of leukocyte-endothelium interactions during the inflammatory response. Rev. Espan. Cardiol. 2009, 62, 552–562. [Google Scholar] [CrossRef]
- Butcher, E.C.; Picker, L.J. Lymphocyte homing and homeostasis. Science 1996, 272, 60–66. [Google Scholar] [CrossRef]
- Geng, Y.; Marshall, J.R.; King, M.R. Glycomechanics of the metastatic cascade: Tumor cell-endothelial cell interactions in the circulation. Ann. Biomed. Eng. 2012, 40, 790–805. [Google Scholar] [CrossRef]
- Ley, K.; Bullard, D.C.; Arbones, M.L.; Bosse, R.; Vestweber, D.; Tedder, T.F.; Beaudet, A.L. Sequential contribution of L- and P-selectin to leukocyte rolling in vivo. J. Exp. Med. 1995, 181, 669–675. [Google Scholar] [CrossRef]
- Jung, U.; Bullard, D.C.; Tedder, T.F.; Ley, K. Velocity differences between L- and P-selectin-dependent neutrophil rolling in venules of mouse cremaster muscle in vivo. Am. J. Physiol. 1996, 271, H2740–H2747. [Google Scholar] [CrossRef]
- Ley, K.; Tedder, T.F.; Kansas, G.S. L-selectin can mediate leukocyte rolling in untreated mesenteric venules in vivo independent of E- or P-selectin. Blood 1993, 82, 1632–1638. [Google Scholar] [CrossRef]
- Bevilacqua, M.P.; Pober, J.S.; Majeau, G.R.; Fiers, W.; Cotran, R.S.; Gimbrone, M.A., Jr. Recombinant tumor necrosis factor induces procoagulant activity in cultured human vascular endothelium: Characterization and comparison with the actions of interleukin 1. Proc. Natl. Acad. Sci. USA 1986, 83, 4533–4537. [Google Scholar] [CrossRef]
- Cotran, R.S.; Gimbrone, M.A., Jr.; Bevilacqua, M.P.; Mendrick, D.L.; Pober, J.S. Induction and detection of a human endothelial activation antigen in vivo. J. Exp. Med. 1986, 164, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Shaw, S.; Graber, N.; Gopal, T.V.; Horgan, K.J.; Van Seventer, G.A.; Newman, W. Activation-independent binding of human memory T cells to adhesion molecule ELAM-1. Nature 1991, 349, 799–802. [Google Scholar] [CrossRef] [PubMed]
- Picker, L.J.; Kishimoto, T.K.; Smith, C.W.; Warnock, R.A.; Butcher, E.C. ELAM-1 is an adhesion molecule for skin-homing T cells. Nature 1991, 349, 796–799. [Google Scholar] [CrossRef] [PubMed]
- Thomas, W. Catch bonds in adhesion. Annu. Rev. Biomed. Eng. 2008, 10, 39–57. [Google Scholar] [CrossRef]
- Cheung, L.S.; Raman, P.S.; Balzer, E.M.; Wirtz, D.; Konstantopoulos, K. Biophysics of selectin-ligand interactions in inflammation and cancer. Phys. Biol. 2011, 8, 015013. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Yago, T.; Lou, J.; Zarnitsyna, V.I.; McEver, R.P. Mechanisms for flow-enhanced cell adhesion. Ann. Biomed. Eng. 2008, 36, 604–621. [Google Scholar] [CrossRef]
- Alon, R.; Chen, S.; Puri, K.D.; Finger, E.B.; Springer, T.A. The kinetics of L-selectin tethers and the mechanics of selectin-mediated rolling. J. Cell Biol. 1997, 138, 1169–1180. [Google Scholar] [CrossRef]
- Finger, E.B.; Puri, K.D.; Alon, R.; Lawrence, M.B.; von Andrian, U.H.; Springer, T.A. Adhesion through L-selectin requires a threshold hydrodynamic shear. Nature 1996, 379, 266–269. [Google Scholar] [CrossRef]
- Lawrence, M.B.; Kansas, G.S.; Kunkel, E.J.; Ley, K. Threshold levels of fluid shear promote leukocyte adhesion through selectins (CD62L,P,E). J. Cell Biol. 1997, 136, 717–727. [Google Scholar] [CrossRef]
- Alon, R.; Hammer, D.A.; Springer, T.A. Lifetime of the P-selectin-carbohydrate bond and its response to tensile force in hydrodynamic flow. Nature 1995, 374, 539–542. [Google Scholar] [CrossRef]
- Alon, R.; Chen, S.; Fuhlbrigge, R.; Puri, K.D.; Springer, T.A. The kinetics and shear threshold of transient and rolling interactions of L-selectin with its ligand on leukocytes. Proc. Natl. Acad. Sci. USA 1998, 95, 11631–11636. [Google Scholar] [CrossRef] [PubMed]
- Puri, K.D.; Finger, E.B.; Springer, T.A. The faster kinetics of L-selectin than of E-selectin and P-selectin rolling at comparable binding strength. J. Immunol. 1997, 158, 405–413. [Google Scholar] [PubMed]
- Kunkel, E.J.; Ley, K. Distinct phenotype of E-selectin-deficient mice. E-selectin is required for slow leukocyte rolling in vivo. Circ. Res. 1996, 79, 1196–1204. [Google Scholar] [CrossRef]
- Hanley, W.D.; Wirtz, D.; Konstantopoulos, K. Distinct kinetic and mechanical properties govern selectin-leukocyte interactions. J. Cell Sci. 2004, 117, 2503–2511. [Google Scholar] [CrossRef]
- Zhu, C.; Long, M.; Chesla, S.E.; Bongrand, P. Measuring receptor/ligand interaction at the single-bond level: Experimental and interpretative issues. Ann. Biomed. Eng. 2002, 30, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Chen, J.; Chesla, S.E.; Yago, T.; Mehta, P.; McEver, R.P.; Zhu, C.; Long, M. Quantifying the effects of molecular orientation and length on two-dimensional receptor-ligand binding kinetics. J. Biol. Chem. 2004, 279, 44915–44923. [Google Scholar] [CrossRef]
- Chen, W.; Evans, E.A.; McEver, R.P.; Zhu, C. Monitoring receptor-ligand interactions between surfaces by thermal fluctuations. Biophys. J. 2008, 94, 694–701. [Google Scholar] [CrossRef]
- Klopocki, A.G.; Yago, T.; Mehta, P.; Yang, J.; Wu, T.; Leppanen, A.; Bovin, N.V.; Cummings, R.D.; Zhu, C.; McEver, R.P. Replacing a lectin domain residue in L-selectin enhances binding to P-selectin glycoprotein ligand-1 but not to 6-sulfo-sialyl Lewis x. J. Biol. Chem. 2008, 283, 11493–11500. [Google Scholar] [CrossRef]
- Bell, G.I. Models for the specific adhesion of cells to cells. Science 1978, 200, 618–627. [Google Scholar] [CrossRef]
- Dembo, M.; Torney, D.C.; Saxman, K.; Hammer, D. The reaction-limited kinetics of membrane-to-surface adhesion and detachment. Proc. R. Soc. Lond. B Biol. Sci. 1988, 234, 55–83. [Google Scholar]
- Sarangapani, K.K.; Yago, T.; Klopocki, A.G.; Lawrence, M.B.; Fieger, C.B.; Rosen, S.D.; McEver, R.P.; Zhu, C. Low force decelerates L-selectin dissociation from P-selectin glycoprotein ligand-1 and endoglycan. J. Biol. Chem. 2004, 279, 2291–2298. [Google Scholar] [CrossRef] [PubMed]
- Marshall, B.T.; Long, M.; Piper, J.W.; Yago, T.; McEver, R.P.; Zhu, C. Direct observation of catch bonds involving cell-adhesion molecules. Nature 2003, 423, 190–193. [Google Scholar] [CrossRef]
- Yago, T.; Wu, J.; Wey, C.D.; Klopocki, A.G.; Zhu, C.; McEver, R.P. Catch bonds govern adhesion through L-selectin at threshold shear. J. Cell Biol. 2004, 166, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Lou, J.; Yago, T.; Klopocki, A.G.; Mehta, P.; Chen, W.; Zarnitsyna, V.I.; Bovin, N.V.; Zhu, C.; McEver, R.P. Flow-enhanced adhesion regulated by a selectin interdomain hinge. J. Cell Biol. 2006, 174, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Springer, T.A. Structural basis for selectin mechanochemistry. Proc. Natl. Acad. Sci. USA 2009, 106, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Waldron, T.T.; Springer, T.A. Transmission of allostery through the lectin domain in selectin-mediated cell adhesion. Proc. Natl. Acad. Sci. USA 2009, 106, 85–90. [Google Scholar] [CrossRef]
- Lou, J.; Zhu, C. A structure-based sliding-rebinding mechanism for catch bonds. Biophys. J. 2007, 92, 1471–1485. [Google Scholar] [CrossRef]
- Gunnerson, K.N.; Pereverzev, Y.V.; Prezhdo, O.V. Atomistic simulation combined with analytic theory to study the response of the P-selectin/PSGL-1 complex to an external force. J. Phys. Chem. B 2009, 113, 2090–2100. [Google Scholar] [CrossRef]
- Furie, B.; Furie, B.C. Role of platelet P-selectin and microparticle PSGL-1 in thrombus formation. Trends Mol. Med. 2004, 10, 171–178. [Google Scholar] [CrossRef]
- Cambien, B.; Wagner, D.D. A new role in hemostasis for the adhesion receptor P-selectin. Trends Mol. Med. 2004, 10, 179–186. [Google Scholar] [CrossRef]
