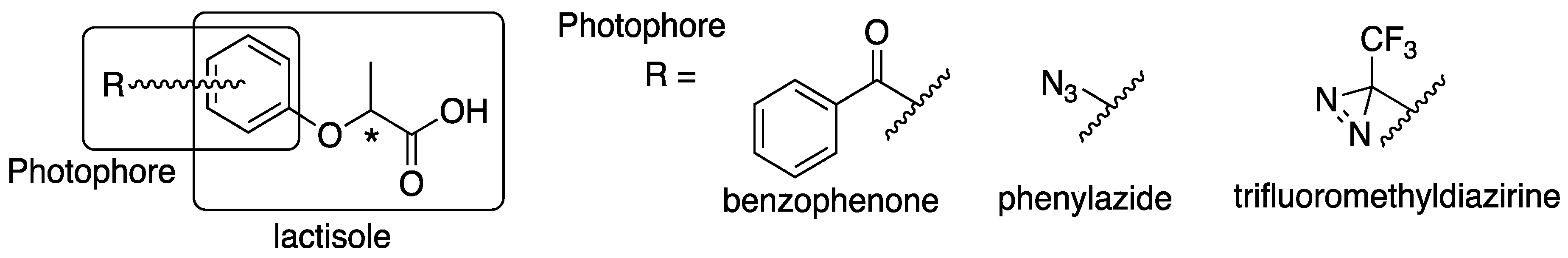
4.2. Synthesis of Benzophenone-Based Lactisole Derivatives
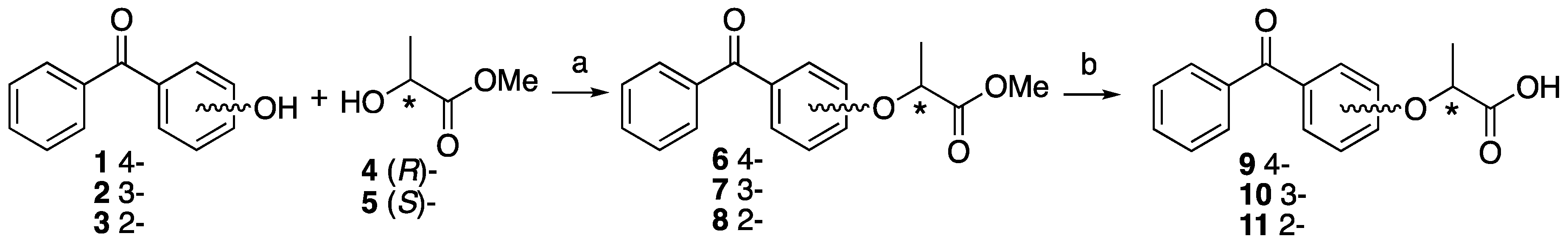
Methyl (S)-2-(4-benzoylphenoxy)propanoate ((S)-6). To 4-hydroxybenzophenone 1 (182 mg, 0.92 mmol) in dry CH2Cl2 (6 mL), methyl D-(+)-lactate 4 (144 mg, 1.38 mmol) and PPh3 (290 mg, 1.10 mmol) was added at 0 °C. After the reaction mixture was stirred for 10 min at 0 °C, DEAD (240 mg, 1.38 mmol) was slowly added at same temperature. The reaction mixture was stirred overnight at room temperature and partitioned between water and CH2Cl2. The organic layer was washed with brine, dried over MgSO4, filtered and concentrated. The residue was purified by column chromatography (ethyl acetate/n-hexane, 1:9) to give methyl (S)-2-(4-benzoylphenoxy)propanoate ((S)-6) (230 mg, 88%). [α]D −42 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.81 (d, J = 8.6 Hz, 2H), 7.75 (d, J = 7.4 Hz, 2H), 7.56 (t, J = 7.4 Hz, 1H), 7.46 (t, J = 7.4 Hz, 2H), 6.93 (d, J = 8.6 Hz, 2H), 4.87 (q, J = 6.8 Hz, 1H), 3.78 (s, 3H), 1.67 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 195.4, 171.9, 161.0, 138.0, 132.5, 131.9, 130.9, 129.7, 128.1, 114.4, 72.4, 52.4, 18.3. HRMS (ESI): m/z calculated for C17H16O4 + H+ [M + H+]: 285.1127. Found: 285.1120. Chiral HPLC (n-hexane/2-propanol 90:10): tR 25.8 min.
Methyl (R)-2-(4-benzoylphenoxy)propanoate ((R)-6). The similar treatment of 4-hydroxybenzophenone 1 (361 mg, 1.82 mmol) and methyl L-(−)-lactate 5 (284 mg, 2.72 mmol) as that just described gave (R)-6 (542 mg, quant). [α]D +42 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.80 (d, J = 8.9 Hz, 2H), 7.74 (d, J = 7.3 Hz, 2H), 7.55 (t, J = 7.3 Hz, 1H), 7.46 (t, J = 7.3 Hz, 2H), 7.46 (t, J = 7.3 Hz, 2H), 6.92 (d, J = 8.9 Hz, 2H), 4.87 (q, J = 6.8 Hz, 1H), 3.77 (s, 3H), 1.66 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 195.2, 171.9, 160.9, 138.0, 132.4, 131.9, 130.8, 129.6, 128.2, 114.3, 72.3, 52.3, 18.3. HRMS (ESI): m/z calculated for C17H16O4 + H+ [M + H+]: 285.1127. Found: 285.1132. Chiral HPLC (n-hexane/2-propanol 90:10): tR 28.0 min.
Methyl (S)-2-(3-benzoylphenoxy)propanoate ((S)-7). The similar treatment of 3-hydroxybenzophenone 2 (200 mg, 1.01 mmol) and methyl D-(+)-lactate 4 (157 mg, 1.51 mmol) as that just described gave (S)-7 (288 mg, quant). [α]D −28 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.79 (d, J = 7.4 Hz, 2H), 7.59 (t, J = 7.4 Hz, 1H), 7.47 (t, J = 7.4 Hz, 2H), 7.38 (m, 2H), 7.30 (m, 1H), 7.12 (m, 1H), 4.83 (q, J = 6.8 Hz, 1H), 3.76 (s, 3H), 1.64 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 196.1, 172.2, 157.4, 138.9, 137.4, 132.4, 130.0, 129.4, 128.2, 123.5, 119.6, 115.8, 72.5, 52.3, 18.4. HRMS (ESI): m/z calculated for C17H16O4 + H+ [M + H+]: 285.1127. Found: 285.1127. Chiral HPLC (n-hexane/2-propanol 90:10): tR 19.6 min.
Methyl (R)-2-(3-benzoylphenoxy)propanoate ((R)-7). The similar treatment of 3-hydroxybenzophenone 2 (360 mg, 1.82 mmol) and methyl L-(−)-lactate 5 (284 mg, 2.72 mmol) as that just described gave (R)-7 (517 mg, quant). [α]D +28 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.71 (d, J = 7.4 Hz, 2H), 7.51 (t, J = 7.4 Hz, 1H), 7.39 (t, J = 7.4 Hz, 2H), 7.31 (m, 2H), 7.22 (m, 1H), 7.04 (m, 1H), 4.75 (q, J = 6.8 Hz, 1H), 3.68 (s, 3H), 1.56 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 196.1, 172.2 157.4, 138.9, 137.4, 132.4, 129.7, 128.2, 123.5, 119.6, 115.9, 72.6, 52.3, 18.5. HRMS (ESI): m/z calculated for C17H16O4 + H+ [M + H+]: 285.1127. Found: 285.1122. Chiral HPLC (n-hexane/2-propanol 90:10): tR 18.9 min.
Methyl (S)-2-(2-benzoylphenoxy)propanoate ((S)-8). The similar treatment of 2-hydroxybenzophenone 3 (210 mg, 1.06 mmol) and methyl D-(+)-lactate 4 (165 mg, 1.59 mmol) as that just described gave (S)-8 (263 mg, 88%). [α]D +11 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.84 (2H, d, J = 8.2 Hz), 7.54 (1H, t, J = 7.4 Hz), 7.43 (1H, d, J = 7.6 Hz), 7.42 (3H, t, J = 7.4 Hz), 7.08 (1H, t, J = 7.4 Hz), 6.80 (1H, d, J = 8.2 Hz), 4.66 (1H, q, J = 6.8 Hz), 3.68 (3H, s), 1.24 (3H, d, J = 6.8 Hz). 13C-NMR (67.5 MHz, CDCl3): δ = 196.3, 172.0, 155.3, 138.0, 132.7, 131.9, 130.0, 129.7, 129.7, 128.0, 121.6, 113.0, 73.2, 52.2, 18.0. HRMS (ESI): m/z calculated for C17H16O4 + H+ [M + H+]: 285.1127. Found: 285.1119. Chiral HPLC (n-hexane:2-propanol = 90:10) tR 15.4 min.
Methyl (R)-2-(2-benzoylphenoxy)propanoate ((R)-8). The similar treatment of 2-hydroxybenzophenone 3 (372 mg, 1.87 mmol) and methyl L-(−)-lactate 5 (293 mg, 2.81 mmol) as that just described gave (R)-8 (435 mg, 82%). [α]D −11 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.83 (d, J = 8.2 Hz, 2H), 7.54 (t, J = 7.4 Hz, 1H), 7.43 (d, J = 7.6 Hz, 1H), 7.42 (t, J = 7.9 Hz, 3H), 7.08 (t, J = 7.4 Hz, 1H), 6.80 (d, J = 8.2 Hz, 1H), 4.66 (q, J = 6.8 Hz, 1H), 3.68 (s, 3H), 1.24 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 196.3, 172.0, 155.3, 138.0, 132.7, 131.8, 130.0, 129.7, 129.7, 128.0, 121.6, 113.0, 73.2, 52.1, 17.9. HRMS (ESI): m/z calculated for C17H16O4 + H+ [M + H+]: 285.1127. Found: 285.1132. Chiral HPLC (n-hexane:2-propanol = 90:10) tR 17.7 min.
(S)-2-(4-benzoylphenoxy)propanoic acid ((S)-9). Methyl (S)-2-(4-benzoylphenoxy)-propanoate (S)-6 (212 mg, 0.75 mmol) was dissolved in MeOH (14.4 mL) and H2O (1.6 mL), and then K2CO3 (309 mg, 2.24 mmol) was added. After the reaction mixture was stirred at reflux for 2 h, cooled to room temperature and then partitioned between ethyl acetate and water. The water layer was acidified by 1 M HCl and extracted by ethyl acetate. The organic layer was washed by H2O and brine, and dried over MgSO4, filtrated and concentrated to give (S)-2-(2-benzoylphenoxy)propanoic acid (S)-9 (206 mg, quant). [α]D −22 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.82 (d, J = 8.9 Hz, 2H), 7.75 (d, J = 7.4 Hz, 2H), 7.57 (t, J = 7.4 Hz, 1H), 7.47 (t, J = 7.4 Hz, 2H), 6.95 (d, J = 8.9 Hz, 2H), 4.91 (q, J = 6.8 Hz, 1H), 1.72 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 195.9, 176.4, 160.9, 137.8, 132.6, 132.1, 130.8, 129.8, 128.2, 114.4, 71.9, 18.3. HRMS (ESI): m/z calculated for C16H14O4 + H+ [M + H+]: 271.0970. Found: 271.0970.
(R)-2-(4-benzoylphenoxy)propanoic acid ((R)-9). The similar treatment of methyl (R)-2-(4-benzoylphenoxy)propanoate (R)-6 (517 mg, 1.82 mmol) as that just described gave (R)-9 (511 mg, quant). [α]D +22 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.82 (d, J = 8.9 Hz, 2H), 7.75 (d, J = 7.4 Hz, 2H), 7.57 (t, J = 7.4 Hz, 1H), 7.47 (t, J = 7.4 Hz, 2H), 6.96 (d, J = 8.9 Hz, 2H), 4.91 (q, J = 6.8 Hz, 1H), 1.72 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 196.0, 175.8, 161.0, 137.8, 132.6, 132.1, 130.8, 129.8, 128.2, 114.5, 72.0, 18.3. HRMS (ESI): m/z calculated for C16H14O4 + H+ [M + H+]: 271.0970. Found: 271.0950.
(S)-2-(3-benzoylphenoxy)propanoic acid ((S)-10). The similar treatment of methyl (S)-2-(3-benzoylphenoxy)propanoate (S)-7 (159 mg, 0.56 mmol) as that just described gave (S)-10 (144 mg, 95%). [α]D −9 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.78 (d, J = 7.3 Hz, 2H), 7.57 (t, J = 7.3 Hz, 1H), 7.46 (t, J = 7.3 Hz, 2H), 7.40 (m, 2H), 7.3 (m, 1H), 7.15 (m, 1H), 4.88 (q, J = 6.8 Hz, 1H), 1.68 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 196.3, 177.0, 157.2, 139.0, 137.3, 132.6, 130.0, 129.6, 128.3, 123.9, 119.7, 116.0, 72.1, 18.3. HRMS (ESI): m/z calculated for C16H14O4 + H+ [M + H+]: 271.0970. Found: 271.0952. Chiral HPLC (n-hexane:2-propanol = 90:10) tR 25.8 min.
(R)-2-(3-benzoylphenoxy)propanoic acid ((R)-10). The similar treatment of methyl (R)-2-(3-benzoylphenoxy)propanoate (R)-7 (413 mg, 1.45 mmol) as that just described gave (R)-10 (465 mg, quant). [α]D +9 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.78 (d, J = 7.4 Hz, 2H), 7.57 (t, J = 7.4 Hz, 1H), 7.45 (t, J = 7.4 Hz, 2H), 7.39 (m, 2H), 7.33 (m, 1H), 7.14 (m, 1H), 4.88 (q, J = 6.8 Hz, 1H), 1.68 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 196.5, 176.5, 157.2, 138.8, 137.2, 132.6, 130.0, 129.5, 128.2, 123.8, 119.7, 115.9, 72.1, 18.3. HRMS (ESI): m/z calculated for C16H14O4 + H+ [M + H+]: 271.0970. Found: 271.0960. Chiral HPLC (n-hexane:2-propanol = 90:10) tR 28.0 min.
