
Valorization of Lignin via Oxidative Depolymerization with Hydrogen Peroxide: Towards Carboxyl-Rich Oligomeric Lignin Fragments

 ,
,
Abstract
1. Introduction
2. Results and Discussion
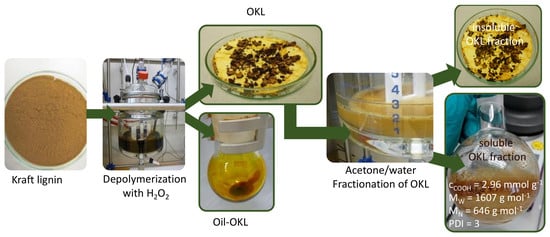
2.1. Oxidative Depolymerization of Kraft Lignin at a Laboratory Scale
2.1.1. Influence of Reaction Parameters on Kraft Lignin Structure
2.1.2. Influence of Reaction Parameters on Molecular Weight of Kraft Lignin
2.2. OKL Solvent Fractionation at a Laboratory Scale
2.3. Oxidative Depolymerization of Kraft Lignin and OKL Solvent Fractionation at a 3 L Scale
2.3.1. Preliminary Studies on Application of Stabilized H2O2 at a Laboratory Scale
2.3.2. Transition of Laboratory-Scale Oxidation and Solvent Fractionation to 3 L Scale
3. Materials and Methods
3.1. Materials
3.2. Oxidative Depolymerization of Kraft Lignin with Hydrogen Peroxide
3.3. Solvent Fractionation of Oligomeric Depolymerization Products
3.4. Analytical Characterization of Kraft Lignin and Reaction Products
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cao, L.; Yu, I.K.M.; Liu, Y.; Ruan, X.; Tsang, D.C.W.; Hunt, A.J.; Ok, Y.S.; Song, H.; Zhang, S. Lignin valorization for the production of renewable chemicals: State-of-the-art review and future prospects. Bioresour. Technol. 2018, 269, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Zhang, Q.; Lee, D.-J. Kraft lignin biorefinery: A perspective. Bioresour. Technol. 2018, 247, 1181–1183. [Google Scholar] [CrossRef] [PubMed]
- Sangchoom, W.; Mokaya, R. Valorization of Lignin Waste: Carbons from Hydrothermal Carbonization of Renewable Lignin as Superior Sorbents for CO2 and Hydrogen Storage. ACS Sustain. Chem. Eng. 2015, 3, 1658–1667. [Google Scholar] [CrossRef]
- Tuck, C.O.; Pérez, E.; Horváth, I.T.; Sheldon, R.A.; Poliakoff, M. Valorization of biomass: Deriving more value from waste. Science 2012, 337, 695–699. [Google Scholar] [CrossRef]
- Chatel, G.; Rogers, R.D. Review: Oxidation of Lignin Using Ionic Liquids—An Innovative Strategy To Produce Renewable Chemicals. ACS Sustain. Chem. Eng. 2014, 2, 322–339. [Google Scholar] [CrossRef]
- Tomani, P. The lignoboost process. Cellul. Chem. Technol. 2010, 44, 53–58. [Google Scholar]
- Huang, S.; Mahmood, N.; Tymchyshyn, M.; Yuan, Z.; Xu, C.C. Reductive de-polymerization of kraft lignin for chemicals and fuels using formic acid as an in-situ hydrogen source. Bioresour. Technol. 2014, 171, 95–102. [Google Scholar] [CrossRef]
- Duval, A.; Lawoko, M. A review on lignin-based polymeric, micro- and nano-structured materials. React. Funct. Polym. 2014, 85, 78–96. [Google Scholar] [CrossRef]
- Vanholme, R.; Demedts, B.; Morreel, K.; Ralph, J.; Boerjan, W. Lignin Biosynthesis and Structure. Plant Physiol. 2010, 153, 895–905. [Google Scholar] [CrossRef]
- Chatterjee, S.; Saito, T. Lignin-Derived Advanced Carbon Materials. ChemSusChem 2015, 8, 3941–3958. [Google Scholar] [CrossRef]
- Gellerstedt, G. Softwood kraft lignin: Raw material for the future. Ind. Crops Prod. 2015, 77, 845–854. [Google Scholar] [CrossRef]
- Jääskeläinen, A.S.; Liitiä, T.; Mikkelson, A. Aqueous organic solvent fractionation as means to improve lignin homogeneity and purity. Ind. Crops Prod. 2017, 103, 51–58. [Google Scholar] [CrossRef]
- Rößiger, B.; Röver, R.; Unkelbach, G.; Pufky-Heinrich, D. Production of Bio-Phenols for Industrial Application: Scale-Up of the Base-Catalyzed Depolymerization of Lignin. Green Sustain. Chem. 2017, 7, 10. [Google Scholar] [CrossRef]