- Vandendries, E.R.; Furie, B.C.; Furie, B. Role of P-selectin and PSGL-1 in coagulation and thrombosis. Thromb. Haemost. 2004, 92, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Polgar, J.; Matuskova, J.; Wagner, D.D. The P-selectin, tissue factor, coagulation triad. J. Thromb. Haemost. 2005, 3, 1590–1596. [Google Scholar] [CrossRef] [PubMed]
- Frenette, P.S.; Denis, C.V.; Weiss, L.; Jurk, K.; Subbarao, S.; Kehrel, B.; Hartwig, J.H.; Vestweber, D.; Wagner, D.D. P-Selectin glycoprotein ligand 1 (PSGL-1) is expressed on platelets and can mediate platelet-endothelial interactions in vivo. J. Exp. Med. 2000, 191, 1413–1422. [Google Scholar] [CrossRef] [PubMed]
- Palabrica, T.; Lobb, R.; Furie, B.C.; Aronovitz, M.; Benjamin, C.; Hsu, Y.M.; Sajer, S.A.; Furie, B. Leukocyte accumulation promoting fibrin deposition is mediated in vivo by P-selectin on adherent platelets. Nature 1992, 359, 848–851. [Google Scholar] [CrossRef]
- Fijnheer, R.; Frijns, C.J.; Korteweg, J.; Rommes, H.; Peters, J.H.; Sixma, J.J.; Nieuwenhuis, H.K. The origin of P-selectin as a circulating plasma protein. Thromb. Haemost. 1997, 77, 1081–1085. [Google Scholar] [CrossRef]
- Celi, A.; Pellegrini, G.; Lorenzet, R.; De Blasi, A.; Ready, N.; Furie, B.C.; Furie, B. P-selectin induces the expression of tissue factor on monocytes. Proc. Natl. Acad. Sci. USA 1994, 91, 8767–8771. [Google Scholar] [CrossRef]
- Weyrich, A.S.; Elstad, M.R.; McEver, R.P.; McIntyre, T.M.; Moore, K.L.; Morrissey, J.H.; Prescott, S.M.; Zimmerman, G.A. Activated platelets signal chemokine synthesis by human monocytes. J. Clin. Investig. 1996, 97, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- Andre, P.; Denis, C.V.; Ware, J.; Saffaripour, S.; Hynes, R.O.; Ruggeri, Z.M.; Wagner, D.D. Platelets adhere to and translocate on von Willebrand factor presented by endothelium in stimulated veins. Blood 2000, 96, 3322–3328. [Google Scholar] [CrossRef]
- Rauch, U.; Nemerson, Y. Tissue factor, the blood, and the arterial wall. Trends Cardiovasc. Med. 2000, 10, 139–143. [Google Scholar] [CrossRef]
- Ishiwata, N.; Takio, K.; Katayama, M.; Watanabe, K.; Titani, K.; Ikeda, Y.; Handa, M. Alternatively spliced isoform of P-selectin is present in vivo as a soluble molecule. J. Biol. Chem. 1994, 269, 23708–23715. [Google Scholar] [PubMed]
- Blann, A.D.; Dobrotova, M.; Kubisz, P.; McCollum, C.N. von Willebrand factor, soluble P-selectin, tissue plasminogen activator and plasminogen activator inhibitor in atherosclerosis. Thromb. Haemost. 1995, 74, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Merten, M.; Thiagarajan, P. P-selectin expression on platelets determines size and stability of platelet aggregates. Circulation 2000, 102, 1931–1936. [Google Scholar] [CrossRef] [PubMed]
- Falati, S.; Liu, Q.; Gross, P.; Merrill-Skoloff, G.; Chou, J.; Vandendries, E.; Celi, A.; Croce, K.; Furie, B.C.; Furie, B. Accumulation of tissue factor into developing thrombi in vivo is dependent upon microparticle P-selectin glycoprotein ligand 1 and platelet P-selectin. J. Exp. Med. 2003, 197, 1585–1598. [Google Scholar] [CrossRef] [PubMed]
- Myers, D., Jr.; Farris, D.; Hawley, A.; Wrobleski, S.; Chapman, A.; Stoolman, L.; Knibbs, R.; Strieter, R.; Wakefield, T. Selectins influence thrombosis in a mouse model of experimental deep venous thrombosis. J. Surg. Res. 2002, 108, 212–221. [Google Scholar] [CrossRef]
- Myers, D.D.; Hawley, A.E.; Farris, D.M.; Wrobleski, S.K.; Thanaporn, P.; Schaub, R.G.; Wagner, D.D.; Kumar, A.; Wakefield, T.W. P-selectin and leukocyte microparticles are associated with venous thrombogenesis. J. Vasc. Surg. 2003, 38, 1075–1089. [Google Scholar] [CrossRef]
- Chong, B.H.; Murray, B.; Berndt, M.C.; Dunlop, L.C.; Brighton, T.; Chesterman, C.N. Plasma P-selectin is increased in thrombotic consumptive platelet disorders. Blood 1994, 83, 1535–1541. [Google Scholar] [CrossRef]
- Wu, G.; Li, F.; Li, P.; Ruan, C. Detection of plasma alpha-granule membrane protein GMP-140 using radiolabeled monoclonal antibodies in thrombotic diseases. Haemostasis 1993, 23, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.; Quarmby, J.W.; Collins, M.; Lockhart, S.M.; Burnand, K.G. Changes in the levels of soluble adhesion molecules and coagulation factors in patients with deep vein thrombosis. Thromb. Haemost. 1999, 82, 1593–1599. [Google Scholar] [CrossRef]
- Frenette, P.S.; Johnson, R.C.; Hynes, R.O.; Wagner, D.D. Platelets roll on stimulated endothelium in vivo: An interaction mediated by endothelial P-selectin. Proc. Natl. Acad. Sci. USA 1995, 92, 7450–7454. [Google Scholar] [CrossRef]
- Wakefield, T.W.; Strieter, R.M.; Downing, L.J.; Kadell, A.M.; Wilke, C.A.; Burdick, M.D.; Wrobleski, S.K.; Phillips, M.L.; Paulson, J.C.; Anderson, D.C.; et al. P-selectin and TNF inhibition reduce venous thrombosis inflammation. J. Surg. Res. 1996, 64, 26–31. [Google Scholar] [CrossRef]
- Ridker, P.M.; Buring, J.E.; Rifai, N. Soluble P-selectin and the risk of future cardiovascular events. Circulation 2001, 103, 491–495. [Google Scholar] [CrossRef]
- Hillis, G.S.; Terregino, C.; Taggart, P.; Killian, A.; Zhao, N.; Dalsey, W.C.; Mangione, A. Elevated soluble P-selectin levels are associated with an increased risk of early adverse events in patients with presumed myocardial ischemia. Am. Heart J. 2002, 143, 235–241. [Google Scholar] [CrossRef]
- Andre, P.; Hartwell, D.; Hrachovinova, I.; Saffaripour, S.; Wagner, D.D. Pro-coagulant state resulting from high levels of soluble P-selectin in blood. Proc. Natl. Acad. Sci. USA 2000, 97, 13835–13840. [Google Scholar] [CrossRef]
- Panicker, S.R.; Mehta-D’souza, P.; Zhang, N.; Klopocki, A.G.; Shao, B.; McEver, R.P. Circulating soluble P-selectin must dimerize to promote inflammation and coagulation in mice. Blood 2017, 130, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Cagnoni, A.J.; Perez Saez, J.M.; Rabinovich, G.A.; Marino, K.V. Turning-Off Signaling by Siglecs, Selectins, and Galectins: Chemical Inhibition of Glycan-Dependent Interactions in Cancer. Front. Oncol. 2016, 6, 109. [Google Scholar] [CrossRef] [PubMed]
- Bendas, G.; Borsig, L. Cancer cell adhesion and metastasis: Selectins, integrins, and the inhibitory potential of heparins. Int. J. Cell Biol. 2012, 2012, 676731. [Google Scholar] [CrossRef] [PubMed]
- Labelle, M.; Hynes, R.O. The initial hours of metastasis: The importance of cooperative host-tumor cell interactions during hematogenous dissemination. Cancer Discov. 2012, 2, 1091–1099. [Google Scholar] [CrossRef] [PubMed]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Poste, G.; Fidler, I.J. The pathogenesis of cancer metastasis. Nature 1980, 283, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Laubli, H.; Spanaus, K.S.; Borsig, L. Selectin-mediated activation of endothelial cells induces expression of CCL5 and promotes metastasis through recruitment of monocytes. Blood 2009, 114, 4583–4591. [Google Scholar] [CrossRef] [PubMed]
- Witz, I.P. Tumor-microenvironment interactions: The selectin-selectin ligand axis in tumor-endothelium cross talk. Cancer Treat. Res. 2006, 130, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Wirtz, D.; Konstantopoulos, K.; Searson, P.C. The physics of cancer: The role of physical interactions and mechanical forces in metastasis. Nat. Rev. Cancer 2011, 11, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Pearce, O.M.T. Cancer glycan epitopes: Biosynthesis, structure and function. Glycobiology 2018, 28, 670–696. [Google Scholar] [CrossRef] [PubMed]
- Hauselmann, I.; Borsig, L. Altered tumor-cell glycosylation promotes metastasis. Front. Oncol. 2014, 4, 28. [Google Scholar] [CrossRef]
- Pinho, S.S.; Reis, C.A. Glycosylation in cancer: Mechanisms and clinical implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef]