(S)-2-(2-benzoylphenoxy)propanoic acid ((S)-11). The similar treatment of methyl (S)-2-(3-benzoylphenoxy)propanoate (S)-8 (229 mg, 0.80 mmol) as that just described gave (S)-11 (187 mg, 87%). [α]D −40 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.87 (d, J = 7.3 Hz, 2H), 7.64 (t, J = 7.3 Hz, 1H), 7.50 (m, 4H), 7.12 (m, 2H), 5.00 (q, J = 6.8 Hz, 1H), 1.65 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 197.1, 173.1, 156.6, 137.0, 133.8, 132.1, 130.7, 128.5, 127.3, 121.9, 115.1, 75.2, 18.6. HRMS (ESI): m/z calculated for C16H14O4 + H+ [M + H+]: 271.0970. Found: 271.0950.
(R)-2-(2-benzoylphenoxy)propanoic acid ((R)-11). The similar treatment of methyl (S)-2-(2-benzoylphenoxy)propanoate (R)-8 (392 mg, 1.38 mmol) as that just described gave (R)-11 (326 mg, 88%). [α]D +40 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.87 (d, J = 6.9 Hz, 2H), 7.64 (t, J = 7.4 Hz, 1H), 7.50 (m, 4H), 7.12(m, 2H), 5.01 (q, J = 6.9 Hz, 1H), 1.66 (d, J = 6.9 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 197.1, 173.0, 156.6, 137.0, 133.8, 132.1, 130.7, 128.5, 127.3, 121.9, 115.0, 75.2, 18.6. HRMS (ESI): m/z calculated for C16H14O4 + H+ [M + H+]: 271.0970. Found: 271.0950.
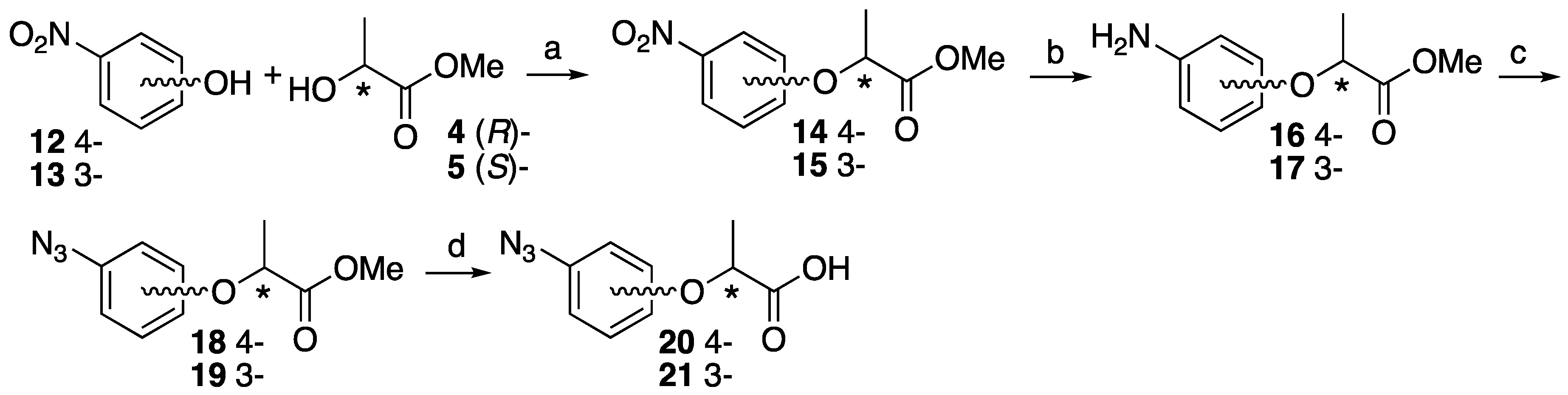
4.3. Synthesis of 3- or 4-Azidephenoxy-Based Lactisole Derivatives
Methyl (S)-2-(4-nitrophenoxy)propanoate ((S)-14). To 4-nitrophenol 12 (437 mg, 3.14 mmol) in dry CH2Cl2 (6 mL), D-(+)-lactate 4 (491 mg, 4.71 mmol) and PPh3 (989 mg, 3.77 mmol) was added at 0 °C After the reaction mixture was stirred for 10 min at 0 °C, DEAD (820 mg, 4.71 mmol) was slowly added at same temperature. The reaction mixture was stirred overnight at room temperature and partitioned between water and CH2Cl2. The organic layer was washed with brine, dried over MgSO4, filtered and concentrated. The residue was purified by column chromatography (ethyl acetate/n-hexane, 1:9) to give (S)-14 (700 mg, 99%). [α]D −64 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 8.20 (d, J = 8.9 Hz, 2H), 6.93 (d, J = 8.9 Hz, 2H), 4.88 (q, J = 6.8 Hz, 1H), 3.78 (s, 3H), 1.68 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 171.4, 162.4, 142.0, 125.9, 114.9, 72.9, 52.6, 18.4. HRMS (ESI): m/z calculated for C10H11NO5 + H+ [M + H+]: 226.0715. Found: 226.0720.
Methyl (R)-2-(4-nitrophenoxy)propanoate ((R)-14). The similar treatment of 4-nitrophenol
12 (128 mg, 0.92 mmol) and methyl L-(−)-lactate
5 (144 mg, 1.38 mmol) as that just described gave (
R)-
14 (213 mg, quant). [α]
D +64 (
c 1, CHCl
3), (ref. [α]
D +64.4 (
c 0.10, CHCl
3) [
29]).
1H-NMR (270 MHz, CDCl
3):
δ = 8.18 (d,
J = 8.6 Hz, 2H), 6.93 (d,
J = 9.2 Hz, 2H), 4.90 (q,
J = 6.8 Hz, 1H), 3.78 (s, 3H), 1.68 d,
J = 6.8 Hz, 3H).
13C-NMR (67.5 MHz, CDCl
3):
δ = 171.2, 162.3, 141.9, 125.8, 114.8, 72.7, 52.5, 18.2. HRMS (ESI):
m/
z calculated for C
10H
11NO
5 + H
+ [M + H
+]: 226.0715. Found: 226.0720.
Methyl (S)-2-(3-nitrophenoxy)propanoate ((S)-15). The similar treatment of 3-nitrophenol 13 (301 mg, 2.16 mmol) and methyl D-(+)-lactate 4 (338 mg, 3.24 mmol) as that just described gave (S)-15 (490 mg, quant). [α]D −59 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.85 (dd, J = 8.1, 2.3 Hz, 1H), 7.69 (t, J = 2.3 Hz, 1H), 7.44 (t, J = 8.1 Hz, 1H), 7.22 (dd, J = 8.1, 2.3 Hz, 1H), 4.87 (q, J = 6.8 Hz, 1H), 3.79 (s, 3H), 1.67 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 171.5, 158.0, 149.1, 130.1, 121.8, 116.5, 109.7, 72.9, 52.5, 18.3. HRMS (ESI): m/z calculated for C10H11NO5 + H+ [M + H+]: 226.0715. Found: 226.0730.
Methyl (R)-2-(3-nitrophenoxy)propanoate ((R)-15). The similar treatment of 3-nitrophenol 13 (422 mg, 3.04 mmol) and methyl L-(−)-lactate 5 (474 mg, 4.55 mmol) as that just described gave (R)-15 (711 mg, quant). [α]D +59 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.85 (dd, J = 8.2, 2.3 Hz, 1H), 7.69 (t, J = 2.3 Hz, 1H), 7.44 (t, J = 8.2 Hz, 1H), 7.22 (dd, J = 8.2, 2.3 Hz, 1H), 4.87 (q, J = 6.8 Hz, 1H), 3.79 (s, 3H), 1.67 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 171.5, 158.0, 149.1, 130.1, 121.8, 116.5, 109.7, 72.9, 52.5, 18.3. HRMS (ESI): m/z calculated for C10H11NO5 + H+ [M + H+]: 226.0715. Found: 226.0710.
Methyl (S)-2-(4-aminophenoxy)propanoate ((S)-16). Methyl (S)-2-(4-nitrophenoxy)-propanoate (S)-15 (700 mg, 3.11 mmol) and 5% Pd/C (35.0 mg) were suspended in MeOH (10 mL). The reaction mixture was stirred vigorously at room temperature for 2 h under H2 atmosphere. The residue was filtrated using Celite and concentrated to give (S)-16 (607 mg, quant). [α]D −50 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 6.73 (d, J = 8.9 Hz, 2H), 6.59 (d, J = 8.9 Hz, 2H), 4.63 (q, J = 6.8 Hz, 1H), 3.73 (s, 3H), 3.46 (s, 2H), 1.57 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 173.0, 150.4, 141.0, 116.7, 116.1, 73.7, 52.1, 18.5. HRMS (ESI): m/z calculated for C10H13NO3 + H+ [M + H+]: 196.0974. Found: 196.0960.
Methyl (R)-2-(4-aminophenoxy)propanoate ((R)-16). The similar treatment of methyl (R)-2-(4-nitrophenoxy)propanoate (R)-15 (575 mg, 2.56 mmol) as that just described gave (R)-16 (500 mg, quant). [α]D +50 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 6.73 (d, J = 8.9 Hz, 2H), 6.66 (d, J = 8.9 Hz, 2H), 4.64 (q, J = 6.8 Hz, 1H), 4.24 (s, 2H), 3.73 (s, 3H), 1.57 (3H, d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 172.9, 151.0, 139.5, 116.9, 116.7, 73.6, 52.1, 18.5. HRMS (ESI): m/z calculated for C10H13NO3 + H+ [M + H+]: 196.0974. Found: 196.0950.
Methyl (S)-2-(3-aminophenoxy)propanoate ((S)-17). The similar treatment of methyl (S)-2-(3-nitrophenoxy)propanoate (S)-15 (490 mg, 2.17 mmol) as that just described gave (S)-17 (367 mg, 87%). [α]D −23 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.01 (t, J = 7.9 Hz, 1H), 6.28 (dd, J = 7.9, 1.0 Hz, 1H), 6.23 (m, 2H), 4.72 (q, J = 6.8 Hz, 1H), 3.73 (s, 3H), 3.68 (s, 3H), 1.58 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 172.7, 158.5, 147.9, 130.0, 108.7, 104.4, 102.1, 72.2, 52.1, 18.5. HRMS (ESI): m/z calculated for C10H13NO3 + H+ [M + H+]: 196.0974. Found: 196.0980.
Methyl (R)-2-(3-aminophenoxy)propanoate ((R)-17). The similar treatment of methyl (R)-2-(3-nitrophenoxy)propanoate (R)-15 (628 mg, 2.78 mmol) as that just described gave (R)-17 (558 mg, quant). [α]D +23 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.05 (t, J = 7.9 Hz, 1H), 6.37 (d, J = 7.9 Hz, 1H), 6.25 (m, 2H), 4.73 (q, J = 6.8 Hz, 1H), 4.19 (s, 2H), 3.75 (s, 3H), 1.59 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 172.8, 158.6, 146.7, 130.2, 109.3, 105.3, 102.9, 72.4, 52.3, 18.5. HRMS (ESI): m/z calculated for C10H13NO3 + H+ [M + H+]: 196.0974. Found: 196.0960.
Methyl (S)-2-(4-azidephenoxy)propanoate ((S)-18). Methyl (S)-2-(4-aminophenoxy)-propanoate (S)-14 (607 mg, 3.11 mmol) was dissolved in water (6.78 mL) and 37% HCl (0.76 mL) at 0 °C. NaNO2 (237 mg, 3.44 mmol) in water (2.26 mL) was added slowly at 0 °C, followed by NaN3 (381 mg, 5.87 mmol) in water (2.26 mL). After 30 min at 0 °C and 30 min at room temperature, the reaction mixture was extracted with ethyl acetate. The organic layer was washed by brine, dried over MgSO4, filtrated and concentrated to give (S)-18 (702 mg, quant). [α]D −45 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 6.94 (d, J = 9.2 Hz, 2H), 6.86 (d, J = 9.2 Hz, 2H), 4.72 (q, J = 6.8 Hz, 1H), 3.76 (s, 3H), 1.61 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 172.4, 154.9, 133.4, 120.1, 116.5, 73.0, 52.3, 18.5. HRMS (ESI): m/z calculated for C10H11N3O3 + H+ [M + H+]: 222.0879. Found: 222.0860. Chiral HPLC (n-hexane/2-propanol 99:1): tR 20.5 min.
Methyl (R)-2-(4-azidephenoxy)propanoate ((R)-18). The similar treatment of methyl (R)-2-(4-aminophenoxy)propanoate (R)-14 (500 mg, 2.56 mmol) as that just described gave (R)-18 (529 mg, 93%). [α]D +45 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 6.94 (d, J = 9.2 Hz, 2H), 6.86 (d, J = 9.2 Hz, 2H), 4.73 (q, J = 6.8 Hz, 1H), 3.76 (s, 3H), 1.61 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 172.4, 154.9, 133.3, 120.1, 116.5, 73.0, 52.3, 18.5. HRMS (ESI): m/z calculated for C10H11N3O3 + H+ [M + H+]: 222.0879. Found: 222.0870. Chiral HPLC (n-hexane/2-propanol 99:1): tR 19.4 min.
