
Application of Post Solid-Phase Oxime Ligation to Fine-Tune Peptide–Protein Interactions




Abstract

1. Introduction
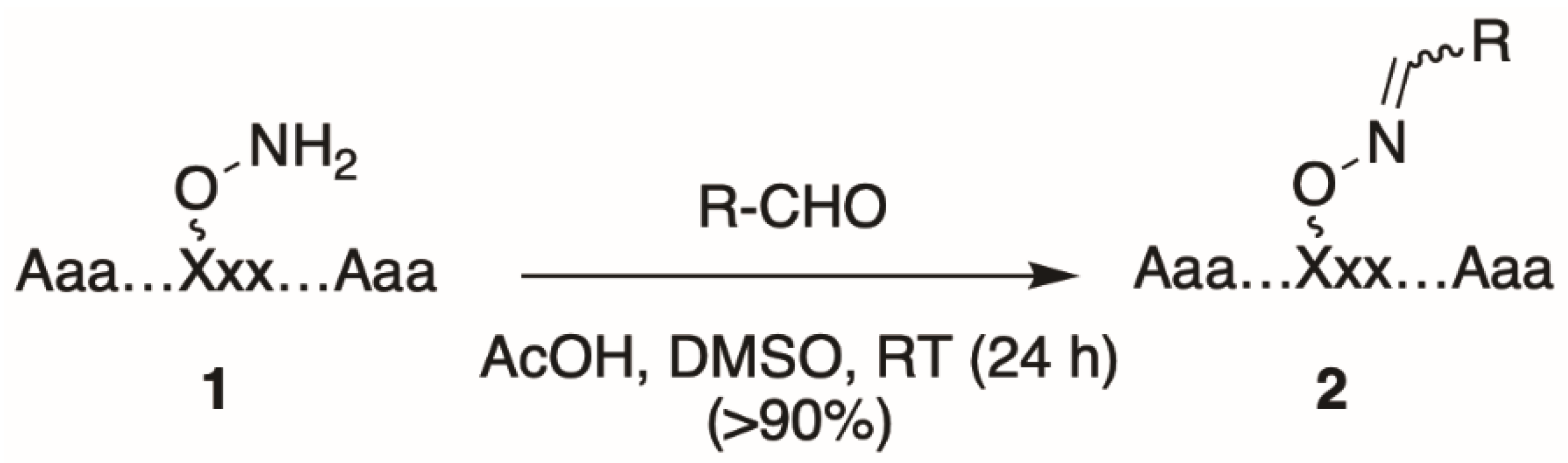
2. Oxime Ligation: General Protocol
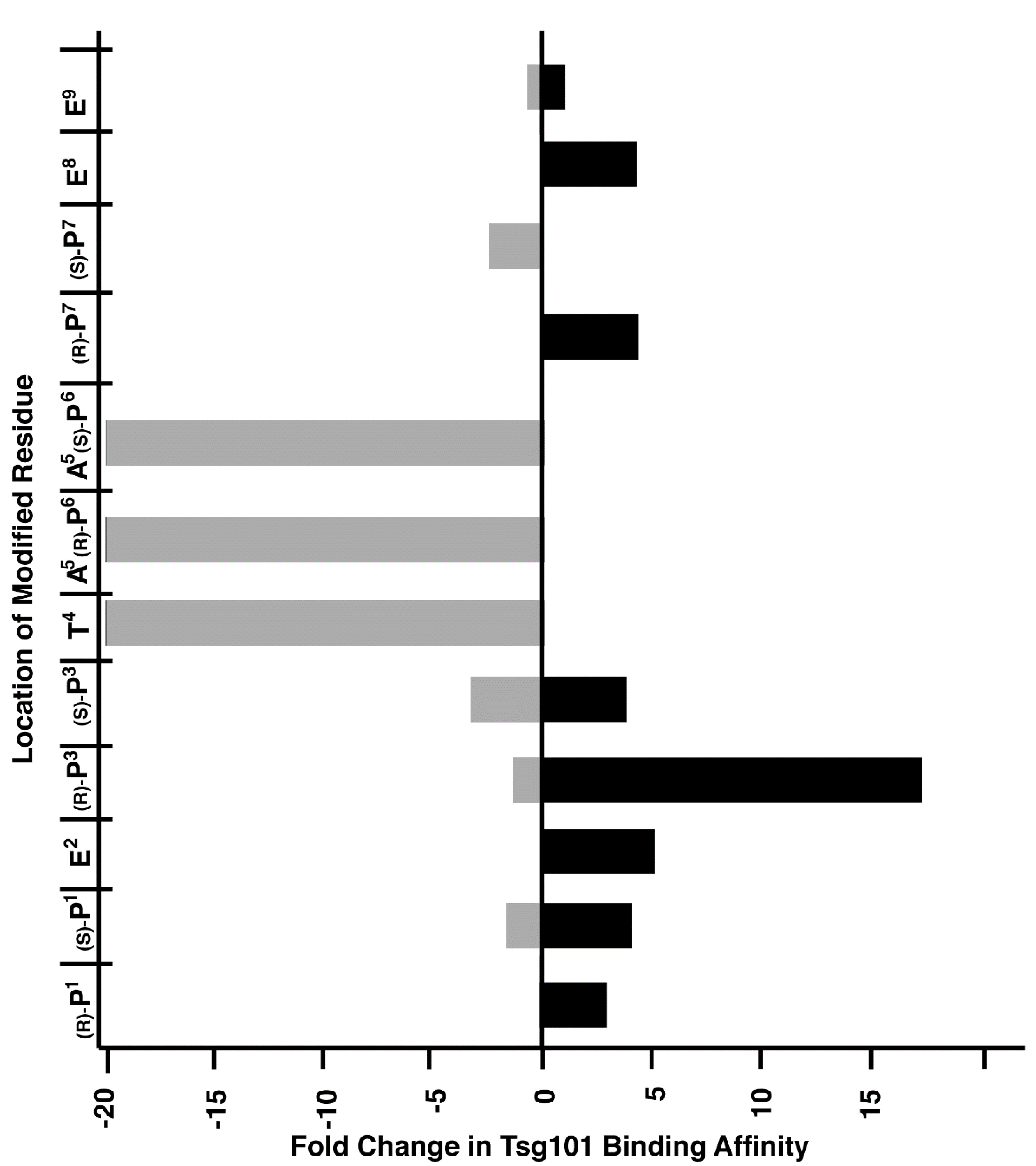
3. Application to Tsg101-Binding Peptides
4. Application to Peptide–Protein Interactions of Protein Tyrosine Phosphatases
5. Application to Plk1 PBD-Binding Peptides
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mosca, R.; Céol, A.; Aloy, P. Interactome3D: Adding structural details to protein networks. Nat. Methods 2013, 10, 47–53. [Google Scholar] [CrossRef]
- Stumpf, M.P.H.; Thorne, T.; de Silva, E.; Stewart, R.; An, H.J.; Lappe, M.; Wiuf, C. Estimating the size of the human interactome. Proc. Nat. Acad. Sci. USA 2008, 105, 6959. [Google Scholar] [CrossRef]
- Ryan, D.P.; Matthews, J.M. Protein–protein interactions in human disease. Curr. Opin. Struct. Biol. 2005, 15, 441–446. [Google Scholar] [CrossRef]
- Wells, J.A.; McClendon, C.L. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 2007, 450, 1001–1009. [Google Scholar] [CrossRef]
- Cukuroglu, E.; Engin, H.B.; Gursoy, A.; Keskin, O. Hot spots in protein–protein interfaces: Towards drug discovery. Prog. Biophys. Mol. Biol. 2014, 116, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Rosell, M.; Fernandez-Recio, J.; Fernandez-Recio, J. Hot-spot analysis for drug discovery targeting protein-protein interactions. Expert Opin. Drug Discov. 2018, 13, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Keskin, O.; Gursoy, A.; Ma, B.; Nussinov, R. Principles of protein-protein interactions: What are the preferred ways for proteins to interact? Chem. Rev. 2008, 108, 1225–1244. [Google Scholar] [CrossRef] [PubMed]
- Mabonga, L.; Kappo, A.P. Protein-protein interaction modulators: Advances, successes and remaining challenges. Biophys Rev. 2019, 11, 559–581. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, P.; Berlicki, L. Peptide-based inhibitors of protein-protein interactions. Bioorg. Med. Chem. Lett. 2016, 26, 707–713. [Google Scholar] [CrossRef]
- Cunningham, A.D.; Qvit, N.; Mochly-Rosen, D. Peptides and peptidomimetics as regulators of protein-protein interactions. Curr. Opin. Struct. Biol. 2017, 44, 59–66. [Google Scholar] [CrossRef]