- Hakomori, S.; Kannagi, R. Glycosphingolipids as tumor-associated and differentiation markers. J. Natl. Cancer Inst. 1983, 71, 231–251. [Google Scholar] [CrossRef]
- Läubli, H.; Borsig, L. Altered Cell Adhesion and Glycosylation Promote Cancer Immune Suppression and Metastasis. Front. Immunol. 2019, 10, 2120. [Google Scholar] [CrossRef]
- Kannagi, R.; Izawa, M.; Koike, T.; Miyazaki, K.; Kimura, N. Carbohydrate-mediated cell adhesion in cancer metastasis and angiogenesis. Cancer Sci. 2004, 95, 377–384. [Google Scholar] [CrossRef]
- Kim, Y.J.; Varki, A. Perspectives on the significance of altered glycosylation of glycoproteins in cancer. Glycoconj. J. 1997, 14, 569–576. [Google Scholar] [CrossRef]
- Kim, Y.J.; Borsig, L.; Varki, N.M.; Varki, A. P-selectin deficiency attenuates tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 1998, 95, 9325–9330. [Google Scholar] [CrossRef]
- Borsig, L.; Wong, R.; Feramisco, J.; Nadeau, D.R.; Varki, N.M.; Varki, A. Heparin and cancer revisited: Mechanistic connections involving platelets, P-selectin, carcinoma mucins, and tumor metastasis. Proc. Natl. Acad. Sci. USA 2001, 98, 3352–3357. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Borsig, L.; Han, H.L.; Varki, N.M.; Varki, A. Distinct selectin ligands on colon carcinoma mucins can mediate pathological interactions among platelets, leukocytes, and endothelium. Am. J. Pathol. 1999, 155, 461–472. [Google Scholar] [CrossRef]
- Borsig, L. Selectin facilitate carcinoma metastasis and heparin can prevent them. News Physiol. Sci. 2014, 19, 16–21. [Google Scholar] [CrossRef]
- Edwards, E.E.; Oh, J.; Anilkumar, A.; Birmingham, K.G.; Thomas, S.N. P-, but not E- or L-, selectin-mediated rolling adhesion persistence in hemodynamic flow diverges between metastatic and leukocytic cells. Oncotarget 2017, 8, 83585–83601. [Google Scholar] [CrossRef] [PubMed]
- Khatib, A.M.; Kontogiannea, M.; Fallavollita, L.; Jamison, B.; Meterissian, S.; Brodt, P. Rapid induction of cytokine and E-selectin expression in the liver in response to metastatic tumor cells. Cancer Res. 1999, 59, 1356–1361. [Google Scholar] [PubMed]
- Brodt, P.; Fallavollita, L.; Bresalier, R.S.; Meterissian, S.; Norton, C.R.; Wolitzky, B.A. Liver endothelial E-selectin mediates carcinoma cell adhesion and promotes liver metastasis. Int. J. Cancer. 1997, 71, 612–619. [Google Scholar] [CrossRef]
- Charpin, C.; Bergeret, D.; Garcia, S.; Andrac, L.; Martini, F.; Horschowski, N.; Choux, R.; Lavaut, M.N. ELAM selectin expression in breast carcinomas detected by automated and quantitative immunohistochemical assays. Int. J. Oncol. 1998, 12, 1041–1048. [Google Scholar] [CrossRef]
- Nguyen, M.; Corless, C.L.; Kraling, B.M.; Tran, C.; Atha, T.; Bischoff, J.; Barsky, S.H. Vascular expression of E-selectin is increased in estrogen-receptor-negative breast cancer: A role for tumor-cell-secreted interleukin-1 alpha. Am. J. Pathol. 1997, 150, 1307–1314. [Google Scholar]
- Staal-van den Brekel, A.J.; Thunnissen, F.B.; Buurman, W.A.; Wouters, E.F. Expression of E-selectin, intercellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1 in non-small-cell lung carcinoma. Virchows Arch. 1996, 428, 21–27. [Google Scholar] [CrossRef]
- Muller, A.M.; Weichert, A.; Muller, K.M. E-cadherin, E-selectin and vascular cell adhesion molecule: Immunohistochemical markers for differentiation between mesothelioma and metastatic pulmonary adenocarcinoma? Virchows Arch. 2002, 441, 41–46. [Google Scholar] [CrossRef]
- Bhaskar, V.; Law, D.A.; Ibsen, E.; Breinberg, D.; Cass, K.M.; DuBridge, R.B.; Evangelista, F.; Henshall, S.M.; Hevezi, P.; Miller, J.C.; et al. E-selectin up-regulation allows for targeted drug delivery in prostate cancer. Cancer Res. 2003, 63, 6387–6394. [Google Scholar]
- Laferriere, J.; Houle, F.; Taher, M.M.; Valerie, K.; Huot, J. Transendothelial migration of colon carcinoma cells requires expression of E-selectin by endothelial cells and activation of stress-activated protein kinase-2 (SAPK2/p38) in the tumor cells. J. Biol. Chem. 2001, 276, 33762–33772. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, P.L.; Auger, F.A.; Huot, J. Regulation of transendothelial migration of colon cancer cells by E-selectin-mediated activation of p38 and ERK MAP kinases. Oncogene 2006, 25, 6563–6573. [Google Scholar] [CrossRef] [PubMed]
- Laubli, H.; Borsig, L. Selectins as mediators of lung metastasis. Cancer Microenviron. 2010, 3, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Stubke, K.; Wicklein, D.; Herich, L.; Schumacher, U.; Nehmann, N. Selectin-deficiency reduces the number of spontaneous metastases in a xenograft model of human breast cancer. Cancer Lett. 2012, 321, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Price, T.T.; Burness, M.L.; Sivan, A.; Warner, M.J.; Cheng, R.; Lee, C.H.; Olivere, L.; Comatas, K.; Magnani, J.; Kim Lyerly, H.; et al. Dormant breast cancer micrometastases reside in specific bone marrow niches that regulate their transit to and from bone. Sci. Transl. Med. 2016, 8, 340ra73. [Google Scholar] [CrossRef]
- Zen, K.; Liu, D.Q.; Guo, Y.L.; Wang, C.; Shan, J.; Fang, M.; Zhang, C.Y.; Liu, Y. CD44v4 is a major E-selectin ligand that mediates breast cancer cell transendothelial migration. PLoS ONE 2008, 3, e1826. [Google Scholar] [CrossRef] [PubMed]
- Laubli, H.; Stevenson, J.L.; Varki, A.; Varki, N.M.; Borsig, L. L-selectin facilitation of metastasis involves temporal induction of Fut7-dependent ligands at sites of tumor cell arrest. Cancer Res. 2006, 66, 1536–1542. [Google Scholar] [CrossRef] [PubMed]
- Mannori, G.; Crottet, P.; Cecconi, O.; Hanasaki, K.; Aruffo, A.; Nelson, R.M.; Varki, A.; Bevilacqua, M.P. Differential colon cancer cell adhesion to E-, P-, and L-selectin: Role of mucin-type glycoproteins. Cancer Res. 1995, 55, 4425–4431. [Google Scholar] [PubMed]
- Borsig, L.; Wong, R.; Hynes, R.O.; Varki, N.M.; Varki, A. Synergistic effects of L- and P-selectin in facilitating tumor metastasis can involve non-mucin ligands and implicate leukocytes as enhancers of metastasis. Proc. Natl. Acad. Sci. USA 2002, 99, 2193–2198. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Sceneay, J.; Smyth, M.J.; Moller, A. The pre-metastatic niche: Finding common ground. Cancer Metastasis Rev. 2013, 32, 449–464. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.A.; Kang, Y. The metastasis-promoting roles of tumor-associated immune cells. J. Mol. Med. (Berl.) 2013, 91, 411–429. [Google Scholar] [CrossRef] [PubMed]
- Hiratsuka, S.; Goel, S.; Kamoun, W.S.; Maru, Y.; Fukumura, D.; Duda, D.G.; Jain, R.K. Endothelial focal adhesion kinase mediates cancer cell homing to discrete regions of the lungs via E-selectin up-regulation. Proc. Natl. Acad. Sci. USA 2011, 108, 3725–3730. [Google Scholar] [CrossRef] [PubMed]
- Vestweber, D.; Blanks, J.E. Mechanisms that regulate the function of the selectins and their ligands. Physiol. Rev. 1999, 79, 181–213. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.L. Signal transduction by cell adhesion receptors and the cytoskeleton: Functions of integrins, cadherins, selectins, and immunoglobulin-superfamily members. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 283–323. [Google Scholar] [CrossRef]
- Lorant, D.E.; Topham, M.K.; Whatley, R.E.; McEver, R.P.; McIntyre, T.M.; Prescott, S.M.; Zimmerman, G.A. Inflammatory roles of P-selectin. J. Clin. Investig. 1993, 92, 559–570. [Google Scholar] [CrossRef]
- Crockett-Torabi, E. Selectins and mechanisms of signal transduction. J. Leukoc. Biol. 1998, 63, 1–14. [Google Scholar] [CrossRef]
- Lefort, C.T.; Ley, K. Neutrophil arrest by LFA-1 activation. Front. Immunol. 2012, 3, 157. [Google Scholar] [CrossRef]