Methyl (S)-2-(3-azidophenoxy)propanoate ((S)-19). The similar treatment of methyl (S)-2-(3-aminophenoxy)propanoate (S)-17 (323 mg, 1.65 mmol) as that just described gave (S)-19 (336 mg, 92%). [α]D −28 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.24 (t, J = 8.2 Hz, 1H), 6.65 (t, J = 8.2 Hz, 2H), 6.55 (s, 1H), 4.76 (q, J = 6.8 Hz, 1H), 3.76 (s, 3H), 1.62 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 172.1, 158.6, 141.3, 130.5, 112.2, 111.1, 106.3, 72.5, 52.3, 18.4. HRMS (ESI): m/z calculated for C10H11N3O3 + H+ [M + H+]: 222.0879. Found: 222.0880. Chiral HPLC (n-hexane/2-propanol 99:1): tR 18.0 min.
Methyl (R)-2-(3-azidophenoxy)propanoate ((R)-19). The similar treatment of methyl (R)-2-(3-aminophenoxy)propanoate (R)-17 (518 mg, 2.65 mmol) as that just described gave (R)-19 (497 mg, 85%). [α]D +28 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.24 (t, J = 8.1 Hz, 1H), 6.65 (t, J = 8.1 Hz, 1H), 6.55 (s, 1H), 4.76 (q, J = 6.8 Hz, 1H), 3.76 (s, 3H), 1.62 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 172.0, 158.5, 141.2, 130.4, 112.0, 111.0, 106.2, 72.3, 52.1, 18.2. HRMS (ESI): m/z calculated for C10H11N3O3 + H+ [M + H+]: 222.0879. Found: 222.0860. Chiral HPLC (n-hexane/2-propanol 99:1): tR 16.7 min.
(S)-2-(4-Azidophenoxy)propanoic acid ((S)-20). Methyl (S)-2-(4-azidophenoxy)propanoate (S)-18 (702 mg, 3.17 mmol) was dissolved in MeOH (15 mL) and 2M NaOH (3.2 mL). After the reaction mixture was stirred at reflux for 2 h, cooled to room temperature and then partitioned between ethyl acetate and water. The water layer was acidified by 1 M HCl aq and extracted by ethyl acetate. The organic layer was washed by H2O and brine, and dried over MgSO4, filtrated and concentrated to give (S)-20 (601 mg, 91%). [α]D +27 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 9.68 (s, 1H), 6.96 (d, J = 8.9 Hz, 4H), 6.89 (d, J = 8.9 Hz, 4H), 4.75 (d, J = 6.9 Hz, 1H), 1.66 (d, J = 6.9 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 177.6, 154.5, 133.7, 120.2, 116.6, 72.5, 18.4. HRMS (ESI): m/z calculated for C9H9N3O3 + H+ [M + H+]: 208.0722. Found: 208.0720.
(R)-2-(4-Azidophenoxy)propanoic acid ((R)-20). The similar treatment of methyl (R)-2-(4-azidophenoxy)propanoate (R)-18 (529 mg, 2.39 mmol) as that just described gave (R)-20 (475 mg, 96%). [α]D −27 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 11.71 (s, 1H), 6.93 (d, J = 8.9 Hz, 2H), 6.87 (d, J = 8.9 Hz, 2H), 4.74 (q, J = 6.8 Hz, 1H), 1.65 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 178.1, 154.5, 133.7, 120.1, 116.6, 72.5, 18.4. HRMS (ESI): m/z calculated for C9H9N3O3 + H+ [M + H+]: 208.0722. Found: 208.0710.
(S)-2-(3-Azidophenoxy)propanoic acid ((S)-21). The similar treatment of methyl (S)-2-(3-azidephenoxy)propanoate (S)-19 (212 mg, 0.96 mmol) as that just described gave (S)-21 (211 mg, quant). [α]D −5 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.26 (t, J = 8.1 Hz, 1H), 6.70 (dd, J = 8.1, 2.1 Hz, 1H), 6.66 (dd, J = 8.1, 2.1 Hz, 1H), 6.57 (t, J = 2.1 Hz, 1H), 4.80 (q, J = 6.8 Hz, 1H), 1.67 (d, J = 6.6 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 177.5, 158.4, 141.6, 130.7, 112.5, 111.2, 106.5, 72.0, 18.3. HRMS (ESI): m/z calculated for C9H9N3O3 + H+ [M + H+]: 208.0722. Found: 208.0750.
(R)-2-(3-Azidophenoxy)propanoic acid ((R)-21). The similar treatment of methyl (R)-2-(3-azidophenoxy)propanoate (R)-19 (386 mg, 1.74 mmol) as that just described gave (R)-21 (294 mg, 81%). [α]D +5 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.25 (t, J = 8.1 Hz, 1H), 6.69 (dd, J = 8.1, 2.1 Hz, 2H), 6.65 (dd, J = 8.1, 2.1 Hz, 2H), 6.57 (t, J = 2.1 Hz, 1H), 4.79 (q, J = 6.8 Hz, 1H), 1.66 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 177.6, 158.4, 141.6, 130.7, 112.5, 111.2, 106.6, 72.0, 18.3. HRMS (ESI): m/z calculated for C9H9N3O3 + H+ [M + H+]: 208.0722. Found: 208.0710.
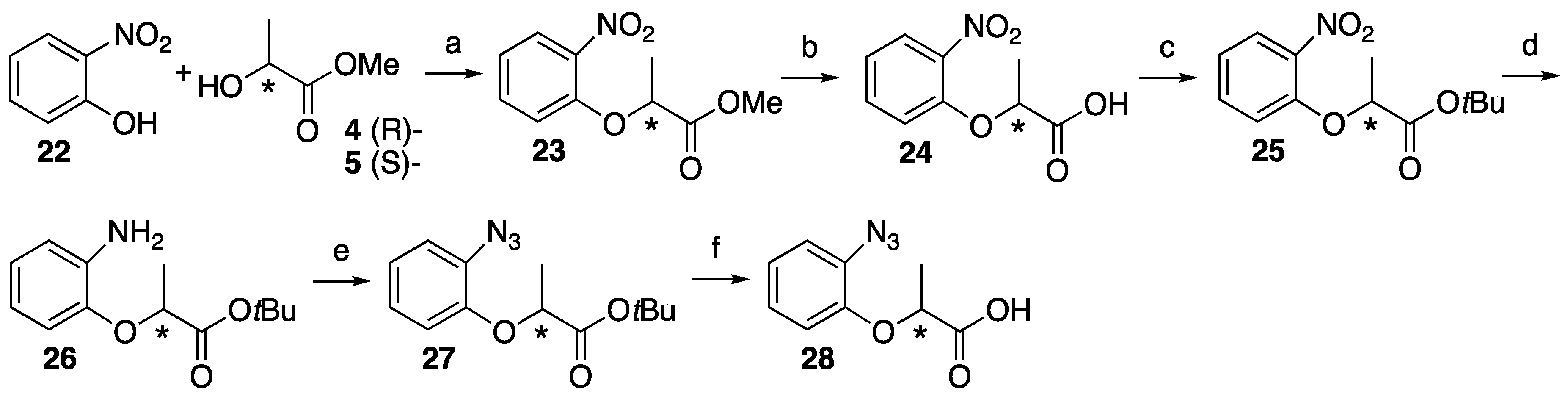
4.5. Synthesis of Trifluoromethyldiazirine-Based Lactisole Derivatives
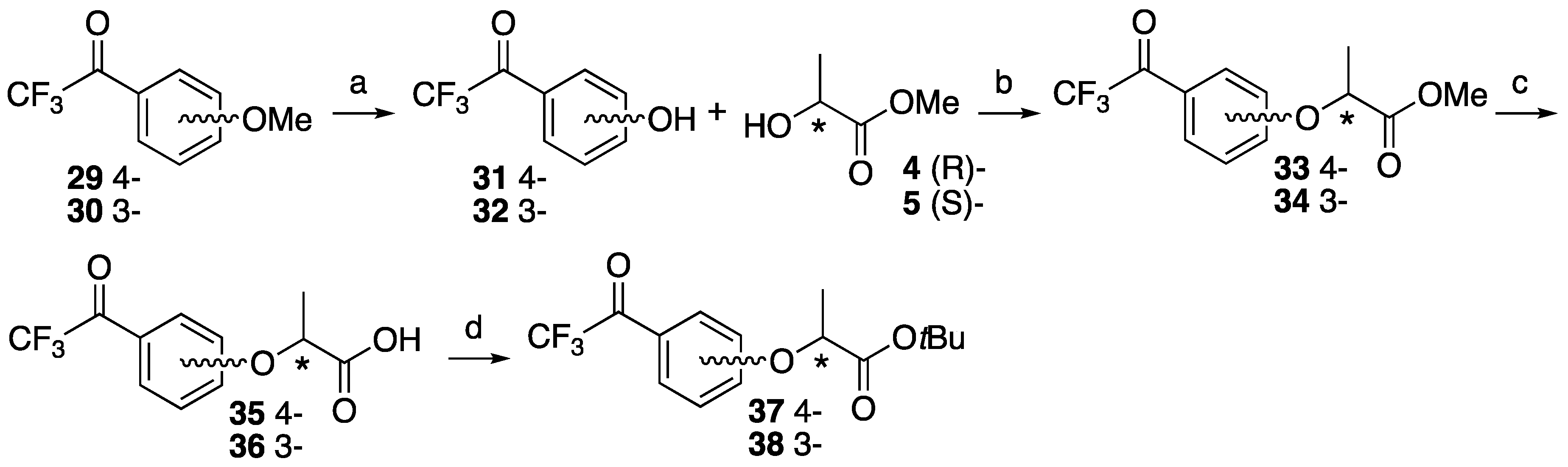
2,2,2-Trifluoro-1-(4-hydroxyphenyl)ethan-1-one 31. 2,2,2-Trifluoro-1-(4-methoxyphenyl)-ethan-1-one 29 (493 mg, 2.41 mmol) and lithium chloride (950 mg, 22.4 mmol) was dissolved in dry DMF (7.5 mL). After the reaction mixture was stirred at reflux for 6 h, the mixture was cooled to room temperature and 1 M HCl was added, and then extracted with ethyl acetate. The organic layer was washed with brine and dried over MgSO4, filtrated and concentrated. The residue was purified by column chromatography (ethyl acetate/n-hexane, 1:8) to give 31 (376 mg, 82%). 1H-NMR (270 MHz, CDCl3): δ = 8.04 (d, J = 8.9 Hz, 2H), 6.96 (d, J = 8.9 Hz, 2H), 5.71 (s, 1H). 13C-NMR (67.5 MHz, CDCl3): δ = 179.0 (q, 2JCF = 34.6 Hz), 133.1, 123.1, 116.9 (q, 1JCF = 291.6 Hz), 116.0. HRMS (ESI): m/z calculated for C8H5F3O2 + H+ [M + H+]: 191.0320. Found: 191.0317.
2,2,2-Trifluoro-1-(3-hydroxyphenyl)ethan-1-one 32. 2,2,2-Trifluoro-1-(4-methoxyphenyl)-ethan-1-one 30 (1.052 g, 5.15 mmol) was dissolved in CH2Cl2 (30 mL) and cooled to −78 °C. BBr3 was added dropwise at −78 °C and the reaction mixture stirred at room temperature for 5 h. The mixture was cooled to 0 °C and 10% NaOH (30 mL) was added slowly, and then HCl was added to make pH 1 at same temperature. NH4OH was also added to make pH 7 at room temperature, then extracted by ethyl acetate. The organic layer was dried over MgSO4, filtrated and concentrated to give 31 (1.018 g, quant). 1H-NMR (270 MHz, CDCl3): δ = 7.63 (d, J = 7.8 Hz, 1H), 7.50 (s, 1H), 7.41 (t, J = 7.8 Hz, 1H), 7.18 (dd, J = 7.8, 2.6 Hz, 1H). 13C-NMR (67.5 MHz, CDCl3): δ = 180.3 (q, 2JCF = 35.2 Hz), 156.1, 131.2, 130.5, 123.0, 122.8, 116.6 (q, 1JCF = 291.6 Hz), 116.2. HRMS (ESI): m/z calculated for C8H5F3O2 + H+ [M + H+]: 191.0320. Found: 191.0316.
Methyl (S)-2-(4-(2,2,2-trifluoroacetyl)phenoxy)propanoate (S)-33. To 2,2,2-trifluoro-1-(4-hydroxyphenyl)ethanone 31 (1.175 g, 7.25 mmol) in dry CH2Cl2 (30 mL), D-(+)-lactate 4 (1.132 g, 10.9 mmol) and PPh3 (2.282 g, 8.70 mmol) was added at 0 °C. After the reaction mixture was stirred for 10 min at 0 °C, DEAD (1.891 g, 10.9 mmol) was slowly added at same temperature. The reaction mixture was stirred overnight at room temperature and partitioned between water and CH2Cl2. The organic layer was washed with brine, dried over MgSO4, filtered and concentrated. The residue was purified by column chromatography (ethyl acetate/n-hexane, 1:9) to give (S)-33 (1.560 g, 78%). [α]D −48 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.96 (d, J = 8.2 Hz, 2H), 6.88 (d, J = 8.2 Hz, 2H), 4.82 (q, J = 6.8 Hz, 1H), 3.70 (s, 3H), 1.59 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 178.8 (q, 2JCF = 34.6 Hz), 171.4, 163.2, 132.7, 123.4, 116.8 (q, 1JCF = 291.4 Hz), 115.2, 72.5, 52.5, 18.3. HRMS (ESI): m/z calculated for C12H11F3O4 + H+ [M + H+]: 277.0688. Found: 277.0674.