- Tsomaia, N. Peptide therapeutics: Targeting the undruggable space. Eur. J. Med. Chem. 2015, 94, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Pelay-Gimeno, M.; Glas, A.; Koch, O.; Grossmann, T.N. Structure-based design of inhibitors of protein–protein interactions: Mimicking peptide binding epitopes. Angew. Chem. Int. Ed. 2015, 54, 8896–8927. [Google Scholar] [CrossRef] [PubMed]
- Robertson, N.S.; Spring, D.R. Using peptidomimetics and constrained peptides as valuable tools for inhibiting protein-protein interactions. Molecules 2018, 23, 959. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angewandte Chem. Int. Ed. Engl. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Kolmel, D.K.; Kool, E.T. Oximes and hydrazones in bioconjugation: Mechanism and catalysis. Chem. Rev. 2017, 117, 10358–10376. [Google Scholar] [CrossRef]
- Ulrich, S.; Boturyn, D.; Marra, A.; Renaudet, O.; Dumy, P. Oxime ligation: A chemoselective click-type reaction for ccessing multifunctional biomolecular constructs. Chem.-Eur. J. 2014, 20, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Bahta, M.; Liu, F.; Kim, S.-E.; Stephen, A.G.; Fisher, R.J.; Burke, T.R., Jr. Oxime-based linker libraries as a general approach for the rapid generation and screening of multidentate inhibitors. Nat. Protocols 2012, 7, 686–702. [Google Scholar] [CrossRef] [PubMed]
- Spetzler, J.C.; Hoeg-Jensen, T. Preparation and application of O-amino-serine, Ams, a new building block in chemoselective ligation chemistry. J. Pept. Sci. 1998, 5, 582–592. [Google Scholar] [CrossRef]
- Lang, I.; Donze, N.; Garrouste, P.; Dumy, P.; Mutter, M. Chemoselectively addressable HCan building blocks in peptide synthesis: L-homocanaline derivatives. J. Pept. Sci. 1998, 4, 72–80. [Google Scholar] [CrossRef]
- Adamczyk, M.; Reddy, R.E. Synthesis of (S)-(+)-2-amino-6-(aminooxy)-hexanoic acid. Synth. Commun. 2001, 31, 579–586. [Google Scholar] [CrossRef]
- Liu, F.; Thomas, J.D.; Burke, T.R., Jr. Synthesis of a homologous series of side chain extended orthogonally protected aminooxy-containing amino acids. Synthesis 2008, 15, 2432–2438. [Google Scholar]
- Liu, F.; Stephen, A.G.; Fisher, R.J.; Burke, T.R. Protected aminooxyprolines for expedited library synthesis: Application to Tsg101-directed proline-oxime containing peptides. Bioorg. Med. Chem. Lett. 2008, 18, 1096–1101. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Garrus, J.E.; von Schwedler, U.K.; Pornillos, O.W.; Morham Scott, G.; Zavitz, K.H.; Wang Hubert, E.; Wettstein, D.A.; Stray Kirsten, M.; Cote, M.; Rich Rebecca, L.; et al. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 2001, 107, 55–65. [Google Scholar] [CrossRef]