- Kuwano, Y.; Spelten, O.; Zhang, H.; Ley, K.; Zarbock, A. Rolling on E- or P-selectin induces the extended but not high-affinity conformation of LFA-1 in neutrophils. Blood 2010, 116, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, V.; Manarini, S.; Sideri, R.; Rotondo, S.; Martelli, N.; Piccoli, A.; Totani, L.; Piccardoni, P.; Vestweber, D.; de Gaetano, G.; et al. Platelet/polymorphonuclear leukocyte interaction: P-selectin triggers protein-tyrosine phosphorylation-dependent CD11b/CD18 adhesion: Role of PSGL-1 as a signaling molecule. Blood 1999, 93, 876–885. [Google Scholar] [CrossRef]
- Kaila, N.; Thomas, B.E. Selectin inhibitors. Expert Opin. Ther. Patents 2003, 13, 305–317. [Google Scholar] [CrossRef]
- Barthel, S.R.; Gavino, J.D.; Descheny, L.; Dimitroff, C.J. Targeting selectins and selectin ligands in inflammation and cancer. Expert Opin. Ther. Targets 2007, 11, 1473–1491. [Google Scholar] [CrossRef] [PubMed]
- Boland, E.W. Clinical observations with 16 alpha-methyl corticosteroid compounds; preliminary therapeutic trials with dexamethasone (16 alpha-methyl 9 alpha-fluoroprednisolone) in patients with rheumatoid arthritis. Ann. Rheum Dis. 1958, 17, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Brostjan, C.; Anrather, J.; Csizmadia, V.; Natarajan, G.; Winkler, H. Glucocorticoids inhibit E-selectin expression by targeting NF-kappaB and not ATF/c-Jun. J. Immunol. 1997, 158, 3836–3844. [Google Scholar] [PubMed]
- Grau, M.; Montero, J.L.; Guasch, J.; Felipe, A.; Carrasco, E.; Julia, S. The pharmacological profile of aceclofenac, a new nonsteroidal antiinflammatory and analgesic drug. Agents Act. Suppl. 1991, 32, 125–129. [Google Scholar]
- Sharma, G.; Singh, J.; Anand, D.; Kumar, M.; Raza, K.; Pareek, A.; Katare, O.P. Aceclofenac: Species-Dependent Metabolism and Newer Paradigm Shift from Oral to Non-oral Delivery. Curr. Top. Med. Chem. 2017, 17, 107–119. [Google Scholar] [CrossRef]
- Raza, K.; Kumar, M.; Kumar, P.; Malik, R.; Sharma, G.; Kaur, M.; Katare, O.P. Topical delivery of aceclofenac: Challenges and promises of novel drug delivery systems. Biomed. Res. Int. 2014, 2014, 406731. [Google Scholar] [CrossRef]
- Gonzalez-Alvaro, I.; Carmona, L.; Diaz-Gonzalez, F.; Gonzalez-Amaro, R.; Mollinedo, F.; Sanchez-Madrid, F.; Laffon, A.; Garcia-Vicuna, R. Aceclofenac, a new nonsteroidal antiinflammatory drug, decreases the expression and function of some adhesion molecules on human neutrophils. J. Rheumatol. 1996, 23, 723–729. [Google Scholar]
- Liu, J.; Zhang, J.; Shi, Y.; Grimsgaard, S.; Alraek, T.; Fonnebo, V. Chinese red yeast rice (Monascus purpureus) for primary hyperlipidemia: A meta-analysis of randomized controlled trials. Chin. Med. 2006, 1, 4. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.J.; Pan, Y.Z.; Liu, Q.J.; Li, X.H. Exposure assessment of lovastatin in Pu-erh tea. Int. J. Food Microbiol. 2013, 164, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Jakobisiak, M.; Golab, J. Potential antitumor effects of statins (Review). Int. J. Oncol. 2003, 23, 1055–1069. [Google Scholar] [CrossRef]
- Chae, Y.K.; Yousaf, M.; Malecek, M.K.; Carneiro, B.; Chandra, S.; Kaplan, J.; Kalyan, A.; Sassano, A.; Platanias, L.C.; Giles, F. Statins as anti-cancer therapy; Can we translate preclinical and epidemiologic data into clinical benefit? Discov. Med. 2015, 20, 413–427. [Google Scholar] [PubMed]
- Nubel, T.; Dippold, W.; Kleinert, H.; Kaina, B.; Fritz, G. Lovastatin inhibits Rho-regulated expression of E-selectin by TNFalpha and attenuates tumor cell adhesion. FASEB J. 2004, 18, 140–142. [Google Scholar] [CrossRef] [PubMed]
- Ostrau, C.; Hulsenbeck, J.; Herzog, M.; Schad, A.; Torzewski, M.; Lackner, K.J.; Fritz, G. Lovastatin attenuates ionizing radiation-induced normal tissue damage in vivo. Radiother. Oncol. 2009, 92, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Hevey, R. Strategies for the Development of Glycomimetic Drug Candidates. Pharmaceuticals (Basel) 2019, 12, 55. [Google Scholar] [CrossRef]
- Kaila, N.; Thomas, B.E.T. Design and synthesis of sialyl Lewis(x) mimics as E- and P-selectin inhibitors. Med. Res. Rev. 2002, 22, 566–601. [Google Scholar] [CrossRef]
- Ernst, B.; Magnani, J.L. From carbohydrate leads to glycomimetic drugs. Nat. Rev. Drug Discov. 2009, 8, 661–677. [Google Scholar] [CrossRef]
- Lefer, D.J. Pharmacology of selectin inhibitors in ischemia/reperfusion states. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 283–294. [Google Scholar] [CrossRef]
- Dube, D.H.; Bertozzi, C.R. Glycans in cancer and inflammation--potential for therapeutics and diagnostics. Nat. Rev. Drug Discov. 2005, 4, 477–488. [Google Scholar] [CrossRef]
- Valverde, P.; Arda, A.; Reichardt, N.-C.; Jimenez-Barbero, J.; Gimeno, A. Glycans in drug discovery. Med. Chem. Commun. 2019, 10, 1678–1691. [Google Scholar] [CrossRef] [PubMed]
- Aydt, E.M.; Bock, D.; Wolff, G. Selectin antagonists and their potential impact for the treatment of inflammatory lung diseases. In New Drugs and Targets for Asthma and COPD; Hansel, T.T., Barnes, P.J., Eds.; S Karger AG: Basel, Switzerland, 2010; Volume 39, pp. 175–184. ISBN 978-3-8055-9566-7. [Google Scholar]
- Tamburrini, A.; Colombo, C.; Bernardi, A. Design and synthesis of glycomimetics: Recent advances. Med. Res. Rev. 2019. [Google Scholar] [CrossRef] [PubMed]
- Kerr, K.M.; Auger, W.R.; Marsh, J.J.; Comito, R.M.; Fedullo, R.L.; Smits, G.J.; Kapelanski, D.P.; Fedullo, P.F.; Channick, R.N.; Jamieson, S.W.; et al. The use of cylexin (CY-1503) in prevention of reperfusion lung injury in patients undergoing pulmonary thromboendarterectomy. Am. J. Respir. Crit. Care Med. 2000, 162, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Ohmoto, H.; Nakamura, K.; Inoue, T.; Kondo, N.; Inoue, Y.; Yoshino, K.; Kondo, H.; Ishida, H.; Kiso, M.; Hasegawa, A. Studies on selectin blocker. 1. Structure-activity relationships of sialyl Lewis X analogs. J. Med. Chem. 1996, 39, 1339–1343. [Google Scholar] [CrossRef]
- Banteli, R.; Ernst, B. Synthesis of sialyl Lewis(x) mimics. Modifications of the 6-position of galactose. Bioorg. Med. Chem. Lett. 2001, 11, 459–462. [Google Scholar] [CrossRef]
- Hanessian, S.; Huynh, H.K.; Reddy, G.V.; McNaughton-Smith, G.; Ernst, B.; Kolb, H.C.; Magnani, J.; Sweeley, C. Exploration of beta-turn scaffolding motifs as components of sialyl Le(X) mimetics and their relevance to P-selectin. Bioorg. Med. Chem. Lett. 1998, 8, 2803–2808. [Google Scholar] [CrossRef]
- Rao, B.N.; Anderson, M.B.; Musser, J.H.; Gilbert, J.H.; Schaefer, M.E.; Foxall, C.; Brandley, B.K. Sialyl Lewis X mimics derived from a pharmacophore search are selectin inhibitors with anti-inflammatory activity. J. Biol. Chem. 1994, 269, 19663–19666. [Google Scholar]
- Kogan, T.P.; Dupre, B.; Keller, K.M.; Scott, I.L.; Bui, H.; Market, R.V.; Beck, P.J.; Voytus, J.A.; Revelle, B.M.; Scott, D. Rational design and synthesis of small molecule, non-oligosaccharide selectin inhibitors: (alpha-D-mannopyranosyloxy)biphenyl-substituted carboxylic acids. J. Med. Chem. 1995, 38, 4976–4984. [Google Scholar] [CrossRef]
- Stewart, A.O.; Bhatia, P.A.; McCarty, C.M.; Patel, M.V.; Staeger, M.A.; Arendsen, D.L.; Gunawardana, I.W.; Melcher, L.M.; Zhu, G.D.; Boyd, S.A.; et al. Discovery of inhibitors of cell adhesion molecule expression in human endothelial cells. 1. Selective inhibition of ICAM-1 and E-selectin expression. J. Med. Chem. 2001, 44, 988–1002. [Google Scholar] [CrossRef]
- Kaila, N.; Chen, L.; Thomas, B.E.T.; Tsao, D.; Tam, S.; Bedard, P.W.; Camphausen, R.T.; Alvarez, J.C.; Ullas, G. Beta-C-mannosides as selectin inhibitors. J. Med. Chem. 2002, 45, 1563–1566. [Google Scholar] [CrossRef]
- Prodger, J.C.; Bamford, M.J.; Bird, M.I.; Gore, P.M.; Holmes, D.S.; Priest, R.; Saez, V. Mimics of the sialyl Lewis X tetrasaccharide. Replacement of the N-acetylglucosamine sugar with simple C2-symmetric 1,2-diols. Bioorg. Med. Chem. 1996, 4, 793–801. [Google Scholar] [CrossRef]