Methyl (R)-2-(4-(2,2,2-trifluoroacetyl)phenoxy)propanoate (R)-33. The similar treatment of 2,2,2-trifluoro-1-(4-hydroxyphenyl)ethenone 31 (484 mg, 2.99 mmol) and methyl L-(−)-lactate 5 (466 mg, 4.48 mmol) as that just described gave (R)-33 (614 mg, 74%). [α]D +48 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 8.04 (d, J = 8.2 Hz, 2H), 6.97 (d, J = 8.2 Hz, 2H), 4.91 (q, J = 6.8 Hz, 1H), 3.78 (s, 3H), 1.68 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 178.8 (q, 2JCF = 34.6 Hz), 171.4, 163.2, 132.7, 123.4, 116.8 (q, 1JCF = 291.4 Hz), 115.2, 72.5, 52.5, 18.3. HRMS (ESI): m/z calculated for C12H11F3O4 + H+ [M + H+]: 277.0688. Found: 277.0660.
Methyl (S)-2-(3-(2,2,2-trifluoroacetyl)phenoxy)propanoate (S)-34. The similar treatment of 2,2,2-trifluoro-1-(3-hydroxyphenyl)ethanone 32 (248 mg, 1.53 mmol) and methyl D-(−)-lactate 4 (248 mg, 2.30 mmol) as that just described gave (S)-34 (300 mg, 71%). [α]D −45 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.69 (d, J = 7.8 Hz, 1H), 7.52 (s, 1H), 7.46 (t, J = 7.8 Hz, 1H), 7.23 (d, J = 7.8 Hz, 1H), 4.84 (q, J = 6.8 Hz, 1H), 3.78 (s, 3H), 1.66 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 180.1 (q, 2JCF = 35.2 Hz), 171.9, 157.9, 131.2, 130.3, 123.5, 123.0, 118.7, 116.6 (q, 1JCF = 291.1 Hz), 115.5, 72.7, 52.5, 18.4. HRMS (ESI): m/z calculated for C12H11F3O4 + H+ [M + H+]: 277.0688. Found: 277.0688.
Methyl (R)-2-(3-(2,2,2-trifluoroacetyl)phenoxy)propanoate (R)-34. The similar treatment of 2,2,2-trifluoro-1-(3-hydroxyphenyl)ethanone 32 (655 mg, 4.04 mmol) and methyl L-(−)-Lactate 5 (631 mg, 6.06 mmol) as that just described gave (R)-34 (835 mg, 75%). [α]D +45 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.69 (d, J = 7.8 Hz, 1H), 7.53 (s, 1H), 7.46 (t, J = 7.8 Hz, 1H), 7.24 (d, J = 7.8 Hz, 1H), 4.85 (q, J = 6.8 Hz, 1H), 3.78 (s, 3H), 1.67 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 180.0 (q, 2JCF = 35.2 Hz), 171.8, 157.9, 131.1, 130.3, 123.4, 122.9, 118.7, 116.5 (q, 2JCF = 291.1 Hz), 115.4, 72.7, 52.4, 18.3. HRMS (ESI): m/z calculated for C12H11F3O4 + H+ [M + H+]: 277.0688. Found: 277.0685.
(S)-2-(4-(2,2,2-Trifluoroacetyl)phenoxy)propanoic acid ((S)-35). Methyl (S)-2-(4-(2,2,2-trifluoroacetyl)phenoxy)propanoate (S)-33 (402 mg, 1.46 mmol) was dissolved in MeOH (18 mL) and H2O (2 mL), and then K2CO3 (605 mg, 4.37 mmol) was added. After the reaction mixture was stirred at reflux for 2 h, cooled to room temperature and then partitioned between ethyl acetate and water. The water layer was acidified by 1 M HCl and extracted by ethyl acetate. The organic layer was washed by H2O and brine, and dried over MgSO4, filtrated and concentrated to give (S)-35 (400 mg, quant). [α]D −31 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 9.05 (1H, s), 7.97 (d, J = 8.9 Hz, 2H), 6.91 (d, J = 8.9 Hz, 2H), 4.85 (q, J = 6.8 Hz, 1H), 1.65 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 178.9 (q, 2JCF = 35.2 Hz), 176.7, 162.9, 132.8, 123.7, 116.8 (q, 1JCF = 291.6 Hz), 115.2, 72.0, 18.2. HRMS (ESI): m/z calculated for C11H9F3O4 + H+ [M + H+]: 263.0531. Found: 263.0543.
(R)-2-(4-(2,2,2-Trifluoroacetyl)phenoxy)propanoic acid ((R)-35). The similar treatment of methyl (R)-2-(4-(2,2,2-trifluoroacetyl)phenoxy)propanoate (R)-33 (476 mg, 1.72 mmol) as that just described gave (R)-35 (428 mg, 95%). [α]D +31 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.99 (d, J = 8.9 Hz, 2H), 6.92 (d, J = 8.9 Hz, 2H), 4.86 (q, J = 6.8 Hz, 1H), 1.66 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 178.9 (q, 2JCF = 34.6 Hz), 176.4, 162.9, 132.8, 123.8, 116.8 (q, 1JCF = 291.3 Hz), 115.2, 71.9, 18.2. HRMS (ESI): m/z calculated for C11H9F3O4 + H+ [M + H+]: 263.0531. Found: 263.0541.
(S)-2-(3-(2,2,2-Trifluoroacetyl)phenoxy)propanoic acid ((S)-36). The similar treatment of methyl (S)-2-(3-(2,2,2-trifluoroacetyl)phenoxy)propanoate (S)-34 (285 mg, 1.03 mmol) as that just described gave (S)-36 (268 mg, 99%). [α]D −28 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.70 (d, J = 7.6 Hz, 1H), 7.56 (s, 1H), 7.47 (t, J = 8.1 Hz, 1H), 7.25 (d, J = 8.6 Hz, 1H), 4.88 (q, J = 6.8 Hz, 1H), 1.70 (d, J = 6.9 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 180.1 (q, 2JCF = 35.2 Hz), 177.4, 157.7, 131.2, 130.4, 123.7, 122.8, 116.5 (q, 1JCF = 291.3 Hz), 115.7, 72.2, 18.2. HRMS (ESI): m/z calculated for C11H9F3O4 + H+ [M + H+]: 263.0531. Found: 263.0543.
(R)-2-(3-(2,2,2-Trifluoroacetyl)phenoxy)propanoic acid ((R)-36). The similar treatment of methyl (R)-2-(3-(2,2,2-trifluoroacetyl)phenoxy)propanoate (R)-34 (288 mg, 1.04 mmol) as that just described gave (R)-36 (265 mg, 97%). [α]D +28 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.59 (d, J = 7.6 Hz, 1H), 7.46 (s, 1H), 7.36 (t, J = 8.1 Hz, 1H), 7.14 (d, J = 8.2 Hz, 1H), 4.78 (q, J = 6.8 Hz, 1H), 1.60 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 180.1 (q, 2JCF = 35.2 Hz), 177.3, 157.7, 131.1, 130.4, 123.6, 122.8, 116.5 (q, 1JCF = 291.6 Hz), 115.7, 72.2, 18.2. HRMS (ESI): m/z calculated for C11H9F3O4 + H+ [M + H+]: 263.0531. Found: 263.0538.
tert-Butyl (S)-2-(4-(2,2,2-trifluoroacetyl)phenoxy)propanoate ((S)-37). To a solution of (S)-2-(4-(2,2,2-trifluoroacetyl)phenoxy)propanoic acid 35 (138 mg, 0.53 mmol), potassium carbonate (1.816 g, 13.1 mmol) and tetrabutylammonium bromide (169 mg, 0.53 mmol) in N,N-dimethylacetamide (2.6 mL), tert-butyl bromide (3.416 g, 24.9 mmol) was added dropwise at 0 °C. The reaction mixture was stirred at 55 °C for 2.5 h. After cooling to room temperature, the mixture was poured into cold water and extracted with ethyl acetate. The water layer was added 1 M HCl to make pH 1 and extracted with ethyl acetate, washed with brine, dried, filtrated and concentrated to recover (S)-36 (21.8 mg, 16%). The organic layer was washed with H2O, brine, dried over MgSO4, filtrated and evaporated. The residue was purified by column chromatography (ethyl acetate/n-hexane, 1:6) to give (S)-37 (111 mg, 66%). [α]D −38 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 8.04 (d, J = 8.9 Hz, 2H), 6.96 (d, J = 8.9 Hz, 2H), 4.75 (q, J = 6.8 Hz, 1H), 1.64 (d, J = 6.8 Hz, 3H), 1.45 (s, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 178.9 (q, 2JCF = 35.2 Hz), 170.1, 163.5, 132.7, 123.3, 116.9 (q, 1JCF = 291.1 Hz), 115.2, 82.7, 73.0, 27.9, 18.2. HRMS (ESI): m/z calculated for C15H17F3O4 + H+ [M + H+]: 319.1157. Found: 319.1158.
tert-Butyl (R)-2-(4-(2,2,2-trifluoroacetyl)phenoxy)propanoate ((R)-37). The similar treatment of (R)-2-(4-(2,2,2-trifluoroacetyl)phenoxy)propanoic acid (R)-35 (428 mg, 1.63 mmol) as that just described gave (R)-37 (363 mg, 70%). [α]D +38 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.96 (d, J = 8.9 Hz, 2H), 6.88 (d, J = 8.9 Hz, 2H), 4.67 (q, J = 6.8 Hz, 1H), 1.55 (d, J = 6.8 Hz, 3H), 1.36 (s, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 178.8 (q, 2JCF = 34.6 Hz), 170.1, 163.5, 132.6, 123.2, 116.8 (q, 1JCF = 291.1 Hz), 115.2, 82.6, 73.0, 27.8, 18.2. HRMS (ESI): m/z calculated for C15H17F3O4 + H+ [M + H+]: 319.1157. Found: 319.1147.
tert-Butyl (S)-2-(3-(2,2,2-trifluoroacetyl)phenoxy)propanoate ((S)-38). The similar treatment of (S)-2-(3-(2,2,2-trifluoroacetyl)phenoxy)propanoic acid (S)-36 (310 mg, 1.18 mmol) as that just described gave (S)-36 (149 mg, 48%) and (S)-38 (141 mg, 38%). [α]D −53 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.67 (d, J = 7.8 Hz, 1H), 7.51 (s, 1H), 7.45 (t, J = 7.8 Hz, 1H), 7.25 (d, J = 7.8 Hz, 1H), 4.69 (q, J = 6.8 Hz, 1H), 1.62 (d, J = 6.8 Hz, 3H), 1.45 (s, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 180.2 (q, 2JCF = 34.6 Hz), 170.6, 158.2, 131.0, 130.2, 123.3, 123.2, 116.6 (q, 1JCF = 291.1 Hz), 115.0, 82.5, 73.0, 27.8, 18.3. HRMS (ESI): m/z calculated for C15H17F3O4 + H+ [M + H+]: 319.1157. Found: 319.1159.
tert-Butyl (R)-2-(3-(2,2,2-trifluoroacetyl)phenoxy)propanoate((R)-38). The similar treatment of (R)-2-(3-(2,2,2-trifluoroacetyl)phenoxy)propanoic acid (R)-36 (344 mg, 1.31 mmol) as that just described gave (R)-36 (167 mg, 48%) and (R)-38 (153 mg, 37%). [α]D +53 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.68 (d, J = 7.8 Hz, 1H), 7.51 (s, 1H), 7.45 (t, J = 7.8 Hz, 1H), 7.24 (d, J = 7.8 Hz, 1H), 4.69 (q, J = 6.8 Hz, 1H), 1.62 (d, J = 6.8 Hz, 3H), 1.45 (s, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 180.1 (q, 2JCF = 35.2 Hz), 170.6, 158.2, 131.0, 130.2, 123.3, 123.2, 116.6 (q, 1JCF = 291.1 Hz), 115.0, 82.5, 73.0, 27.8, 18.3. HRMS (ESI): m/z calculated for C15H17F3O4 + H+ [M + H+]: 319.1157. Found: 319.1183.
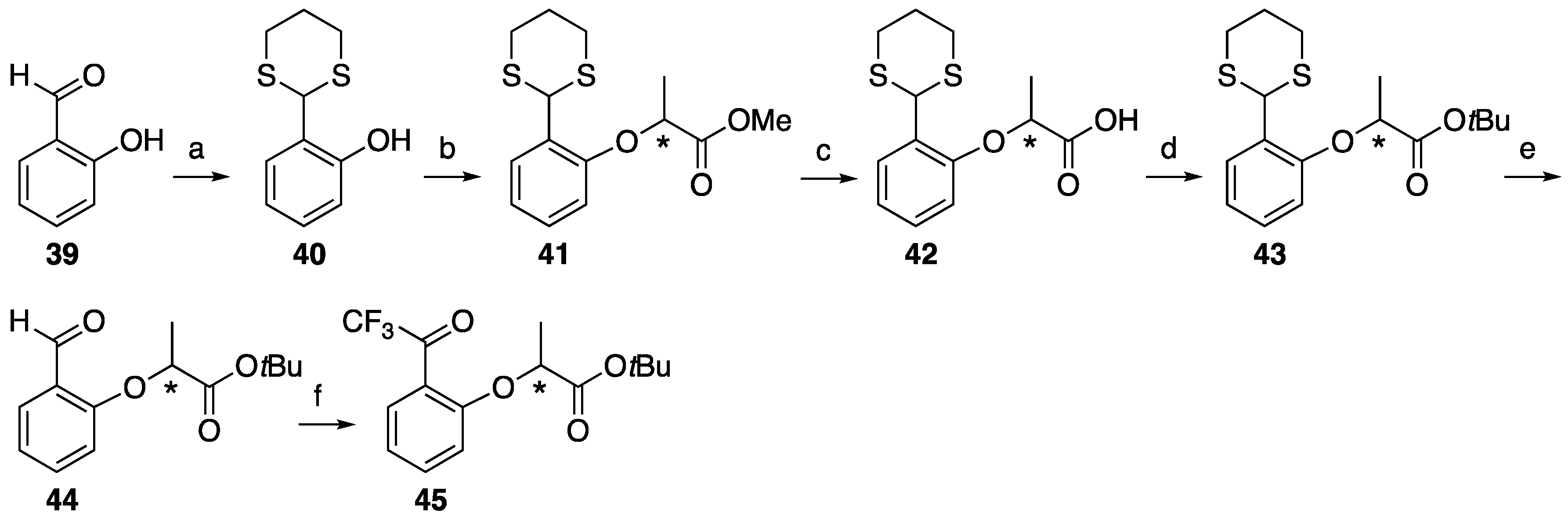
2-(1,3-Dithian-2-yl)phenol 40. Salicylaldehyde 39 (0.62 g, 5.06 mmol) and iodine (130 mg, 0.51 mmol) were dissolved in CH2Cl2 (25 mL) and then 1,3-propanedithiol (0.65 g, 6.00 mmol) was added. After the reaction mixture was stirred at room temperature for 1 h, then quenched aqueous sodium thiosulfate (0.5 M, 10 mL). The mixture was extracted with CH2Cl2, washed with brine, dried over MgSO4, filtrated and concentrated to give 2-(1,3-dithian-2-yl)phenol 40 (1.02 g, 95%). 1H-NMR (270 MHz, CDCl3): δ = 7.29 (dd, J = 7.9, 1.6 Hz, 1H), 7.21 (td, J = 7.7, 1.6 Hz, 1H), 6.88 (dq, J = 7.4, 2.0 Hz, 2H), 6.33 (s, 1H), 5.40 (s, 1H), 3.07 (td, J = 13.6, 3.0 Hz, 2H), 2.91 (dt, J = 14.2, 3.6 Hz, 2H), 2.26–2.13 (m, 1H), 2.00–1.87 (m, 1H). 13C-NMR (67.5 MHz, CDCl3): δ = 154.5, 130.1, 129.1, 123.5, 120.8, 117.3, 47.4, 31.6, 24.8. HRMS (ESI): m/z calculated for C10H12O2S2 + H+ [M + H+]: 213.0408. Found: 213.0383.