- Azadi, P.; Inderwildi, O.R.; Farnood, R.; King, D.A. Liquid fuels, hydrogen and chemicals from lignin: A critical review. Renew. Sustain. Energy Rev. 2013, 21, 506–523. [Google Scholar] [CrossRef]
- Pandey, M.P.; Kim, C.S. Lignin Depolymerization and Conversion: A Review of Thermochemical Methods. Chem. Eng. Technol. 2011, 34, 29–41. [Google Scholar] [CrossRef]
- Wang, H.; Tucker, M.; Ji, Y. Recent Development in Chemical Depolymerization of Lignin: A Review. J. Appl. Chem. 2013, 2013, 9. [Google Scholar] [CrossRef]
- Lange, H.; Decina, S.; Crestini, C. Oxidative upgrade of lignin—Recent routes reviewed. Eur. Polym. J. 2013, 49, 1151–1173. [Google Scholar] [CrossRef]
- Vangeel, T.; Schutyser, W.; Renders, T. Perspective on Lignin Oxidation: Advances, Challenges, and Future Directions. Top. Curr. Chem. 2018, 376, 30. [Google Scholar] [CrossRef]
- Li, C.; Zhao, X.; Wang, A.; Huber, G.W.; Zhang, T. Catalytic transformation of lignin for the production of chemicals and fuels. Chem. Rev. 2015, 115, 11559–11624. [Google Scholar] [CrossRef]
- Xu, C.; Arancon, R.A.D.; Labidi, J.; Luque, R. Lignin depolymerisation strategies: Towards valuable chemicals and fuels. Chem. Soc. Rev. 2014, 43, 7485–7500. [Google Scholar] [CrossRef]
- Dos Santos, P.S.B.; Erdocia, X.; Gatto, D.A.; Labidi, J. Bio-oil from base-catalyzed depolymerization of organosolv lignin as an antifungal agent for wood. Wood Sci. Technol. 2016, 50, 599–615. [Google Scholar] [CrossRef]
- Mahmood, N.; Yuan, Z.; Schmidt, J.; Xu, C. Depolymerization of lignins and their applications for the preparation of polyols and rigid polyurethane foams: A review. Renew. Sustain. Energy Rev. 2016, 60, 317–329. [Google Scholar] [CrossRef]
- He, W.; Gao, W.; Fatehi, P. Oxidation of Kraft Lignin with Hydrogen Peroxide and its Application as a Dispersant for Kaolin Suspensions. ACS Sustain. Chem. Eng. 2017, 5, 10597–10605. [Google Scholar] [CrossRef]
- Napoly, F.; Kardos, N.; Jean-Gérard, L.; Goux-Henry, C.; Andrioletti, B.; Draye, M. H2O2-Mediated Kraft Lignin Oxidation with Readily Available Metal Salts: What about the Effect of Ultrasound? Ind. Eng. Chem. Res. 2015, 54, 6046–6051. [Google Scholar] [CrossRef]
- Rößiger, B.; Unkelbach, G.; Pufky-Heinrich, D. Base-Catalyzed Depolymerization of Lignin: History, Challenges and Perspectives. In Lignin-Trends and Applications; Poletto, M., Ed.; IntechOpen: London, UK, 2018; pp. 99–120. [Google Scholar] [CrossRef]
- Heitner, C. Lignin and Lignans Advances in Chemistry; CRC Press: Boca Raton, FL, USA, 2010; ISBN 978-1-57444-486-5. [Google Scholar]
- Zeronian, S.H.; Inglesby, M.K. Bleaching of cellulose by hydrogen peroxide. Cellulose 1995, 2, 265–272. [Google Scholar] [CrossRef]
- Ma, R.; Guo, M.; Zhang, X. Recent advances in oxidative valorization of lignin. Catal. Today 2018, 302, 50–60. [Google Scholar] [CrossRef]
- Liu, C.; Wu, S.; Zhang, H.; Xiao, R. Catalytic oxidation of lignin to valuable biomass-based platform chemicals: A review. Fuel Process. Technol. 2019, 191, 181–201. [Google Scholar] [CrossRef]
- Qin, J.; Woloctt, M.; Zhang, J. Use of Polycarboxylic Acid Derived from Partially Depolymerized Lignin as a Curing Agent for Epoxy Application. ACS Sustain. Chem. Eng. 2014, 2, 188–193. [Google Scholar] [CrossRef]
- Liu, W.; Zhou, R.; Goh, H.L.S.; Huang, S.; Lu, X. From Waste to Functional Additive: Toughening Epoxy Resin with Lignin. Appl. Mater. Interfaces ACS 2014, 6, 5810–5817. [Google Scholar] [CrossRef]