- Thoma, G.; Magnani, J.L.; Patton, J.T.; Ernst, B.; Jahnke, W. Preorganization of the Bioactive Conformation of Sialyl Lewis(X) Analogues Correlates with Their Affinity to E-Selectin. Angew. Chem. Int. Ed. Engl. 2001, 40, 1941–1945. [Google Scholar] [CrossRef]
- Thoma, G.; Magnani, J.L.; Patton, J.T. Synthesis and biological evaluation of a sialyl Lewis X mimic with significantly improved E-selectin inhibition. Bioorg. Med. Chem. Lett. 2001, 11, 923–925. [Google Scholar] [CrossRef]
- De Vleeschauwer, M.; Vaillancourt, M.; Goudreau, N.; Guindon, Y.; Gravel, D. Design and synthesis of a new sialyl Lewis X mimetic: How selective are the selectin receptors? Bioorg. Med. Chem. Lett. 2001, 11, 1109–1112. [Google Scholar] [CrossRef]
- Hanessian, S.; Mascitti, V.; Rogel, O. Synthesis of a potent antagonist of E-selectin. J. Org. Chem. 2002, 67, 3346–3354. [Google Scholar] [CrossRef]
- Schwizer, D.; Patton, J.T.; Cutting, B.; Smiesko, M.; Wagner, B.; Kato, A.; Weckerle, C.; Binder, F.P.; Rabbani, S.; Schwardt, O.; et al. Pre-organization of the core structure of E-selectin antagonists. Chem. Eur. J. 2012, 18, 1342–1351. [Google Scholar] [CrossRef]
- Kogan, T.P.; Dupre, B.; Bui, H.; McAbee, K.L.; Kassir, J.M.; Scott, I.L.; Hu, X.; Vanderslice, P.; Beck, P.J.; Dixon, R.A. Novel synthetic inhibitors of selectin-mediated cell adhesion: Synthesis of 1,6-bis[3-(3-carboxymethylphenyl)-4-(2-alpha-D- mannopyranosyloxy)phenyl]hexane (TBC1269). J. Med. Chem. 1998, 41, 1099–1111. [Google Scholar] [CrossRef]
- Tsai, C.Y.; Park, W.K.; Weitz-Schmidt, G.; Ernst, B.; Wong, C.H. Synthesis of sialyl Lewis X mimetics using the Ugi four-component reaction. Bioorg. Med. Chem. Lett. 1998, 8, 2333–2338. [Google Scholar] [CrossRef]
- Thoma, G.; Banteli, R.; Jahnke, W.; Magnani, J.L.; Patton, J.T. A Readily Available, Highly Potent E-Selectin Antagonist. Angew. Chem. Int. Ed. Engl. 2001, 40, 3644–3647. [Google Scholar] [CrossRef]
- Hiramatsu, Y.; Tsujishita, H.; Kondo, H. Studies on selectin blocker. 3. Investigation of the carbohydrate ligand sialyl Lewis X recognition site of P-selectin. J. Med. Chem. 1996, 39, 4547–4553. [Google Scholar] [CrossRef]
- Calosso, M.; Charpentier, D.; Vaillancourt, M.; Bencheqroun, M.; St-Pierre, G.; Wilkes, B.C.; Guindon, Y. A new approach to explore the binding space of polysaccharide-based ligands: Selectin antagonists. ACS Med. Chem. Lett. 2012, 3, 1045–1049. [Google Scholar] [CrossRef] [PubMed]
- Calosso, M.; Tambutet, G.; Charpentier, D.; St-Pierre, G.; Vaillancourt, M.; Bencheqroun, M.; Gratton, J.P.; Prevost, M.; Guindon, Y. Acyclic tethers mimicking subunits of polysaccharide ligands: Selectin antagonists. ACS Med. Chem. Lett. 2014, 5, 1054–1059. [Google Scholar] [CrossRef]
- Egger, J.; Weckerle, C.; Cutting, B.; Schwardt, O.; Rabbani, S.; Lemme, K.; Ernst, B. Nanomolar E-selectin antagonists with prolonged half-lives by a fragment-based approach. J. Am. Chem. Soc. 2013, 135, 9820–9828. [Google Scholar] [CrossRef] [PubMed]
- Bruehl, R.E.; Dasgupta, F.; Katsumoto, T.R.; Tan, J.H.; Bertozzi, C.R.; Spevak, W.; Ahn, D.J.; Rosen, S.D.; Nagy, J.O. Polymerized liposome assemblies: Bifunctional macromolecular selectin inhibitors mimicking physiological selectin ligands. Biochemistry 2001, 40, 5964–5974. [Google Scholar] [CrossRef] [PubMed]
- Sakagami, M.; Horie, K.; Nakamoto, K.; Kawaguchi, T.; Hamana, H. Synthesis of sialyl Lewis X-polysaccharide conjugates. Chem. Pharm. Bull. (Tokyo) 2000, 48, 1256–1263. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Murohara, T.; Margiotta, J.; Phillips, L.M.; Paulson, J.C.; DeFrees, S.; Zalipsky, S.; Guo, L.S.; Lefer, A.M. Cardioprotection by liposome-conjugated sialyl Lewisx-oligosaccharide in myocardial ischaemia and reperfusion injury. Cardiovasc. Res. 1995, 30, 965–974. [Google Scholar] [CrossRef]
- Spevak, W.; Foxall, C.; Charych, D.H.; Dasgupta, F.; Nagy, J.O. Carbohydrates in an acidic multivalent assembly: Nanomolar P-selectin inhibitors. J. Med. Chem. 1996, 39, 1018–1020. [Google Scholar] [CrossRef][Green Version]
- Moog, K.E.; Barz, M.; Bartneck, M.; Beceren-Braun, F.; Mohr, N.; Wu, Z.; Braun, L.; Dernedde, J.; Liehn, E.A.; Tacke, F.; et al. Polymeric Selectin Ligands Mimicking Complex Carbohydrates: From Selectin Binders to Modifiers of Macrophage Migration. Angew. Chem. Int. Ed. Engl. 2017, 56, 1416–1421. [Google Scholar] [CrossRef]
- Thoma, G.; Duthaler, R.O.; Magnani, J.L.; Patton, J.T. Nanomolar E-selectin inhibitors: 700-fold potentiation of affinity by multivalent ligand presentation. J. Am. Chem. Soc. 2001, 123, 10113–10114. [Google Scholar] [CrossRef]
- Esko, J.D.; Lindahl, U. Molecular diversity of heparan sulfate. J. Clin. Investig. 2001, 108, 169–173. [Google Scholar] [CrossRef]
- Wang, L.; Brown, J.R.; Varki, A.; Esko, J.D. Heparin’s anti-inflammatory effects require glucosamine 6-O-sulfation and are mediated by blockade of L- and P-selectins. J. Clin. Investig. 2002, 110, 127–136. [Google Scholar] [CrossRef]
- Aydt, E.; Wolff, G. Development of synthetic pan-selectin antagonists: A new treatment strategy for chronic inflammation in asthma. Pathobiology 2002, 70, 297–301. [Google Scholar] [CrossRef]
- Kozlowski, E.O.; Pavao, M.S.; Borsig, L. Ascidian dermatan sulfates attenuate metastasis, inflammation and thrombosis by inhibition of P-selectin. J. Thromb. Haemost. 2011, 9, 1807–1815. [Google Scholar] [CrossRef]
- Kawashima, H.; Hirose, M.; Hirose, J.; Nagakubo, D.; Plaas, A.H.; Miyasaka, M. Binding of a large chondroitin sulfate/dermatan sulfate proteoglycan, versican, to L-selectin, P-selectin, and CD44. J. Biol. Chem. 2000, 275, 35448–35456. [Google Scholar] [CrossRef]
- Wang, R.; Huang, J.; Wei, M.; Zeng, X. The synergy of 6-O-sulfation and N- or 3-O-sulfation of chitosan is required for efficient inhibition of P-selectin-mediated human melanoma A375 cell adhesion. Biosci. Biotechnol. Biochem. 2010, 74, 1697–1700. [Google Scholar] [CrossRef]
- van Weelden, G.; Bobinski, M.; Okla, K.; van Weelden, W.J.; Romano, A.; Pijnenborg, J.M.A. Fucoidan Structure and Activity in Relation to Anti-Cancer Mechanisms. Mar. Drugs 2019, 17, 32. [Google Scholar] [CrossRef] [PubMed]
- Atashrazm, F.; Lowenthal, R.M.; Woods, G.M.; Holloway, A.F.; Dickinson, J.L. Fucoidan and cancer: A multifunctional molecule with anti-tumor potential. Mar. Drugs 2015, 13, 2327–2346. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.Y.; Hwang, P.A. Clinical applications of fucoidan in translational medicine for adjuvant cancer therapy. Clin. Transl. Med. 2019, 8, 15. [Google Scholar] [CrossRef]
- Kwak, J.Y. Fucoidan as a marine anticancer agent in preclinical development. Mar. Drugs 2014, 12, 851–870. [Google Scholar] [CrossRef] [PubMed]
- Cumashi, A.; Ushakova, N.A.; Preobrazhenskaya, M.E.; D’Incecco, A.; Piccoli, A.; Totani, L.; Tinari, N.; Morozevich, G.E.; Berman, A.E.; Bilan, M.I.; et al. A comparative study of the anti-inflammatory, anticoagulant, antiangiogenic, and antiadhesive activities of nine different fucoidans from brown seaweeds. Glycobiology 2007, 17, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Bachelet, L.; Bertholon, I.; Lavigne, D.; Vassy, R.; Jandrot-Perrus, M.; Chaubet, F.; Letourneur, D. Affinity of low molecular weight fucoidan for P-selectin triggers its binding to activated human platelets. Biochim. Biophys. Acta 2009, 1790, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Weyrich, A.S.; Ma, X.Y.; Lefer, D.J.; Albertine, K.H.; Lefer, A.M. In vivo neutralization of P-selectin protects feline heart and endothelium in myocardial ischemia and reperfusion injury. J. Clin. Investig. 1993, 91, 2620–2629. [Google Scholar] [CrossRef] [PubMed]