- Demirov, D.G.; Ono, A.; Orenstein, J.M.; Freed, E.O. Overexpression of the N-terminal domain of TSG101 inhibits HIV-1 budding by blocking late domain function. Proc. Nat. Acad. Sci. USA 2002, 99, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Stephen, A.G.; Waheed, A.A.; Aman, M.J.; Freed, E.O.; Fisher, R.J.; Burke, T.R., Jr. SAR by oxime-containing peptide libraries: Application to Tsg101 ligand optimization. ChemBioChem 2008, 9, 2000–2004. [Google Scholar] [CrossRef]
- Kim, S.-E.; Liu, F.; Im, Y.J.; Stephen, A.G.; Fivash, M.J.; Waheed, A.A.; Freed, E.O.; Fisher, R.J.; Hurley, J.H.; Burke, T.R. Elucidation of new binding interactions with the human Tsg101 protein using modified HIV-1 Gag-p6 derived peptide ligands. ACS Med. Chem. Lett. 2011, 2, 337–341. [Google Scholar] [CrossRef]
- Im, Y.J.; Kuo, L.; Ren, X.; Burgos, P.V.; Zhao, X.-Z.; Liu, F.; Burke, T.R., Jr.; Bonifacino, J.S.; Freed, E.O.; Hurley, J.H. Structure-based validation of the ESCRT-I /HIV-1 Gag PTAP interaction as an antiviral target. Structure 2010, 18, 1536–1547. [Google Scholar] [CrossRef]
- Hogan, M.; Bahta, M.; Tsuji, K.; Nguyen, T.X.; Cherry, S.; Lountos, G.T.; Tropea, J.E.; Zhao, B.M.; Zhao, X.Z.; Waugh, D.S.; et al. Targeting protein–protein interactions of tyrosine phosphatases with microarrayed fragment libraries displayed on phosphopeptide substrate acaffolds. ACS Comb. Sci. 2019, 21, 158–170. [Google Scholar] [CrossRef]
- Lee, K.S.; Burke, T.R.; Park, J.-E.; Bang, J.K.; Lee, E. Recent advances and new strategies in trgeting Plk1 for anticancer therapy. Trends Pharmacol. Sci. 2015, 36, 858–877. [Google Scholar] [CrossRef]
- Park, J.-E.; Hymel, D.; Burke, T.R., Jr.; Lee, K.S. Current progress and future perspectives in the development of anti-polo-like kinase 1 therapeutic agents [version 1; peer review: 4 approved]. F1000Research 2017, 6, 1024. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.-M.; Moulaei, T.; Lim, D.; Bang, J.K.; Park, J.-E.; Shenoy, S.R.; Liu, F.; Kang, Y.H.; Liao, C.; Soung, N.-K.; et al. Structural and functional analyses of minimal phosphopeptides targeting the polo-box domain of polo-like kinase 1. Nat. Struct. Mol. Biol. 2009, 16, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Park, J.-E.; Qian, W.-J.; Lim, D.; Scharow, A.; Berg, T.; Yaffe, M.B.; Lee, K.S.; Burke, T.R., Jr. Identification of high affinity polo-like kinase 1 (Plk1) polo-box domain binding peptides using oxime-based diversification. ACS Chem. Biol. 2012, 7, 805–810. [Google Scholar] [CrossRef]