Methyl (S)-2-(2-(1,3-dithian-2-yl)phenoxy)propanoate ((S)-41). 2-(1,3-Dithian-2-yl)phenol 40 (1.27 g) phenol, methyl D-(+)-lactate 4 (0.94 g, 8.99 mmol) and PPh3 (1.90 g, 7.22 mmol) were dissolved in dry CH2Cl2 (30 mL). After stirring for 10 min at 0 °C, DEAD (1.57 g, 9.02 mmol) was slowly added. The reaction mixture was stirred overnight at room temperature, and then partitioned between water and CH2Cl2. The organic layer was washed with brine, dried over MgSO4, filtered and concentrated. The residue was purified by column chromatography (ethyl acetate/n-hexane, 1:9) to give (S)-41 (1.48 g, 98%). [α]D +31 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.60 (d, J = 7.6 Hz, 1H), 7.19 (t, J = 7.6 Hz, 1H), 7.00 (t, J = 7.6 Hz, 1H), 6.75 (d, J = 7.6 Hz, 1H), 5.76 (s, 1H), 4.78 (q, J = 6.8 Hz, 1H), 3.75 (s, 3H), 3.11 (m, 2H), 2.88 (m, 2H), 2.16 (m, 1H), 1.95 (m, 1H), 1.67 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 172.3, 153.8, 129.4, 129.2, 128.6, 122.2, 113.4, 73.9, 52.2, 43.8, 32.3, 32.2, 25.3, 18.5. HRMS (ESI): m/z calculated for C14H18O3S2 + Na+ [M + Na+]: 321.0595. Found: 321.0597.
Methyl (R)-2-(2-(1,3-dithian-2-yl)phenoxy)propanoate ((R)-41). 2-(1,3-Dithian-2-yl)phenol 40 (1.27 g) as that just described gave (R)-41 (1.47 g, 98%). [α]D −31 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.60 (d, J = 7.6 Hz, 1H), 7.19 (t, J = 7.6 Hz, 1H), 7.00 (t, J = 7.6 Hz, 1H), 6.75 (d, J = 7.6 Hz, 1H), 5.76 (s, 1H), 4.78 (q, J = 6.8 Hz, 1H), 3.75 (s, 3H), 3.11 (m, 2H), 2.90 (m, 2H), 2.16 (m, 1H), 1.95 (m, 1H), 1.67 (d, J = 6.9 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 172.3, 153.8, 129.4, 129.2, 128.7, 122.3, 113.4, 74.0, 52.2, 43.8, 32.3, 32.2, 25.3, 18.6. HRMS (ESI): m/z calculated for C14H18O3S2 + Na+ [M + Na+]: 321.0595. Found: 321.0598.
(S)-2-(2-(1,3-Dithian-2-yl)phenoxy)propanoic acid (S)-42. Methyl (S)-2-(2-(1,3-dithian-2-yl)phenoxy)propanoate (S)-41 (1.51 g, 5.06 mmol) was dissolved in MeOH (30 mL) and H2O (3.3 mL), and then K2CO3 (700 mg, 5.06 mmol) was added. After the reaction mixture was stirred at reflux for 2 h, cooled to room temperature and then partitioned between ethyl acetate and water. The water layer was acidified by 1 M HCl aq and extracted by ethyl acetate. The organic layer was washed by H2O and brine, and dried over MgSO4, filtrated and concentrated to give (S)-42 (1.63 g, quant). [α]D +9 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.55 (d, J = 7.6 Hz, 1H), 7.26 (t, J = 7.6 Hz, 1H), 7.03 (t, J = 7.6 Hz, 1H), 6.82 (d, J = 7.6 Hz, 1H), 5.62 (s, 1H), 4.88 (q, J = 6.8 Hz, 1H), 3.12 (m, 2H), 2.92 (m, 2H), 2.19 (m, 1H), 1.96 (m, 1H), 1.71 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 174.9, 153.3, 129.6, 129.5, 128.1, 122.6, 112.8, 73.0, 45.4, 32.2, 32.1, 25.3, 18.0. HRMS (ESI): m/z calculated for C13H16O3S2 + H+ [M + H+]: 285.0619. Found: 285.0591.
(R)-2-(2-(1,3-dithian-2-yl)phenoxy)propanoic acid (R)-42. The similar treatment of methyl (R)-2-(2-(1,3-dithian-2-yl)phenoxy)propanoate (R)-41 (1.56 g, 5.22 mmol) as that just described gave (S)-42 (1.69 g, quant). [α]D −9 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.56 (d, J = 7.6 Hz, 1H), 7.24 (t, J = 7.6 Hz, 1H), 7.02 (t, J = 7.6 Hz, 1H), 6.81 (d, J = 7.6 Hz, 1H), 5.65 (s, 1H), 4.85 (q, J = 6.8 Hz, 1H), 3.11 (m, 2H), 2.90 (m, 2H), 2.18 (m, 1H), 1.95 (m, 1H), 1.71 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 175.8, 153.3, 129.5, 129.5, 128.2, 122.5, 112.9, 73.1, 45.0, 32.2, 32.1, 25.2, 18.1. HRMS (ESI): m/z calculated for C13H16O3S2 + H+ [M + H+]: 285.0619. Found: 285.0608.
tert-Butyl (S)-2-(2-(1,3-dithian-2-yl)phenoxy)propanoate (S)-43. To a solution of (S)-2-(2-(1,3-dithian-2-yl)phenoxy)propanoic acid (1.63 g, 5.72 mmol) in N,N-dimethylacetamide (29 mL) in the presence of potassium carbonate (19.8 g, 143 mmol) and tetrabutylammonium bromide (1.85 g, 5.73 mmol) at 0 °C, tert-butyl bromide (37.6 g, 274 mmol) was added dropwise and the reaction mixture was stirred at 55 °C for 2.5 h. After cooling to room temperature, the mixture was added into cold water and extracted with ethyl acetate. The organic layer was washed with H2O, brine, dried over MgSO4, filtrated and evaporated. The residue was purified by column chromatography (ethyl acetate/n-hexane, 1:6) to give (S)-43 (1.61 g, 83%). [α]D +25 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.59 (d, J = 7.8 Hz, 1H), 7.18 (t, J = 7.8 Hz, 1H), 6.97 (t, J = 7.8 Hz, 1H), 6.74 (d, J = 7.8 Hz, 1H), 5.75 (s, 1H), 4.64 (q, J = 6.8 Hz, 1H), 3.11 (m, 2H), 2.88 (m, 2H), 2.16 (m, 1H), 1.93 (m, 1H), 1.63 (d, J = 6.8Hz, 3H), 1.43 (s, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 171.0, 153.9, 129.3, 129.0, 128.3, 121.8, 112.8, 81.8, 73.9, 43.8, 32.3, 32.2, 27.9, 25.4, 18.4. HRMS (ESI): m/z calculated for C17H24O3S2 + H+ [M + H+]: 341.1245. Found: 341.1237.
tert-Butyl (R)-2-(2-(1,3-dithian-2-yl)phenoxy)propanoate (R)-43. The similar treatment of (R)-2-(2-(1,3-dithian-2-yl)phenoxy)propanoic acid 42 (1.69 g, 5.93 mmol) as that just described gave (R)-43 (1.65 g, 82%). [α]D −25 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.59 (d, J = 7.8 Hz, 1H), 7.18 (t, J = 7.8 Hz, 1H), 6.97 (t, J = 7.8 Hz, 1H), 6.73 (d, J = 7.8 Hz, 1H), 5.75 (s, 1H), 4.64 (q, J = 6.8 Hz, 1H), 3.11 (m, 2H), 2.88 (m, 2H), 2.16 (m, 1H), 1.95 (m, 1H), 1.63 (d, J = 6.8 Hz, 3H), 1.43 (s, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 171.0, 153.9, 129.3, 129.0, 128.3, 121.8, 112.8, 81.8, 73.9, 43.8, 32.4, 32.2, 27.9, 25.4, 18.4. HRMS (ESI): m/z calculated for C17H24O3S2 + H+ [M + H+]: 341.1245. Found: 341.1239.
tert-Butyl (S)-2-(2-formylphenoxy)propanoate (S)-44.tert-Butyl (S)-2-(2-(1,3-dithian-2-yl)phenoxy)propanoate (S)-43 (94.3 mg, 0.28 mmol) and sodium hydrogen carbonate (465 mg, 5.54 mmol) were dissolved in acetonitrile (10 mL) and water (2 mL), and then iodomethane (392 mg, 2.8 mmol)was added. After the reaction mixture was stirred at room temperature for 24 h, iodomethane (196 mg, 1.4 mmol) was added. After stirred at room temperature for 24 h, the reaction mixture extracted with ethyl acetate. The organic layer was washed with brine, dried over MgSO4, filtrated and concentrated. The residue was purified by column chromatography (ethyl acetate/n-hexane, 1:6) to give (S)-44 (52.3 mg, 78%). [α]D +14 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 10.57 (s, 1H), 7.85 (d, J = 7.9 Hz, 1H), 7.49 (t, J = 7.9 Hz, 1H), 7.04 (t, J = 7.9 Hz, 1H), 6.85 (d, J = 7.9 Hz, 1H), 4.77 (q, J = 6.8 Hz, 1H), 1.66 (d, J = 6.8 Hz, 3H), 1.42 (s, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 189.7, 170.4, 160.2, 135.5, 128.2, 125.4, 121.4, 113.2, 82.3, 73.5, 27.8, 18.2. HRMS (ESI): m/z calculated for C14H18O4 + H+ [M + H+]: 251.1283. Found: 251.1312.
tert-Butyl (R)-2-(2-formylphenoxy)propanoate (R)-44. The similar treatment of tert-butyl (R)-2-(2-(1,3-dithian-2-yl)phenoxy)propanoate (R)-43 (156 mg, 0.46 mmol) as that just described gave (R)-44 (90.4 mg, 79%). [α]D −14 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 10.58 (s, 1H), 7.84 (d, J = 7.6 Hz, 1H), 7.50 (t, J = 7.6 Hz, 1H), 7.04 (t, J = 7.6 Hz, 1H), 6.85 (d, J = 7.6 Hz, 1H), 4.78 (q, J = 6.8 Hz, 1H), 1.66 (d, J = 6.8 Hz, 3H), 1.42 (s, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 189.7, 170.4, 160.2, 135.5, 128.2, 125.4, 121.4, 113.2, 82.3, 73.5, 27.8, 18.2. HRMS (ESI): m/z calculated for C14H18O4 + H+ [M + H+]: 251.1283. Found: 251.1292.
tert-Butyl (S)-2-(2-(2,2,2-trifluoroacetyl)phenoxy)propanoate (S)-45. To the solution of tert-butyl (S)-2-(2-formylphenoxy)propanoate (S)-44 (591 mg, 2.04 mmol) in dimethylacetamide (7.9 mL), (trifluoromethyl)trimetylsilane (579 mg, 4.07 mmol) and potassium carbonate (16.3 mg, 0.118 mmol) were added. The reaction mixture was and stirred at room temperature for 4 h and treated with 1 M HCl (1 mL) at room temperature for 4 h, then extracted with ethyl acetate. The organic layer was washed with brine twice, dried over MgSO4, filtrated and concentrated to afford crude material. To crude phenylethanol derivative (784 mg) was dissolved in CH2Cl2 (13 mL), Dess-Martin periodinane (1.096 g, 2.58 mmol) was added at room temperature. The reaction mixture was stirred at room temperature for 3 h, then washed with water and brine. The organic layer was dried over MgSO4, filtrated and concentrated. The residue was subjected to column chromatography (hexane/AcOEt, 8:1) to give (S)-45 (636 mg, 85%). [α]D +25 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.54 (d, J = 7.6 Hz, 1H), 7.41 (t, J = 7.6 Hz, 1H), 6.95 (t, J = 7.6 Hz, 1H), 6.71 (d, J = 7.6 Hz, 1H), 4.61 (q, J = 6.8 Hz, 1H), 1.51 (d, J = 6.8 Hz, 3H), 1.30 (s, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 183.6 (q, 2JCF = 37.4 Hz), 170.2, 157.5, 135.3, 131.5, 122.6, 121.2, 116.1 (q, 1JCF = 290.5 Hz), 112.8, 82.4, 73.7, 27.8, 17.8. HRMS (ESI): m/z calculated for C15H17F3O4 + H+ [M + H+]: 319.1157. Found: 319.1171.
tert-Butyl (R)-2-(2-(2,2,2-trifluoroacetyl)phenoxy)propanoate (R)-45. The similar treatment of tert-butyl (R)-2-(2-formylphenoxy)propanoate (R)-44 (577 mg, 2.30 mmol) as that just described gave (R)-45 (651 mg, 89%). [α]D −25 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.65 (d, J = 7.6 Hz, 1H), 7.53 (t, J = 7.6 Hz, 1H), 7.06 (t, J = 7.6 Hz, 1H), 6.83 (d, J = 7.6 Hz, 1H), 4.72 (q, J = 6.8 Hz, 1H), 1.62 (d, J = 6.8 Hz, 3H), 1.41 (s, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 183.6 (q, 2JCF = 37.4 Hz), 170.2, 157.5, 135.3, 131.6, 122.6, 121.2, 116.1 (q, 1JCF = 290.5 Hz), 112.8, 82.4, 73.7, 27.8, 17.8. HRMS (ESI): m/z calculated for C15H17F3O4 + H+ [M + H+]: 319.1157. Found: 319.1141.