- Bhushan, M.; Bleiker, T.O.; Ballsdon, A.E.; Allen, M.H.; Sopwith, M.; Robinson, M.K.; Clarke, C.; Weller, R.P.; Graham-Brown, R.A.; Keefe, M.; et al. Anti-E-selectin is ineffective in the treatment of psoriasis: A randomized trial. Br. J. Dermatol. 2002, 146, 824–831. [Google Scholar] [CrossRef] [PubMed]
- Eniola, A.O.; Hammer, D.A. Artificial polymeric cells for targeted drug delivery. J. Control. Release 2003, 87, 15–22. [Google Scholar] [CrossRef]
- Shamay, Y.; Elkabets, M.; Li, H.; Shah, J.; Brook, S.; Wang, F.; Adler, K.; Baut, E.; Scaltriti, M.; Jena, P.V.; et al. P-selectin is a nanotherapeutic delivery target in the tumor microenvironment. Sci. Transl. Med. 2016, 8, 345ra87. [Google Scholar] [CrossRef]
- Spragg, D.D.; Alford, D.R.; Greferath, R.; Larsen, C.E.; Lee, K.D.; Gurtner, G.C.; Cybulsky, M.I.; Tosi, P.F.; Nicolau, C.; Gimbrone, M.A., Jr. Immunotargeting of liposomes to activated vascular endothelial cells: A strategy for site-selective delivery in the cardiovascular system. Proc. Natl. Acad. Sci. USA 1997, 94, 8795–8800. [Google Scholar] [CrossRef]
- Bendas, G.; Krause, A.; Schmidt, R.; Vogel, J.; Rothe, U. Selectins as new targets for immunoliposome-mediated drug delivery. A potential way of anti-nflammatory therapy. Pharm. Acta Helv. 1998, 73, 19–26. [Google Scholar] [CrossRef]
- Slee, D.H.; Romano, S.J.; Yu, J.; Nguyen, T.N.; John, J.K.; Raheja, N.K.; Axe, F.U.; Jones, T.K.; Ripka, W.C. Development of potent non-carbohydrate imidazole-based small molecule selectin inhibitors with antiinflammatory activity. J. Med. Chem. 2001, 44, 2094–2107. [Google Scholar] [CrossRef]
- Kranich, R.; Busemann, A.S.; Bock, D.; Schroeter-Maas, S.; Beyer, D.; Heinemann, B.; Meyer, M.; Schierhorn, K.; Zahlten, R.; Wolff, G.; et al. Rational design of novel, potent small molecule pan-selectin antagonists. J. Med. Chem. 2007, 50, 1101–1115. [Google Scholar] [CrossRef]
- Kaila, N.; Somers, W.S.; Thomas, B.E.; Thakker, P.; Janz, K.; DeBernardo, S.; Tam, S.; Moore, W.J.; Yang, R.; Wrona, W.; et al. Quinic acid derivatives as sialyl Lewis(x)-mimicking selectin inhibitors: Design, synthesis, and crystal structure in complex with E-selectin. J. Med. Chem. 2005, 48, 4346–4357. [Google Scholar] [CrossRef] [PubMed]
- Schon, M.P.; Krahn, T.; Schon, M.; Rodriguez, M.L.; Antonicek, H.; Schultz, J.E.; Ludwig, R.J.; Zollner, T.M.; Bischoff, E.; Bremm, K.D.; et al. Efomycine M, a new specific inhibitor of selectin, impairs leukocyte adhesion and alleviates cutaneous inflammation. Nat. Med. 2002, 8, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Ulbrich, H.K.; Luxenburger, A.; Prech, P.; Eriksson, E.E.; Soehnlein, O.; Rotzius, P.; Lindbom, L.; Dannhardt, G. A novel class of potent nonglycosidic and nonpeptidic pan-selectin inhibitors. J. Med. Chem. 2006, 49, 5988–5999. [Google Scholar] [CrossRef]
- Hiramatsu, Y.; Tsukida, T.; Nakai, Y.; Inoue, Y.; Kondo, H. Study on selectin blocker. 8. Lead discovery of a non-sugar antagonist using a 3D-pharmacophore model. J. Med. Chem. 2000, 43, 1476–1483. [Google Scholar] [CrossRef] [PubMed]
- von Bonin, A.; Buchmann, B.; Bader, B.; Rausch, A.; Venstrom, K.; Schafer, M.; Grundemann, S.; Gunther, J.; Zorn, L.; Nubbemeyer, R.; et al. Efomycine M: An inhibitor of selectins? Nat. Med. 2006, 12, 873. [Google Scholar] [CrossRef]
- Wienrich, B.G.; Krahn, T.; Schon, M.; Rodriguez, M.L.; Kramer, B.; Busemann, M.; Boehncke, W.H.; Schon, M.P. Structure-function relation of efomycines, a family of small-molecule inhibitors of selectin functions. J. Investig. Dermatol. 2006, 126, 882–889. [Google Scholar] [CrossRef]
- Barth, R.; Mulzer, J. Two-directional total synthesis of efomycine M and formal total synthesis of elaiolide. Tetrahedron 2008, 64, 4718–4735. [Google Scholar] [CrossRef]
- Fukuda, M.N.; Ohyama, C.; Lowitz, K.; Matsuo, O.; Pasqualini, R.; Ruoslahti, E.; Fukuda, M. A peptide mimic of E-selectin ligand inhibits sialyl Lewis X-dependent lung colonization of tumor cells. Cancer Res. 2000, 60, 450–456. [Google Scholar]
- Molenaar, T.J.; Appeldoorn, C.C.; de Haas, S.A.; Michon, I.N.; Bonnefoy, A.; Hoylaerts, M.F.; Pannekoek, H.; van Berkel, T.J.; Kuiper, J.; Biessen, E.A. Specific inhibition of P-selectin-mediated cell adhesion by phage display-derived peptide antagonists. Blood 2002, 100, 3570–3577. [Google Scholar] [CrossRef]
- Ye, Z.; Zhang, S.; Liu, Y.; Wang, S.; Zhang, J.; Huang, R. A Peptide Analogue of Selectin Ligands Attenuated Atherosclerosis by Inhibiting Monocyte Activation. Mediat. Inflamm. 2019, 2019, 8709583. [Google Scholar] [CrossRef]
- Watz, H.; Bock, D.; Meyer, M.; Schierhorn, K.; Vollhardt, K.; Woischwill, C.; Pedersen, F.; Kirsten, A.; Beeh, K.M.; Meyer-Sabellek, W.; et al. Inhaled pan-selectin antagonist Bimosiamose attenuates airway inflammation in COPD. Pulm Pharmacol. Ther. 2013, 26, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.; Vohra, Y.; Myers, D.D.; Locatelli-Hoops, S.; Magnani, J.L. A Novel Glycomimetic Compound (GMI-1757) with Dual Functional Antagonism to E-Selectin and Galectin-3 Demonstrates Inhibition of Thrombus Formation in an Inferior Vena Cava Model. Blood 2018, 132 (Suppl. 1), 2211. [Google Scholar] [CrossRef]
- DeAngelo, D.J.; Erba, H.P.; Jonas, B.A.; O’Dwyer, M.; Marlton, P.; Hul, G.A.; Liesveld, J.; Cooper, B.W.; Bhatnagar, B.; Armstrong, M.; et al. A phase III trial to evaluate the efficacy of uproleselan (GMI-1271) with chemotherapy in patients with relapsed/refractory acute myeloid leukemia. J. Clin. Oncol. 2019, 37. [Google Scholar] [CrossRef]
- Telen, M.J.; Wun, T.; McCavit, T.L.; De Castro, L.M.; Krishnamurti, L.; Lanzkron, S.; Hsu, L.L.; Smith, W.R.; Rhee, S.; Magnani, J.L.; et al. Randomized phase 2 study of GMI-1070 in SCD: Reduction in time to resolution of vaso-occlusive events and decreased opioid use. Blood 2015, 125, 2656–2664. [Google Scholar] [CrossRef] [PubMed]
- Compain, P.; Martin, O.R. Design, synthesis and biological evaluation of iminosugar-based glycosyltransferase inhibitors. Curr. Top. Med. Chem. 2003, 3, 541–560. [Google Scholar] [CrossRef] [PubMed]
- Compain, P.; Martin, O.R. Carbohydrate mimetics-based glycosyltransferase inhibitors. Bioorg. Med. Chem. 2001, 9, 3077–3092. [Google Scholar] [CrossRef]
- Wang, S.; Vidal, S. Recent design of glycosyltransferase inhibitors. Carbohydr. Chem. 2013, 39, 78–101. [Google Scholar] [CrossRef]
- Szabo, R.; Skropeta, D. Advancement of Sialyltransferase Inhibitors: Therapeutic Challenges and Opportunities. Med. Res. Rev. 2017, 37, 219–270. [Google Scholar] [CrossRef]
- Tu, Z.; Lin, Y.N.; Lin, C.H. Development of fucosyltransferase and fucosidase inhibitors. Chem. Soc. Rev. 2013, 42, 4459–4475. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Y.; Wu, L.; Sun, X.L. Sialyltransferase inhibition and recent advances. Biochim. Biophys. Acta 2016, 1864, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Kajimoto, T.; Node, M. Synthesis of Glycosyltransferase Inhibitors. Synthesis 2009, 3179–3210. [Google Scholar] [CrossRef]
- Videira, P.A.; Marcelo, F.; Grewal, R.K. Glycosyltransferase inhibitors: A promising strategy to pave a path from laboratory to therapy. Carbohydr. Chem. 2018, 43, 135–158. [Google Scholar] [CrossRef]
- Tedaldi, L.; Wagner, G.K. Beyond substrate analogues: New inhibitor chemotypes for glycosyltransferases. MedChemComm 2014, 5, 1106–1125. [Google Scholar] [CrossRef]
- Butters, T.D.; Dwek, R.A.; Platt, F.M. Imino sugar inhibitors for treating the lysosomal glycosphingolipidoses. Glycobiology 2005, 15, 43R–52R. [Google Scholar] [CrossRef] [PubMed]