- Liu, F.; Park, J.-E.; Qian, W.-J.; Lim, D.; Graber, M.; Berg, T.; Yaffe, M.B.; Lee, K.S.; Burke, T.R., Jr. Serendipitous alkylation of a Plk1 ligand uncovers a new binding channel. Nat. Chem. Biol. 2011, 7, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Liu, F.; Burke, T.R. Investigation of unanticipated alkylation at the N(π) position of a histidyl residue under Mitsunobu conditions and synthesis of orthogonally protected histidine analogues. J. Org. Chem. 2011, 76, 8885–8890. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Z.; Hymel, D.; Burke, T.R., Jr. Application of oxime-diversification to optimize ligand interactions within a cryptic pocket of the polo-like kinase 1 polo-box domain. Bioorg. Med. Chem. Lett. 2016, 26, 5009–5012. [Google Scholar] [CrossRef][Green Version]
- Zhao, X.Z.; Hymel, D.; Burke, T.R., Jr. Enhancing polo-like kinase 1 selectivity of polo-box domain-binding peptides. Bioorg. Med. Chem. 2017, 25, 5041–5049. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| No. | R | IC50(nM) a | ||
|---|---|---|---|---|
| Plk1 | Plk2 | Plk3 | ||
| 12 | n/a | 5 | 130 (26×) b | 280 (56×) |
| 17 |  | 7 | 290 (44×) | 1500 (220×) |
| 18 |  | 6 | 3400 (620×) | 1900 (345×) |
| 19 |  | 11 | 3500 (312×) | 29,500 (2600×) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, X.Z.; Liu, F.; Burke, T.R., Jr. Application of Post Solid-Phase Oxime Ligation to Fine-Tune Peptide–Protein Interactions. Molecules 2020, 25, 2807. https://doi.org/10.3390/molecules25122807
Zhao XZ, Liu F, Burke TR Jr. Application of Post Solid-Phase Oxime Ligation to Fine-Tune Peptide–Protein Interactions. Molecules. 2020; 25(12):2807. https://doi.org/10.3390/molecules25122807
Chicago/Turabian StyleZhao, Xue Zhi, Fa Liu, and Terrence R. Burke, Jr. 2020. "Application of Post Solid-Phase Oxime Ligation to Fine-Tune Peptide–Protein Interactions" Molecules 25, no. 12: 2807. https://doi.org/10.3390/molecules25122807
APA StyleZhao, X. Z., Liu, F., & Burke, T. R., Jr. (2020). Application of Post Solid-Phase Oxime Ligation to Fine-Tune Peptide–Protein Interactions. Molecules, 25(12), 2807. https://doi.org/10.3390/molecules25122807