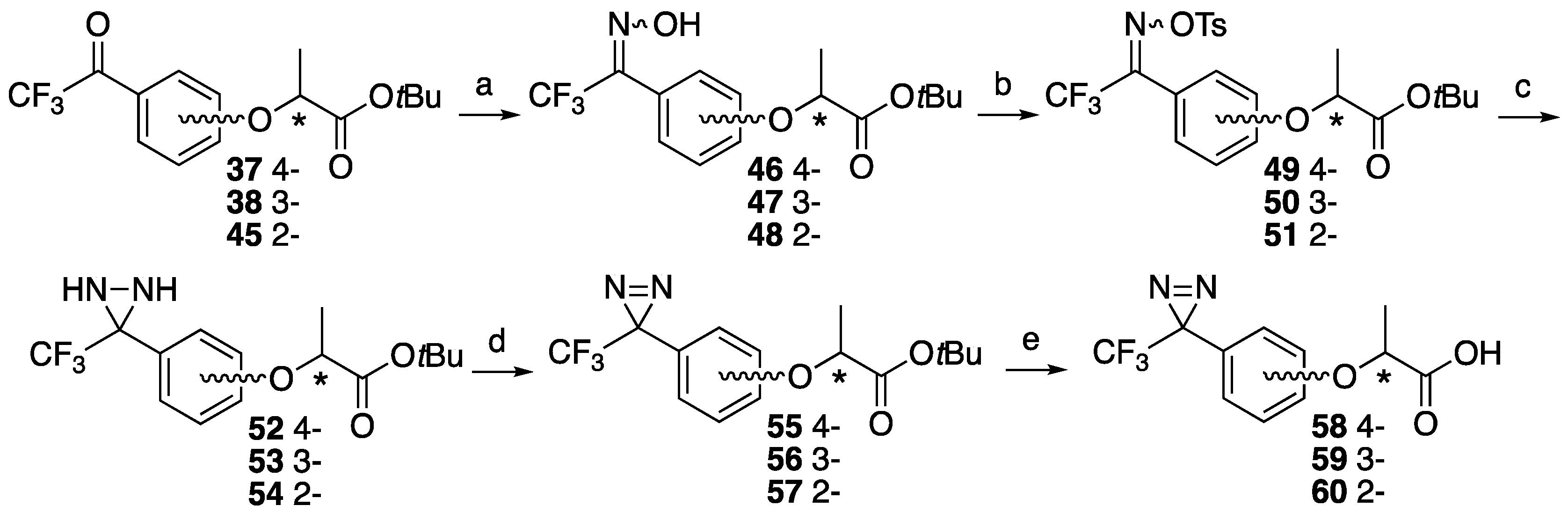
tert-Butyl (S)-2-(4-(2,2,2-trifluoro-1-(hydroxyimino)ethyl)phenoxy)propanoate ((S)-46). A solution of tert-butyl (S)-2-(4-(2,2,2-trifluoroacetyl)phenoxy)propanoate (S)-37 (252 mg, 0.79 mmol) and hydroxylamine hydrochloride (60.7 mg, 0.87 mmol) in ethanol (0.44 mL) and dry pyridine (0.79 mL) was heated at 60 °C for 8 h. The reaction mixture was partitioned between water and ether, and the organic layer was washed with 1 M HCl, saturated NaHCO3 and brine. The organic layer was dried over MgSO4, filtrated and concentrated. The residue was purified by column chromatography (ethyl acetate/n-hexane, 1:6) to give (S)-46 (275 mg, quant). The product was mixture of syn- and anti- isomers. [α]D −38 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 8.89 and 8.75 (s, 1H), 7.44 and 7.34 (d × 2, J = 8.9 Hz, 2H), 6.86 and 6.81 (d × 2, J = 8.9 Hz, 2H), 4.60 and 4.59 (q × 2, J = 6.8 Hz, 1H), 1.53 (d, J = 6.8 Hz, 3H), 1.37 (s, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 171.0, 159.3, 146.9 (q, 2JCF = 32.4 Hz), 130.5, 120.8 (q, 1JCF = 274.9 Hz), 118.6, 114.9, 82.4, 72.9, 27.9, 18.3. HRMS (ESI): m/z calculated for C15H18F3NO4 + H+ [M + H+]: 334.1266. Found: 334.1256.
tert-Butyl (R)-2-(4-(2,2,2-trifluoro-1-(hydroxyimino)ethyl)phenoxy)propanoate ((R)-46). The similar treatment of tert-butyl (R)-2-(4-(2,2,2-trifluoroacetyl)phenoxy)propanoate (R)-37 (459 mg, 1.44 mmol) as that just described gave (R)-46 (490 mg, quant). [α]D +38 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 8.36 and 8.19 (s, 1H), 7.50 and 7.42 (d × 2, J = 7.8 Hz, 2H), 6.93 and 6.88 (d × 2, J = 8.9 Hz, 2H), 4.67 and 4.66 (q × 2, J = 6.8 Hz, 1H), 1.60 (d, J = 6.8 Hz, 3H), 1.44 (s, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 171.0, 159.3, 146.9 (q, 2JCF = 32.4 Hz), 130.5, 120.8 (q, 1JCF = 274.9 Hz), 118.6, 114.9, 82.4, 72.9, 27.9, 18.3. HRMS (ESI): m/z calculated for C15H18F3NO4 + H+ [M + H+]: 334.1266. Found: 334.1236.
tert-Butyl (S)-2-(3-(2,2,2-trifluoro-1-(hydroxyimino)ethyl)phenoxy)propanoate ((S)-47). The similar treatment of tert-butyl (S)-2-(3-(2,2,2-trifluoroacetyl)phenoxy)propanoate (S)-38 (81.7 mg, 0.26 mmol) as that just described gave (S)-47 (85.7 mg, quant). [α]D −40 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.36 and 7.30 t × 2, J = 7.8 Hz, 1H), 7.09 (d, J = 7.8 Hz, 1H), 6.97 (m, 2H), 4.66 (q, J = 6.8 Hz, 1H), 1.60 and 1.59 (d × 2, J = 6.8 Hz, 3H), 1.43 and 1.42 (s × 2, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 171.3, 157.6, 147.3 (q, 2JCF = 32.4 Hz), 129.7 and 129.6, 127.2, 121.5, 120.6 (q, 1JCF = 274.3 Hz), 117.4 and 117.0, 115.4 and 115.2, 82.5, 73.1, 27.8, 18.4. HRMS (ESI): m/z calculated for C15H18F3NO4 + H+ [M + H+]: 334.1266. Found: 334.1265.
tert-Butyl (R)-2-(3-(2,2,2-trifluoro-1-(hydroxyimino)ethyl)phenoxy)propanoate ((R)-47). The similar treatment of tert-butyl (R)-2-(3-(2,2,2-trifluoroacetyl)phenoxy)propanoate (R)-38 (97.2 mg, 0.31 mmol) as that just described gave (R)-47 (86.6 mg, 85%). [α]D +40 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.37 and 7.30 (t × 2, J = 7.8 Hz, 2H), 7.08 (d, J = 7.8 Hz, 1H), 6.97 (m, 2H), 4.65 (q, J = 6.8 Hz, 1H), 1.60 (d, J = 6.8 Hz, 3H), 1.43 and 1.42 (s × 2, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 171.1, 157.6, 147.6 (q, 2JCF = 32.4 Hz), 129.7 and 129.5, 127.1, 121.4, 120.5 (q, 1JCF = 274.9 Hz), 117.4 and 117.1, 115.3 and 115.1, 82.3, 73.1, 27.8, 18.4. HRMS (ESI): m/z calculated for C15H18F3NO4 + H+ [M + H+]: 334.1266. Found: 334.1257.
tert-Butyl (S)-2-(2-(2,2,2-trifluoro-1-(hydroxyimino)ethyl)phenoxy)propanoate ((S)-48). The similar treatment of tert-butyl (S)-2-(2-(2,2,2-trifluoroacetyl)phenoxy)propanoate (S)-45 (309 mg, 0.97 mmol) as that just described gave (S)-48 (331 mg, quant). [α]D +8 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.37 and 7.36 (t × 2, J = 7.6 Hz, 1H), 7.27 and 7.21 (d × 2, J = 7.6 Hz, 1H), 7.03 and 6.99 (t × 2, J = 7.6 Hz, 1H), 6.80 and 6.75 (d × 2, J = 7.6 Hz, 1H), 4.62 (q, J = 6.8 Hz, 1H), 1.57 and 1.54 (d × 2, J = 6.8 Hz, 3H), 1.41 (s, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 171.2 and 171.0, 156.5, 155.1, 147.6 and 146.7 (q × 2, 2JCF = 33.5 Hz), 131.6, 131.0, 129.8, 121.0, 117.9 (q, 1JCF = 287.2 Hz), 116.3, 112.2 and 111.7, 82.3 and 82.2, 73.5 and 73.4, 27.8, 18.2 and 18.0. HRMS (ESI): m/z calculated for C15H18F3NO4 + H+ [M + H+]: 334.1266. Found: 334.1246.
tert-Butyl (R)-2-(2-(2,2,2-trifluoro-1-(hydroxyimino)ethyl)phenoxy)propanoate ((R)-48). The similar treatment of tert-butyl (R)-2-(2-(2,2,2-trifluoroacetyl)phenoxy)propanoate (R)-45 (291 mg, 0.92 mmol) as that just described gave (R)-48 (299 mg, 98%). [α]D −8 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.37 (m, 1H), 7.27 and 7.21 (d, J = 7.6 Hz, 1H), 7.03 and 6.98 (t, J = 7.6 Hz, 1H), 6.80 and 6.74 (d, J = 7.6 Hz, 1H), 4.62 (q, J = 6.8 Hz, 1H), 1.57 and 1.54 (d, J = 6.8 Hz, 3H), 1.41 (s, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 171.2 and 171.1, 156.5, 155.1, 147.5 and 146.5 (q × 2, 2JCF = 34.1 Hz), 131.6, 131.0, 129.8, 121.0, 120.5 and 118.2 (q × 2, 1JCF = 287.2 Hz), 116.3, 112.2 and 111.7, 82.3 and 82.2, 73.5 and 73.4, 27.8, 18.1 and 18.0. HRMS (ESI): m/z calculated for C15H18F3NO4 + H+ [M + H+]: 334.1266. Found: 334.1261.
tert-Butyl (S)-2-(4-(2,2,2-trifluoro-1-((tosyloxy)imino)ethyl)phenoxy)propanoate ((S)-49).tert-Butyl (S)-2-(4-(2,2,2-trifluoro-1-(hydroxyimino)ethyl)phenoxy)propanoate (S)-46 (268 mg, 0.80 mmol), triethylamine (203 mg, 2.01 mmol) and DMAP (4.9 mg, 0.040 mmol) were suspended in CH2Cl2 (1.5 mL) at 0 °C. p-Toluenesulfonyl chloride (168 mg, 0.88 mmol) was added at 0 °C. The reaction mixture was stirred at room temperature for 45 min, then washed with water. The organic layer was dried over MgSO4, filtrated and concentrated to give (S)-49 (418 mg, quant). The product was mixture of syn- and anti- isomers. [α]D −20 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.88 (d, J = 8.2 Hz, 2H), 7.45–7.35 (m, 4H), 6.92 and 6.87 (d, J = 8.2 Hz, 2H), 4.68 (q, J = 6.8 Hz, 1H), 2.46 and 2.45 (s × 2, 3H), 1.61 and 1.60 (d × 2, J = 6.8 Hz, 3H), 1.44 (s, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 170.5 and 170.4, 160.6 and 160.2, 152.9 (q, 2JCF = 27.9 Hz), 146.0 and 145.9, 131.4 and 131.1, 130.5, 129.8, 129.1, 129.0, 128.9, 120.3, 119.7 and 117.4 (q, 1JCF = 276.0 Hz), 116.9, 115.0, 82.3, 72.8 and 72.7, 27.8 and 27.7, 21.6, 18.2. HRMS (ESI): m/z calculated for C22H24F3NO6S + H+ [M + H+]: 488.1355. Found: 488.1372.