- Li, Z.; Ge, Y. Application of Lignin and Its Derivatives in Adsorption of Heavy Metal Ions in Water: A Review. ACS Sustain. Chem. Eng. 2018, 6, 7181–7192. [Google Scholar] [CrossRef]
- Quintana, G.C.; Rocha, G.J.M.; Goncalves, A.R.; Velasquez, J.A. Evaluation of heavy metal removal by oxidised lignins in acid media from various sources. BioResources 2008, 3, 1092–1102. [Google Scholar] [CrossRef]
- Cui, C.; Sun, R.; Argyropoulos, D.S. Fractional Precipitation of Softwood Kraft Lignin: Isolation of Narrow Fractions Common to a Variety of Lignins. ACS Sustain. Chem. Eng. 2014, 2, 959–968. [Google Scholar] [CrossRef]
- Duval, A.; Vilaplana, F.; Crestini, C.; Lawoko, M. Solvent screening for the fractionation of industrial kraft lignin. Holzforschung 2016, 70, 11–20. [Google Scholar] [CrossRef]
- Ropponen, J.; Räsänen, L.; Rovio, S. Solvent extraction as a means of preparing homogeneous lignin fractions. Holzforschung 2011, 65, 543–549. [Google Scholar] [CrossRef]
- Ajao, O.; Jeaidi, J.; Benali, M.; Abdelaziz, O.Y.; Hulteberg, C.P. Green solvents-based fractionation process for kraft lignin with controlled dispersity and molecular weight. Bioresour. Technol. 2019, 291, 121799. [Google Scholar] [CrossRef]
- Boeriu, C.G.; Fiţigău, F.I.; Gosselink, R.J.A.; Frissen, A.E.; Stoutjesdijk, J.; Peter, F. Fractionation of five technical lignins by selective extraction in green solvents and characterisation of isolated fractions. Ind. Crops Prod. 2014, 62, 481–490. [Google Scholar] [CrossRef]
- Liang, S.; Wan, C. Biorefinery Lignin to Renewable Chemicals via Sequential Fractionation and Depolymerization. Waste Biomass Valorization 2017, 8, 393–400. [Google Scholar] [CrossRef]
- Legrini, O.; Oliveros, E.; Braun, A.M. Photochemical processes for water treatment. Chem. Rev. 1993, 93, 671–698. [Google Scholar] [CrossRef]
- Kadla, J.F.; Chang, H.-M.; Jameel, H. The Reactions of Lignins with Hydrogen Peroxide at High Temperature. Part I. The Oxidation of Lignin Model Compounds. Holzforschung 1997, 5, 428. [Google Scholar] [CrossRef]
- Kadla, J.F.; Chang, H.M. The Reactions of Peroxides with Lignin and Lignin Model Compounds. In Oxidative Delignification Chemistry; American Chemical Society: Washington, DC, USA, 2001; pp. 108–129. ISBN 0-8412-3738-7. [Google Scholar]
- Faix, O. Fourier Transform Infrared Spectroscopy. In Methods in Lignin Chemistry, 1st ed.; Lin, S.Y., Dence, C.W., Eds.; Springer: Berlin/Heidelberg, Germany, 1992; pp. 83–109. ISBN 3642740650. [Google Scholar]
- Boeriu, C.G.; Bravo, D.; Gosselink, R.J.A. Characterisation of structure-dependent functional properties of lignin with infrared spectroscopy. Ind. Crops Prod. 2004, 20, 205–218. [Google Scholar] [CrossRef]
- Hesse, M.; Meier, H.; Zeeh, B. Spektroskopische Methoden in der Organischen Chemie; Thieme: Stuttgart, NY, USA, 2005; ISBN 978-3135761077. [Google Scholar]
- Kozhevnikov, A.; Semushina, M.; Podrukhina, E. Modification of Hydrolysis Lignin by Hydrogen Peroxide to Obtain an Effective Adsorbent of Highly Toxic Rocket Fuel. Eurasian Chem.-Technol. J. 2017, 19, 155. [Google Scholar] [CrossRef]
- Kadla, J.F.; Chang, H.-M.; Jameel, H. The Reactions of Lignins with High Temperature Hydrogen Peroxide Part 2. The Oxidation of Kraft Lignin. Holzforschung 1999, 53, 277–284. [Google Scholar] [CrossRef]