- Belanger, A.E.; Besra, G.S.; Ford, M.E.; Mikusova, K.; Belisle, J.T.; Brennan, P.J.; Inamine, J.M. The embAB genes of Mycobacterium avium encode an arabinosyl transferase involved in cell wall arabinan biosynthesis that is the target for the antimycobacterial drug ethambutol. Proc. Natl. Acad. Sci. USA 1996, 93, 11919–11924. [Google Scholar] [CrossRef] [PubMed]
- Tvaroska, I.; Andre, I.; Carver, J.P. Ab Initio Molecular Orbital Study of the Catalytic Mechanism of Glycosyltransferases: Description of Reaction Pthways and Determination of Transition-State Structures for Inverting N-Acetylglucosaminyltranferases. J. Am. Chem. Soc. 2000, 122, 8762–8776. [Google Scholar] [CrossRef]
- Merino, P.; Tejero, T.; Delso, I.; Hurtado-Guerrero, R.; Gomez-SanJuan, A.; Sadaba, D. Recent progress on fucosyltransferase inhibitors. Mini Rev. Med. Chem. 2012, 12, 1455–1464. [Google Scholar] [CrossRef]
- Lin, Y.-H.; Stein, D.; Liin, S.-M.; Chang, T.-C.; Lin, Y.-R.; Chuang, J.; Gervey-Hague, H.; Narimatsu, H.; Lin, C.-H. Chemoenzymatic Synthesis of GDP-l-Fucose Derivatives as Potent and Selective a-1,3-Fucosyltransferase Inhibitors. Adv. Synth. Catal. 2012, 354, 1750–1758. [Google Scholar] [CrossRef]
- Lee, L.V.; Mitchell, M.L.; Huang, S.J.; Fokin, V.V.; Sharpless, K.B.; Wong, C.H. A potent and highly selective inhibitor of human alpha-1,3-fucosyltransferase via click chemistry. J. Am. Chem. Soc. 2003, 125, 9588–9589. [Google Scholar] [CrossRef]
- Williams, S.J.; Withers, S.G. Glycosyl fluorides in enzymatic reactions. Carbohydr. Res. 2000, 327, 27–46. [Google Scholar] [CrossRef]
- Burkart, M.D.; Vincent, S.P.; Duffels, A.; Murray, B.W.; Ley, S.V.; Wong, C.H. Chemo-enzymatic synthesis of fluorinated sugar nucleotide: Useful mechanistic probes for glycosyltransferases. Bioorg. Med. Chem. 2000, 8, 1937–1946. [Google Scholar] [CrossRef]
- Rillahan, C.D.; Antonopoulos, A.; Lefort, C.T.; Sonon, R.; Azadi, P.; Ley, K.; Dell, A.; Haslam, S.M.; Paulson, J.C. Global metabolic inhibitors of sialyl- and fucosyltransferases remodel the glycome. Nat. Chem. Biol. 2012, 8, 661–668. [Google Scholar] [CrossRef]
- Macauley, M.S.; Arlian, B.M.; Rillahan, C.D.; Pang, P.C.; Bortell, N.; Marcondes, M.C.; Haslam, S.M.; Dell, A.; Paulson, J.C. Systemic blockade of sialylation in mice with a global inhibitor of sialyltransferases. J. Biol. Chem. 2014, 289, 35149–35158. [Google Scholar] [CrossRef] [PubMed]
- Heise, T.; Pijnenborg, J.F.A.; Bull, C.; van Hilten, N.; Kers-Rebel, E.D.; Balneger, N.; Elferink, H.; Adema, G.J.; Boltje, T.J. Potent Metabolic Sialylation Inhibitors Based on C-5-Modified Fluorinated Sialic Acids. J. Med. Chem. 2019, 62, 1014–1021. [Google Scholar] [CrossRef] [PubMed]
- Yuasa, H.; Palcic, M.M.; Hindsgaul, O. Synthesis of the carbocyclic analog of uridine 5′-(a-galactopyranosyl diphosphate) (UDP-Gal) as an inhibitor of b(1-4)-galactosyltransferase. Can. J. Chem. 1995, 73, 2190–2195. [Google Scholar] [CrossRef]
- Takaya, K.; Nagahori, N.; Kurogochi, M.; Furuike, T.; Miura, N.; Monde, K.; Lee, Y.C.; Nishimura, S. Rational design, synthesis, and characterization of novel inhibitors for human beta-1,4-galactosyltransferase. J. Med. Chem. 2005, 48, 6054–6065. [Google Scholar] [CrossRef]
- Schaub, C.; Muller, B.; Schmidt, R.R. New sialyltransferase inhibitors based on CMP-quinic acid: Development of a new sialyltransferase assay. Glycoconj. J. 1998, 15, 345–354. [Google Scholar] [CrossRef]
- Hosoguchi, K.; Maeda, T.; Furukawa, J.; Shinohara, Y.; Hinou, H.; Sekiguchi, M.; Togame, H.; Takemoto, H.; Kondo, H.; Nishimura, S. An efficient approach to the discovery of potent inhibitors against glycosyltransferases. J. Med. Chem. 2010, 53, 5607–5619. [Google Scholar] [CrossRef]
- Dimitroff, C.J.; Bernacki, R.J.; Sackstein, R. Glycosylation-dependent inhibition of cutaneous lymphocyte-associated antigen expression: Implications in modulating lymphocyte migration to skin. Blood 2003, 101, 602–610. [Google Scholar] [CrossRef]
- Dimitroff, C.J.; Kupper, T.S.; Sackstein, R. Prevention of leukocyte migration to inflamed skin with a novel fluorosugar modifier of cutaneous lymphocyte-associated antigen. J. Clin. Investig. 2003, 112, 1008–1018. [Google Scholar] [CrossRef]
- Zandberg, W.F.; Kumarasamy, J.; Pinto, B.M.; Vocadlo, D.J. Metabolic inhibition of sialyl-Lewis X biosynthesis by 5-thiofucose remodels the cell surface and impairs selectin-mediated cell adhesion. J. Biol. Chem. 2012, 287, 40021–40030. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, L.H.; Ye, X.S. Recent development in the design of sialyltransferase inhibitors. Med. Res. Rev. 2003, 23, 32–47. [Google Scholar] [CrossRef] [PubMed]
- Brockhausen, I.; Reck, F.; Kuhns, W.; Khan, S.; Matta, K.L.; Meinjohanns, E.; Paulsen, H.; Shah, R.N.; Baker, M.A.; Schachter, H. Substrate specificity and inhibition of UDP-GlcNAc:GlcNAc beta 1-2Man alpha 1-6R beta 1,6-N-acetylglucosaminyltransferase V using synthetic substrate analogues. Glycoconj. J. 1995, 12, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Pesnot, T.; Jorgensen, R.; Palcic, M.M.; Wagner, G.K. Structural and mechanistic basis for a new mode of glycosyltransferase inhibition. Nat. Chem. Biol. 2010, 6, 321–323. [Google Scholar] [CrossRef]
- Pesnot, T.; Wagner, G.K. Novel derivatives of UDP-glucose: Concise synthesis and fluorescent properties. Org. Biomol. Chem. 2008, 6, 2884–2891. [Google Scholar] [CrossRef] [PubMed]
- Descroix, K.; Pesnot, T.; Yoshimura, Y.; Gehrke, S.S.; Wakarchuk, W.; Palcic, M.M.; Wagner, G.K. Inhibition of galactosyltransferases by a novel class of donor analogues. J. Med. Chem. 2012, 55, 2015–2024. [Google Scholar] [CrossRef]
- Jorgensen, R.; Pesnot, T.; Lee, H.J.; Palcic, M.M.; Wagner, G.K. Base-modified donor analogues reveal novel dynamic features of a glycosyltransferase. J. Biol. Chem. 2013, 288, 26201–26208. [Google Scholar] [CrossRef]
- Qasba, P.K.; Ramakrishnan, B.; Boeggeman, E. Substrate-induced conformational changes in glycosyltransferases. Trends Biochem. Sci. 2005, 30, 53–62. [Google Scholar] [CrossRef]
- Jiang, J.; Kanabar, V.; Padilla, B.; Man, F.; Pitchford, S.C.; Page, C.P.; Wagner, G.K. Uncharged nucleoside inhibitors of beta-1,4-galactosyltransferase with activity in cells. Chem. Commun. (Camb.) 2016, 52, 3955–3958. [Google Scholar] [CrossRef]
- Hindsgaul, O.; Kaurz, K.J.; Srivastavaz, G.; Balaszczyk-ThurinlI, M.; CrawleyII, S.C.; Heerzell, L.D.; Palcic, M.M. Evaluation of Deoxygenated Oligosaccharide Specific Inhibitors of Glycosyltransferases. J. Biol. Chem. 1991, 266, 17858–17862. [Google Scholar]
- Brown, J.R.; Fuster, M.M.; Li, R.; Varki, N.; Glass, C.A.; Esko, J.D. A disaccharide-based inhibitor of glycosylation attenuates metastatic tumor cell dissemination. Clin. Cancer Res. 2006, 12, 2894–2901. [Google Scholar] [CrossRef] [PubMed]
- Fuster, M.M.; Brown, J.R.; Wang, L.; Esko, J.D. A disaccharide precursor of sialyl Lewis X inhibits metastatic potential of tumor cells. Cancer Res. 2003, 63, 2775–2781. [Google Scholar] [PubMed]
- Sarkar, A.K.; Fritz, T.A.; Taylor, W.H.; Esko, J.D. Disaccharide uptake and priming in animal cells: Inhibition of sialyl Lewis X by acetylated Gal beta 1-->4GlcNAc beta-O-naphthalenemethanol. Proc. Natl. Acad. Sci. USA 1995, 92, 3323–3327. [Google Scholar] [CrossRef]
- Sarkar, A.K.; Rostand, K.S.; Jain, R.K.; Matta, K.L.; Esko, J.D. Fucosylation of disaccharide precursors of sialyl LewisX inhibit selectin-mediated cell adhesion. J. Biol. Chem. 1997, 272, 25608–25616. [Google Scholar] [CrossRef]
- Brown, J.R.; Fuster, M.M.; Whisenant, T.; Esko, J.D. Expression patterns of alpha 2,3-sialyltransferases and alpha 1,3-fucosyltransferases determine the mode of sialyl Lewis X inhibition by disaccharide decoys. J. Biol. Chem. 2003, 278, 23352–23359. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R.; Yang, F.; Sinha, A.; Ramakrishnan, B.; Tor, Y.; Qasba, P.K.; Esko, J.D. Deoxygenated disaccharide analogs as specific inhibitors of beta-1-4-galactosyltransferase 1 and selectin-mediated tumor metastasis. J. Biol. Chem. 2009, 284, 4952–4959. [Google Scholar] [CrossRef]