tert-Butyl (R)-2-(4-(2,2,2-trifluoro-1-((tosyloxy)imino)ethyl)phenoxy)propanoate ((R)-49). The similar treatment of tert-butyl (R)-2-(4-(2,2,2-trifluoro-1-(hydroxyimino)ethyl)phenoxy)-propanoate (R)-46 (331 mg, 0.99 mmol) as that just described gave (R)-49 (473 mg, 98%). [α]D +20 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.89 (d, J = 7.8 Hz, 2H), 7.38 (m, 4H), 6.87 (d, J = 7.8 Hz, 2H), 4.65 (q, J = 6.8 Hz, 1H), 2.46 (s, 3H), 1.60 (d, J = 6.8 Hz, 3H), 1.44 (s, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 170.5 and 170.4, 160.6 and 160.2, 153.2 (q, 2JCF = 29.9 Hz), 146.0 and 145.9, 131.4 and 131.1, 130.5, 129.8, 129.1, 128.9, 120.3, 118.6 (q, 1JCF = 283.8 Hz), 116.9, 115.0, 82.3, 72.8 and 72.7, 27.7 and 27.8, 21.6, 18.2. HRMS (ESI): m/z calculated for C22H24F3NO6S + H+ [M + H+]: 488.1355. Found: 488.1380.
tert-Butyl (S)-2-(3-(2,2,2-trifluoro-1-((tosyloxy)imino)ethyl)phenoxy)propanoate ((S)-50). The similar treatment of tert-butyl (S)-2-(3-(2,2,2-trifluoro-1-(hydroxyimino)ethyl)phenoxy)-propanoate (S)-47 (277 mg, 0.83 mmol) as that just described gave (S)-50 (431 mg, quant). [α]D −34 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.88 (d, J = 8.6 Hz, 2H), 7.35 (m, 3H), 7.00 (d, J = 7.8 Hz, 2H), 6.97 (d, J = 7.8 Hz, 2H), 6.85 (s, 1H), 4.62 (q, J = 6.8 Hz, 1H), 2.48 (s, 3H), 1.60 (d, J = 6.8 Hz, 3H), 1.42 (s, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 170.7, 157.8 and 157.7, 153.6 (q, 2JCF = 32.4 Hz), 146.1 and 146.0, 131.5 and 131.2, 130.0, 129.9 and 129.8, 129.3 and 129.1, 128.8, 125.6, 121.8 and 121.3, 119.5 (q, 1JCF = 274.3 Hz), 118.4 and 118.2, 115.4 and 114.9, 82.3, 73.0, 27.8, 21.7, 18.3. HRMS (ESI): m/z calculated for C22H24F3NO6S + H+ [M + H+]: 488.1355. Found: 488.1330.
tert-Butyl (R)-2-(3-(2,2,2-trifluoro-1-((tosyloxy)imino)ethyl)phenoxy)propanoate ((R)-50). The similar treatment of tert-butyl (R)-2-(3-(2,2,2-trifluoro-1-(hydroxyimino)ethyl)phenoxy)-propanoate (R)-47 (348 mg, 1.04 mmol) as that just described gave (R)-50 (471 mg, 93%). [α]D +34 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.89 (d, J = 8.2 Hz, 2H), 7.35 (m, 3H), 7.02 (m, 2H), 6.93 (s, 1H), 4.61 (q, J = 6.7 Hz, 1H), 2.46 (s, 3H), 1.59 (d, J = 6.6 Hz, 3H), 1.42 (s, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 170.7, 157.8 and 157.7, 153.7 (q, 2JCF = 33.5 Hz), 146.1 and 146.0, 131.5 and 131.2, 130.0, 129.9 and 129.8, 129.3 and 129.1, 127.8, 125.6, 121.8 and 121.3, 119.5 (q, 1JCF = 280.5 Hz), 118.4 and 118.2, 115.4 and 114.9, 82.3, 73.1 and 73.0, 27.8, 21.8, 18.3. HRMS (ESI): m/z calculated for C22H24F3NO6S + H+ [M + H+]: 488.1355. Found: 488.1330.
tert-Butyl (S)-2-(2-(2,2,2-trifluoro-1-((tosyloxy)imino)ethyl)phenoxy)propanoate ((S)-51). The similar treatment of tert-butyl (S)-2-(2-(2,2,2-trifluoro-1-(hydroxyimino)ethyl)phenoxy)-propanoate (S)-48 (300 mg, 0.90 mmol) as that just described gave (S)-51 (412 mg, 94%). [α]D +24 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.88 (d, J = 8.2 Hz, 2H), 7.39 (m, 3H), 7.12 (d, J = 7.6 Hz, 1H), 6.97 (t, J = 7.6 Hz, 1H), 6.72 (d, J = 7.6 Hz, 1H), 4.56 (q, J = 6.8 Hz, 1H), 2.45 (s, 3H), 1.47 (d, J = 6.8 Hz, 3H), 1.39 (s, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 170.3, 156.6, 155.3 (q, 2JCF = 34.6 Hz), 145.7, 132.9, 132.4, 131.7, 131.1, 129.8, 129.0, 121.1, 117.8, 116.8 (q, 1JCF = 283.2 Hz), 111.8, 82.2, 73.6, 27.7, 21.7, 17.8. HRMS (ESI): m/z calculated for C22H24F3NO6S + H+ [M + H+]: 488.1355. Found: 488.1339.
tert-Butyl (R)-2-(2-(2,2,2-trifluoro-1-((tosyloxy)imino)ethyl)phenoxy)propanoate ((R)-51). The similar treatment of tert-butyl (R)-2-(2-(2,2,2-trifluoro-1-(hydroxyimino)ethyl)phenoxy)-propanoate (R)-48 (254 mg, 0.76 mmol) as that just described gave (R)-51 (373 mg, quant). [α]D −24 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.88 (d, J = 8.2 Hz, 2H), 7.39 (m, 3H), 7.12 (d, J = 7.6 Hz, 1H), 6.97 (t, J = 7.6 Hz, 1H), 6.72 (d, J = 7.6 Hz, 1H), 4.55 (q, J = 6.8 Hz, 1H), 2.46 (s, 3H), 1.47 (d, J = 6.8 Hz, 3H), 1.39 (s, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 170.3, 156.6, 155.3 (q, 2JCF = 35.2 Hz), 145.7, 132.9, 132.4, 131.7, 131.1, 129.8, 129.1, 121.1, 117.8, 116.8 (q, 1JCF = 282.7 Hz), 111.8, 82.2, 73.6, 27.8, 21.7, 17.8. HRMS (ESI): m/z calculated for C22H24F3NO6S + H+ [M + H+]: 488.1355. Found: 488.1362.
tert-Butyl (S)-2-(4-(3-(trifluoromethyl)diaziridin-3-yl)phenoxy)propanoate ((S)-52). To liquid NH3 (10 mL) at −78 °C in a sealed tube, tert-butyl (S)-2-(2-(2,2,2-trifluoro-1-((tosyloxy)imino)-ethyl)phenoxy)propanoate (S)-49 (336 mg, 0.69 mmol) in dry ether (3 mL) was added. The reaction mixture was stirred at room temperature for 3 h. After evaporation of NH3 gas, the reaction mixture was partitioned between ether and water. The organic layer was dried over MgSO4, filtrated and concentrated. The residue was purified by column chromatography (ethyl acetate/n-hexane, 1:5) to give (S)-52 (200 mg, 88%). [α]D −32 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.51 (d, J = 8.6 Hz, 2H), 6.88 (d, J = 8.6 Hz, 2H), 4.64 (q, J = 6.8 Hz, 1H), 2.76 (d, J = 8.6 Hz, 1H), 2.18 (d, J = 8.6 Hz, 1H), 1.59 (d, J = 6.8 Hz, 3H), 1.44 (s, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 170.9, 159.0, 129.4, 124.3, 123.6 (q, 1JCF = 278.2 Hz), 115.1, 82.1, 72.8, 57.5 (q, 2JCF = 35.8 Hz), 27.9, 18.3. HRMS (ESI): m/z calculated for C15H19F3N2O3 + H+ [M + H+]: 333.1426. Found: 333.1414.
tert-Butyl (R)-2-(4-(3-(trifluoromethyl)diaziridin-3-yl)phenoxy)propanoate ((R)-52). The similar treatment of tert-butyl (R)-2-(4-(2,2,2-trifluoro-1-((tosyloxy)imino)ethyl)phenoxy)-propanoate (R)-49 (433 mg, 0.89 mmol) as that just described gave (R)-52 (259 mg, 88%). [α]D +32 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.52 (d, J = 8.6 Hz, 2H), 6.88 (d, J = 8.6 Hz, 2H), 4.64 (q, J = 6.8 Hz, 1H), 2.75 (s, 1H), 2.16 (s, 1H), 1.59 (d, J = 6.8 Hz, 3H), 1.44 (s, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 170.9, 159.0, 129.5, 124.3, 123.6 (q, 1JCF = 277.7 Hz), 115.1, 82.2, 72.8, 57.6 (q, 2JCF = 35.2 Hz), 27.9, 18.3. HRMS (ESI): m/z calculated for C15H19F3N2O3 + H+ [M + H+]: 333.1426. Found: 333.1416.
tert-Butyl (S)-2-(3-(3-(trifluoromethyl)diaziridin-3-yl)phenoxy)propanoate ((S)-53). The similar treatment of tert-butyl (S)-2-(3-(2,2,2-trifluoro-1-((tosyloxy)imino)ethyl)phenoxy)-propanoate (S)-50 (431 mg, 0.88 mmol) as that just described gave (S)-53 (269 mg, 92%). [α]D −38 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.32 (t, J = 7.6 Hz, 1H), 7.21 (d, J = 7.6 Hz, 1H), 7.12 (s, 1H), 6.94 (d, J = 7.6 Hz, 1H), 4.64 (q, J = 6.8 Hz, 1H), 2.75 (s, 1H), 2.20 (s, 1H), 1.59 (d, J = 6.8 Hz, 3H), 1.44 (s, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 170.9, 157.9, 133.1, 129.9, 123.5 (q, 1JCF = 278.2 Hz), 120.9, 116.9, 114.6, 82.2, 72.9, 57.9 (q, 2JCF = 35.8 Hz), 27.9, 18.3. HRMS (ESI): m/z calculated for C15H19F3N2O3 + H+ [M + H+]: 333.1426. Found: 333.1414.
tert-Butyl (R)-2-(3-(3-(trifluoromethyl)diaziridin-3-yl)phenoxy)propanoate ((R)-53). The similar treatment of tert-butyl (R)-2-(3-(2,2,2-trifluoro-1-((tosyloxy)imino)ethyl)phenoxy)-propanoate (R)-50 (471 mg, 0.97 mmol) as that just described gave (R)-53 (347 mg, quant). [α]D +38 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.32 (t, J = 7.6 Hz, 1H), 7.21 (d, J = 7.6 Hz, 1H), 7.12 (s, 1H), 6.94 (d, J = 7.6 Hz, 1H), 4.64 (q, J = 6.8 Hz, 1H), 2.74 (s, 1H), 2.18 (s, 1H), 1.59 (d, J = 6.8 Hz, 3H), 1.44 (s, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 170.9, 157.9, 133.1, 129.9, 123.5 (q, 1JCF = 278.2 Hz), 120.9, 116.8, 114.6, 82.1, 72.8, 57.9 (q, 2JCF = 35.8 Hz), 27.9, 18.4. HRMS (ESI): m/z calculated for C15H19F3N2O3 + H+ [M + H+]: 333.1426. Found: 333.1421.
tert-Butyl (S)-2-(2-(3-(trifluoromethyl)diaziridin-3-yl)phenoxy)propanoate ((S)-54). The similar treatment of tert-butyl (S)-2-(2-(2,2,2-trifluoro-1-((tosyloxy)imino)ethyl)phenoxy)-propanoate (S)-51 (354 mg, 0.73 mmol) as that just described gave (S)-54 (182 mg, 76%). [α]D −17 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.54 (d, J = 7.6 Hz, 1H), 7.34 (t, J = 7.6 Hz, 1H), 7.01 (t, J = 7.6 Hz, 1H), 6.80 (d, J = 7.6 Hz, 1H), 4.79 (q, J = 6.8 Hz, 1H), 1.63 (d, J = 6.8 Hz, 3H), 1.36 (s, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 170.9, 156.4, 131.5, 130.9, 123.6 (q, 1JCF = 278.2 Hz), 121.1, 120.8, 111.9, 82.3, 72.7, 55.8 (q, 2JCF = 37.6 Hz), 27.6, 18.2. HRMS (ESI): m/z calculated for C15H19F3N2O3 + H+ [M + H+]: 333.1426. Found: 333.1425.
tert-Butyl (R)-2-(2-(3-(trifluoromethyl)diaziridin-3-yl)phenoxy)propanoate ((R)-54). The similar treatment of tert-butyl (R)-2-(2-(2,2,2-trifluoro-1-((tosyloxy)imino)ethyl)phenoxy)-propanoate (R)-51 (317 mg, 0.65 mmol) as that just described gave (R)-54 (173 mg, 80%). [α]D +17 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.54 (d, J = 7.6 Hz, 1H), 7.34 (t, J = 7.6 Hz, 1H), 7.01 (t, J = 7.6 Hz, 1H), 6.80 (d, J = 7.6 Hz, 1H), 4.79 (q, J = 6.8 Hz, 1H), 1.63 (d, J = 6.8 Hz, 3H), 1.36 (s, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 170.9, 156.4, 131.5, 130.9, 123.6 (q, 1JCF = 278.8 Hz), 121.1, 120.8, 111.9, 82.3, 72.7, 55.8 (q, 2JCF = 37.4 Hz), 27.6, 18.2. HRMS (ESI): m/z calculated for C15H19F3N2O3 + H+ [M + H+]: 333.1426. Found: 333.1415.
tert-Butyl (S)-2-(4-(3-(trifluoromethyl)-3H-diazirin-3-yl)phenoxy)propanoate ((S)-55).tert-Butyl (S)-2-(4-(3-(trifluoromethyl)diaziridin-3-yl)phenoxy)propanoate (S)-52 (151 mg, 0.45 mmol) was dissolved in CH2Cl2 (2 mL). MnO2 (197 mg, 2.27 mmol) was added to the solution, and the reaction mixture was stirred at room temperature for 2 h, followed by filtration and then concentrated. The residue was purified by column chromatography (ethyl acetate/n-hexane, 1:9) to give (S)-55 (136 mg, 91%). [α]D −35 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.12 (d, J = 8.6 Hz, 2H), 6.86 (d, J = 8.6 Hz, 2H), 4.62 (q, J = 6.8 Hz, 1H), 1.58 (d, J = 6.8 Hz, 3H), 1.43 (s, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 170.8, 158.8, 128.1, 122.2 (q, 1JCF = 274.9 Hz), 121.6, 115.3, 82.2, 72.9, 28.2 (q, 2JCF = 40.8 Hz), 27.9, 18.3. HRMS (ESI): m/z calculated for C15H17F3N2O3 + H+ [M + H+]: 331.1270. Found: 333.1261. Chiral HPLC (n-hexane/2-propanol 90:10): tR 15.4 min.
tert-Butyl (R)-2-(4-(3-(trifluoromethyl)-3H-diazirin-3-yl)phenoxy)propanoate ((R)-55). The similar treatment of tert-butyl (R)-2-(4-(3-(trifluoromethyl)diaziridin-3-yl)phenoxy)-propanoate (R)-52 (201 mg, 0.60 mmol) as that just described gave (R)-55 (184 mg, 92%). [α]D +35 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.12 (d, J = 8.6 Hz, 2H), 6.86 (d, J = 8.6 Hz, 2H), 4.61 (q, J = 6.8 Hz, 1H), 1.58 (d, J = 6.8 Hz, 3H), 1.43 (s, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 170.8, 158.8, 128.1, 122.2 (q, 1JCF = 274.9 Hz), 121.6, 115.3, 82.2, 72.9, 28.2 (q, 2JCF = 40.8 Hz), 27.9, 18.3. HRMS (ESI): m/z calculated for C15H17F3N2O3 + H+ [M + H+]: 331.1270. Found: 333.1260. Chiral HPLC (n-hexane/2-propanol 90:10): tR 17.1 min.
tert-Butyl (S)-2-(3-(3-(trifluoromethyl)-3H-diazirin-3-yl)phenoxy)propanoate ((S)-56). The similar treatment of tert-butyl (S)-2-(3-(3-(trifluoromethyl)diaziridin-3-yl)phenoxy)-propanoate (S)-53 (269 mg, 0.81 mmol) as that just described gave (S)-56 (220 mg, 82%). [α]D −38 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.3 (t, J = 7.6 Hz, 1H), 6.9 (d, J = 7.6 Hz, 1H), 6.8 (d, J = 7.6 Hz, 1H), 6.7 (s, 1H), 4.6 (q, J = 6.8 Hz, 1H), 1.6 (d, J = 6.8 Hz, 3H), 1.4 (s, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 170.8, 158.0, 130.5, 130.0, 122.1 (q, 1JCF = 274.9 Hz), 119.3, 116.3, 113.2, 82.3, 72.9, 28.3 (q, 2JCF = 40.8 Hz), 27.9, 18.3. HRMS (ESI): m/z calculated for C15H17F3N2O3 + H+ [M + H+]: 331.1270. Found: 333.1268. Chiral HPLC (n-hexane/2-propanol 90:10): tR 9.4 min.
tert-Butyl (R)-2-(3-(3-(trifluoromethyl)-3H-diazirin-3-yl)phenoxy)propanoate ((R)-56). The similar treatment of tert-butyl (R)-2-(3-(3-(trifluoromethyl)diaziridin-3-yl)phenoxy)-propanoate (R)-53 (347 mg, 1.05 mmol) as that just described gave (R)-56 (283 mg, 82%). [α]D +38 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.3 (t, J = 7.6 Hz, 1H), 6.9 (d, J = 7.6 Hz, 1H), 6.8 (d, J = 7.6 Hz, 1H), 6.7 (s, 1H), 4.6 (q, J = 6.8 Hz, 1H), 1.6 (d, J = 6.8 Hz, 3H), 1.4 (s, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 170.8, 158.0, 130.5, 130.0, 122.1 (q, 1JCF = 274.9 Hz), 119.3, 116.3, 113.2, 82.3, 72.9, 28.3 (q, 2JCF = 40.2 Hz), 27.9, 18.3. HRMS (ESI): m/z calculated for C15H17F3N2O3 + H+ [M + H+]: 331.1270. Found: 333.1263. Chiral HPLC (n-hexane/2-propanol 90:10): tR 8.8 min.
tert-Butyl (S)-2-(2-(3-(trifluoromethyl)-3H-diazirin-3-yl)phenoxy)propanoate ((S)-57). The similar treatment of tert-butyl (S)-2-(2-(3-(trifluoromethyl)diaziridin-3-yl)phenoxy)-propanoate (S)-54 (105 mg, 0.32 mmol) as that just described gave (S)-57 (93.4 mg, 89%). [α]D −10 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.37 (d, J = 7.6 Hz, 1H), 7.24 (t, J = 7.6 Hz, 1H), 6.87 (t, J = 7.6 Hz, 1H), 6.66 (d, J = 7.6 Hz, 1H), 4.60 (q, J = 6.8 Hz, 1H), 1.61 (d, J = 6.8 Hz, 3H), 1.30 (s, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 170.7, 158.0, 131.7, 131.1, 122.1 (q, 1JCF = 274.9 Hz), 121.3, 117.1, 112.2, 82.1, 73.3, 27.8, 26.4 (q, 2JCF = 43.0 Hz), 18.2. HRMS (ESI): m/z calculated for C15H17F3N2O3 + H+ [M + H+]: 331.1270. Found: 333.1285. Chiral HPLC (n-hexane/2-propanol 90:10): tR 9.9 min.
tert-Butyl (R)-2-(2-(3-(trifluoromethyl)-3H-diazirin-3-yl)phenoxy)propanoate ((R)-57). The similar treatment of tert-butyl (R)-2-(2-(3-(trifluoromethyl)diaziridin-3-yl)phenoxy)-propanoate (R)-54 (85.8 mg, 0.26 mmol) as that just described gave (R)-57 (81.7 mg, 96%). [α]D +10 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.45 (d, J = 7.6 Hz, 1H), 7.33 (t, J = 7.6 Hz, 1H), 6.96 (t, J = 7.6 Hz, 1H), 6.74 (d, J = 7.6 Hz, 1H), 4.68 (q, J = 6.8 Hz, 1H), 1.69 (d, J = 6.8 Hz, 3H), 1.39 (s, 9H). 13C-NMR (67.5 MHz, CDCl3): δ = 170.7, 158.0, 131.7, 131.1, 122.1 (q, 1JCF = 274.9 Hz), 121.3, 117.1, 112.2, 82.1, 73.2, 27.8, 26.4 (q, 2JCF = 43.0 Hz), 18.2. HRMS (ESI): m/z calculated for C15H17F3N2O3 + H+ [M + H+]: 331.1270. Found: 333.1276. Chiral HPLC (n-hexane/2-propanol 90:10): tR 10.3 min.
(S)-2-(4-(3-(Trifluoromethyl)-3H-diazirin-3-yl)phenoxy)propanoic acid ((S)-58).tert-Butyl (S)-2-(4-(3-(trifluoromethyl)-3H-diazirin-3-yl)phenoxy)propanoate (S)-55 (165 mg, 0.50 mmol) was dissolved in CH2Cl2 (1 mL) and then trifluoroacetic acid (2 mL) was added to the solution. After the reaction mixture was stirred at room temperature for 2 h, the reaction mixture was poured into the cold water and extracted by CH2Cl2. The organic layer was dried over MgSO4, filtrated and concentrated to give (S)-58 (135 mg, 98%). [α]D −20 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 11.33 (s, 1H), 7.14 (d, J = 8.6 Hz, 2H), 6.89 (d, J = 8.6 Hz, 2H), 4.79 (q, J = 6.8 Hz, 1H), 1.66 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 177.8, 158.2, 128.3, 122.3, 122.2 (q, 1JCF = 274.3 Hz), 115.4, 71.9, 28.1 (q, 2JCF = 40.8 Hz), 18.3. HRMS (ESI): m/z calculated for C11H9F3N2O3 + H+ [M + H+]: 275.0644. Found: 275.0648.
(R)-2-(4-(3-(Trifluoromethyl)-3H-diazirin-3-yl)phenoxy)propanoic acid ((R)-58). The similar treatment of tert-butyl (R)-2-(4-(3-(trifluoromethyl)-3H-diazirin-3-yl)phenoxy)-propanoate (R)-55 (201 mg, 0.61 mmol) as that just described gave (R)-58 (184 mg, 92%). [α]D +20 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 10.28 (s, 1H), 7.15 (d, J = 8.6 Hz, 2H), 6.89 (d, J = 8.6 Hz, 2H), 4.79 (q, J = 6.8 Hz, 1H), 1.67 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 177.4, 158.2, 128.3, 122.3, 122.2 (q, 1JCF = 274.3 Hz), 115.4, 72.0, 28.1 (q, 2JCF = 40.8 Hz), 18.3. HRMS (ESI): m/z calculated for C11H9F3N2O3 + H+ [M + H+]: 275.0644. Found: 275.0614.
(S)-2-(3-(3-(Trifluoromethyl)-3H-diazirin-3-yl)phenoxy)propanoic acid ((S)-59). The similar treatment of tert-butyl (S)-2-(3-(3-(trifluoromethyl)-3H-diazirin-3-yl)phenoxy)-propanoate (S)-56 (220 mg, 0.67 mmol) as that just described gave (S)-59 (209 mg, quant). [α]D −13 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 11.47 (s, 1H), 7.29 (t, J = 7.8 Hz, 1H), 6.89 (d, J = 7.8 Hz, 1H), 6.79 (d, J = 7.8 Hz, 1H), 6.74 (s, 1H), 4.78 (q, J = 6.8 Hz, 1H), 1.66 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 178.0, 157.5, 130.9, 130.2, 122.0 (q, 1JCF = 274.7 Hz), 119.8, 115.7, 114.0, 72.1, 28.3 (q, 2JCF = 39.9 Hz), 18.2. HRMS (ESI): m/z calculated for C11H9F3N2O3 + H+ [M + H+]: 275.0644. Found: 275.0661.
(R)-2-(3-(3-(Trifluoromethyl)-3H-diazirin-3-yl)phenoxy)propanoic acid ((R)-59). The similar treatment of tert-butyl (R)-2-(3-(3-(trifluoromethyl)-3H-diazirin-3-yl)phenoxy)-propanoate (R)-56 (283 mg, 0.86 mmol) as that just described gave (R)-59 (208 mg, 89%). [α]D +13 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 11.32 (s, 1H), 7.30 (t, J = 8 Hz, 1H), 6.90 (d, J = 7.8 Hz, 1H), 6.80 (d, J = 7.8 Hz, 1H), 6.74 (s, 1H), 4.78 (q, J = 6.8 Hz, 1H), 1.66 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 177.9, 157.5, 130.9, 130.2, 122.0 (q, 1JCF = 274.5 Hz), 119.8, 115.7, 114.0, 72.1, 28.3 (q, 2JCF = 39.7 Hz), 18.2. HRMS (ESI): m/z calculated for C11H9F3N2O3 + H+ [M + H+]: 275.0644. Found: 275.0627.
(S)-2-(2-(3-(Trifluoromethyl)-3H-diazirin-3-yl)phenoxy)propanoic acid ((S)-60). The similar treatment of tert-butyl (S)-2-(2-(3-(trifluoromethyl)-3H-diazirin-3-yl)phenoxy)-propanoate (S)-57 (95.4 mg, 0.29 mmol) as that just described gave (S)-60 (89.0 mg, quant). [α]D −10 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.49 (d, J = 7.6 Hz, 1H), 7.37 (t, J = 7.6 Hz, 1H), 7.02 (t, J = 7.6 Hz, 1H), 6.78 (d, J = 7.6 Hz, 1H), 4.86 (q, J = 6.8 Hz, 1H), 1.78 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 177.1, 157.5, 132.0, 131.4 122.0 (q, 1JCF = 275.4 Hz), 122.0, 117.4, 112.4, 72.3, 26.3 (q, 2JCF = 42.5 Hz), 18.2. HRMS (ESI): m/z calculated for C11H9F3N2O3 + H+ [M + H+]: 275.0644. Found: 275.0663.
(R)-2-(2-(3-(Trifluoromethyl)-3H-diazirin-3-yl)phenoxy)propanoic acid ((R)-60). The similar treatment of tert-butyl (R)-2-(2-(3-(trifluoromethyl)-3H-diazirin-3-yl)phenoxy)-propanoate (R)-57 (166 mg, 0.50 mmol) as that just described gave (R)-60 (151 mg, quant). [α]D +10 (c 1, CHCl3). 1H-NMR (270 MHz, CDCl3): δ = 7.50 (d, J = 7.6 Hz, 1H), 7.38 (t, J = 7.6 Hz, 1H), 7.03 (t, J = 7.6 Hz, 1H), 6.80 (d, J = 7.6 Hz, 1H), 4.88 (q, J = 6.8 Hz, 1H), 1.77 (d, J = 6.8 Hz, 3H). 13C-NMR (67.5 MHz, CDCl3): δ = 176.6, 157.4, 132.0, 131.3, 122.0, 122.0 (q, 1JCF = 274.9 Hz), 117.3, 112.3, 72.2, 26.3 (q, 2JCF = 42.5 Hz), 18.1. HRMS (ESI): m/z calculated for C11H9F3N2O3 + H+ [M + H+]: 275.0644. Found: 275.0654.

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}