- Mong, T.K.; Lee, L.V.; Brown, J.R.; Esko, J.D.; Wong, C.H. Synthesis of N-acetyllactosamine derivatives with variation in the aglycon moiety for the study of inhibition of sialyl Lewis x expression. ChemBioChem 2003, 4, 835–840. [Google Scholar] [CrossRef]
- Izumi, M.; Yuasa, H.; Hashimoto, H. Bisubstrate analogues as glycosyltransferase inhibitors. Curr. Top. Med. Chem. 2009, 9, 87–105. [Google Scholar] [CrossRef]
- Hashimoto, H.; Endo, T.; Kajihara, Y. Synthesis of the First Tricomponent Bisubstrate Analogue That Exhibits Potent Inhibition against GlcNAc:beta-1,4-Galactosyltransferase. J. Org. Chem. 1997, 62, 1914–1915. [Google Scholar] [CrossRef]
- Izumi, M.; Kaneko, S.; Yuasa, H.; Hashimoto, H. Synthesis of bisubstrate analogues targeting alpha-1,3-fucosyltransferase and their activities. Org. Biomol. Chem. 2006, 4, 681–690. [Google Scholar] [CrossRef]
- Hinou, H.; Sun, X.L.; Ito, Y. Systematic syntheses and inhibitory activities of bisubstrate-type inhibitors of sialyltransferases. J. Org. Chem. 2003, 68, 5602–5613. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.Y.; Hsu, C.C.; Chen, S.T.; Tsai, Y.C. Soyasaponin I, a potent and specific sialyltransferase inhibitor. Biochem. Biophys. Res. Commun. 2001, 284, 466–469. [Google Scholar] [CrossRef]
- Chang, W.W.; Yu, C.Y.; Lin, T.W.; Wang, P.H.; Tsai, Y.C. Soyasaponin I decreases the expression of alpha2,3-linked sialic acid on the cell surface and suppresses the metastatic potential of B16F10 melanoma cells. Biochem. Biophys. Res. Commun. 2006, 341, 614–619. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.H.; Wang, C.H.; Chang, H.C.; More, S.V.; Li, W.S.; Hung, W.C. A novel sialyltransferase inhibitor AL10 suppresses invasion and metastasis of lung cancer cells by inhibiting integrin-mediated signaling. J. Cell Physiol. 2010, 223, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.H.; Lee, L.; Chen, J.; Li, W.S. Lithocholic acid analogues, new and potent alpha-2,3-sialyltransferase inhibitors. Chem. Commun. 2006, 629–631. [Google Scholar] [CrossRef]
- Niu, X.; Fan, X.; Sun, J.; Ting, P.; Narula, S.; Lundell, D. Inhibition of fucosyltransferase VII by gallic acid and its derivatives. Arch. Biochem. Biophys. 2004, 425, 51–57. [Google Scholar] [CrossRef]
- Lee, K.Y.; Kim, H.G.; Hwang, M.R.; Chae, J.I.; Yang, J.M.; Lee, Y.C.; Choo, Y.K.; Lee, Y.I.; Lee, S.S.; Do, S.I. The Hexapeptide inhibitor of Galbeta 1,3GalNAc-specific alpha-2,3-sialyltransferase as a generic inhibitor of sialyltransferases. J. Biol. Chem. 2002, 277, 49341–49351. [Google Scholar] [CrossRef]
- Badhani, B.; Sharma, N.; Kakkar, R. Gallic acid: A versatile antioxidant with promising therapeutic and industrial applications. RSC Adv. 2015, 5, 27540–27557. [Google Scholar] [CrossRef]
- Gao, Y.; Vlahakis, J.Z.; Szarek, W.A.; Brockhausen, I. Selective inhibition of glycosyltransferases by bivalent imidazolium salts. Bioorg. Med. Chem. 2013, 21, 1305–1311. [Google Scholar] [CrossRef]
- Takayama, S.; Chung, S.J.; Igarashi, Y.; Ichikawa, Y.; Sepp, A.; Lechler, R.I.; Wu, J.; Hayashi, T.; Siuzdak, G.; Wong, C.H. Selective inhibition of beta-1,4- and alpha-1,3-galactosyltransferases: Donor sugar-nucleotide based approach. Bioorg. Med. Chem. 1999, 7, 401–409. [Google Scholar] [CrossRef]
- Pauling, L. Chemical achievement and hope for the future. Am. Sci. 1948, 36, 51–58. [Google Scholar] [PubMed]
- Wolfenden, R. Transition state analogues for enzyme catalysis. Nature 1969, 223, 704–705. [Google Scholar] [CrossRef] [PubMed]
- Schramm, V.L. Enzymatic transition states and transition state analog design. Annu. Rev. Biochem. 1998, 67, 693–720. [Google Scholar] [CrossRef] [PubMed]
- Schramm, V.L. Transition States and transition state analogue interactions with enzymes. Acc. Chem. Res. 2015, 48, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Gloster, T.M.; Vocadlo, D.J. Developing inhibitors of glycan processing enzymes as tools for enabling glycobiology. Nat. Chem. Biol. 2012, 8, 683–694. [Google Scholar] [CrossRef]
- Truhlar, D.G. Transition state theory for enzyme kinetics. Arch. Biochem. Biophys. 2015, 582, 10–17. [Google Scholar] [CrossRef]
- Schwartz, S.D.; Schramm, V.L. Enzymatic transition states and dynamic motion in barrier crossing. Nat. Chem. Biol. 2009, 5, 551–558. [Google Scholar] [CrossRef]
- Schmidt, R.R.; Frische, K. A new Galactosyl Transferase Inhibitor. Bioorg. Med. Chem. Lett. 1993, 3, 1747–1750. [Google Scholar] [CrossRef]
- Muller, B.; Schaub, C.; Schmidt, R.R. Efficient Sialyltransferase Inhibitors Based on Transition-State Analogues of the Sialyl Donor. Angew. Chem. Int. Ed. Engl. 1998, 37, 2893–2897. [Google Scholar] [CrossRef]
- Amann, F.; Schaub, C.; Muller, B.; Schmidt, R.R. New Potent Sialyltransferase Inhibitors—Synthesis of Donor and of Transition-State Analogues of Sialyl Donor CMP-Neu5Ac. Chem. Eur. J. 1998, 4, 1106–1115. [Google Scholar] [CrossRef]
- Guo, J.; Li, W.; Xue, W.; Ye, X.S. Transition State-Based Sialyltransferase Inhibitors: Mimicking Oxocarbenium Ion by Simple Amide. J. Med. Chem. 2017, 60, 2135–2141. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.T.; Yang, J.; Amaral, K.E.; Horenstein, N.A. Synthesis of a new transition-state analog of the sialyl donor. Inhibition of sialyltransferases. Tetrahedron Lett. 2001, 42, 2451–2453. [Google Scholar] [CrossRef]
- Mitchell, M.L.; Tian, F.; Lee, L.V.; Wong, C.H. Synthesis and evaluation of transition-state analogue inhibitors of alpha-1,3-fucosyltransferase. Angew. Chem. Int. Ed. Engl. 2002, 41, 3041–3044. [Google Scholar] [CrossRef]
- Raab, M.; Kozmon, S.; Tvaroska, I. Potential transition-state analogs for glycosyltransferases. Design and DFT calculations of conformational behavior. Carbohydr. Res. 2005, 340, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Sihelnikova, L.; Kozmon, S.; Tvaroska, I. DFT and Docking Study of Potential Transition State Analogue Inhibitors of Glycosyltransferases. Collect. Czech. Chem. Commun. 2008, 73, 591–607. [Google Scholar] [CrossRef]
- Barath, M.; Koos, M.; Tvaroska, I.; Hirsch, J.A. Synthesis of potential inhibitors of glycosyltransferases representing UDP-GlcNAc. Chem. Pap. 2015, 69, 339–347. [Google Scholar] [CrossRef]
- Barath, M.; Lin, C.-H.; Tvaroska, I.; Hirsch, J. Development of transition state analogue inhibitors for N-acetylglycosyltransferases bearing D-psico- or D-tagatofuranose scaffolds. Chem. Pap. 2015, 69, 348–357. [Google Scholar] [CrossRef]
- Hirsch, J.A.; Koos, M.; Tvaroska, I. Synthesis of saccharide precursors for preparation of potential inhibitors of glycosyltransferases. Chem. Pap. 2009, 63, 329–333. [Google Scholar] [CrossRef]























© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tvaroška, I.; Selvaraj, C.; Koča, J. Selectins—The Two Dr. Jekyll and Mr. Hyde Faces of Adhesion Molecules—A Review. Molecules 2020, 25, 2835. https://doi.org/10.3390/molecules25122835
Tvaroška I, Selvaraj C, Koča J. Selectins—The Two Dr. Jekyll and Mr. Hyde Faces of Adhesion Molecules—A Review. Molecules. 2020; 25(12):2835. https://doi.org/10.3390/molecules25122835
Chicago/Turabian StyleTvaroška, Igor, Chandrabose Selvaraj, and Jaroslav Koča. 2020. "Selectins—The Two Dr. Jekyll and Mr. Hyde Faces of Adhesion Molecules—A Review" Molecules 25, no. 12: 2835. https://doi.org/10.3390/molecules25122835
APA StyleTvaroška, I., Selvaraj, C., & Koča, J. (2020). Selectins—The Two Dr. Jekyll and Mr. Hyde Faces of Adhesion Molecules—A Review. Molecules, 25(12), 2835. https://doi.org/10.3390/molecules25122835