- Beauchet, R.; Monteil-Rivera, F.; Lavoie, J.M. Conversion of lignin to aromatic-based chemicals (L-chems) and biofuels (L-fuels). Bioresour. Technol. 2012, 121, 328–334. [Google Scholar] [CrossRef]
- Hildebrand, J.H.; Scott, R.L. The Solubility of Nonelectrolytes; Reinhold Pub. Corp.: New York, NY, USA, 1950. [Google Scholar]
- Sarkanen, S.; Teller, D.C.; Stevens, C.R.; McCarthy, J.L. Lignin. 20. Associative Interactions between Kraft Lignin Components. Macromolecules 1984, 17, 2588–2597. [Google Scholar] [CrossRef]
- Upton, B.M.; Kasko, A.M. Strategies for the Conversion of Lignin to High-Value Polymeric Materials: Review and Perspective. Chem. Rev. 2016, 116, 2275–2306. [Google Scholar] [CrossRef]
- Jawerth, M.; Johansson, M.; Lundmark, S.; Gioia, C.; Lawoko, M. Renewable Thiol–Ene Thermosets Based on Refined and Selectively Allylated Industrial Lignin. ACS Sustain. Chem. Eng. 2017, 5, 10918–10925. [Google Scholar] [CrossRef]
- Gioia, C.; Lo Re, G.; Lawoko, M.; Berglund, L. Tunable Thermosetting Epoxies Based on Fractionated and Well-Characterized Lignins. J. Am. Chem. Soc. 2018, 140, 4054–4061. [Google Scholar] [CrossRef]
- Semke, H. Charakterisierung, Funktionalisierung und Verarbeitung von Ligninen aus Dikotyledonen. Ph.D. Thesis, Albert-Ludwigs-Universität Freiburg, Freiburg im Breisgau, Germany, 2001. [Google Scholar]
- Xie, S.; Li, Q.; Karki, P.; Zhou, F.; Yuan, J.S. Lignin as Renewable and Superior Asphalt Binder Modifier. ACS Sustain. Chem. Eng. 2017, 5, 2817–2823. [Google Scholar] [CrossRef]
- Glasser, W.G.; Northey, R.A.; Schultz, T.P. Lignin: Historical, Biological, and Materials Perspectives; ACS Publications: Washington, DC, USA, 1999; ISBN 0841236119. [Google Scholar]
- Sluiter, A.; Hames, B.; Ruiz, R.; Scarlata, C.; Sluiter, J.; Templeton, D. Tetermination of Ash in Biomass NREL/TP-510-42622; National Renewable Energy Laboratory: Golden, CO, USA, 2008. [Google Scholar]
- Dence, C.W. Determination of Carboxyl Groups. In Methods in Lignin Chemistry; Lin, S.Y., Dence, C.W., Eds.; Springer: Berlin/Heidelberg, Germany, 1992; pp. 458–464. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample/Treatment/Scale | MN (g mol−1) | MW (g mol−1) | PDI | Y (%) |
|---|---|---|---|---|
| Kraft lignin | 1177 ± 153 | 6920 ± 216 | 5.9 ± 0.6 | n.a.a |
| H2O2lab scale | 1362 ± 62 | 5170 ± 365 | 3.7 ± 0.1 | 69 ± 9 |
| H2O2stabilized, lab scale | 801 ± 153 | 2802 ± 216 | 3.5 ± 0.6 | 47 ± 14 |
| H2O2stabilized, 3 L scale | 1058 ± 121 | 4625 ± 322 | 4.4 ± 0.3 | 73 ± 3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Junghans, U.; Bernhardt, J.J.; Wollnik, R.; Triebert, D.; Unkelbach, G.; Pufky-Heinrich, D. Valorization of Lignin via Oxidative Depolymerization with Hydrogen Peroxide: Towards Carboxyl-Rich Oligomeric Lignin Fragments. Molecules 2020, 25, 2717. https://doi.org/10.3390/molecules25112717
Junghans U, Bernhardt JJ, Wollnik R, Triebert D, Unkelbach G, Pufky-Heinrich D. Valorization of Lignin via Oxidative Depolymerization with Hydrogen Peroxide: Towards Carboxyl-Rich Oligomeric Lignin Fragments. Molecules. 2020; 25(11):2717. https://doi.org/10.3390/molecules25112717
Chicago/Turabian StyleJunghans, Ulrike, Justin J. Bernhardt, Ronja Wollnik, Dominik Triebert, Gerd Unkelbach, and Daniela Pufky-Heinrich. 2020. "Valorization of Lignin via Oxidative Depolymerization with Hydrogen Peroxide: Towards Carboxyl-Rich Oligomeric Lignin Fragments" Molecules 25, no. 11: 2717. https://doi.org/10.3390/molecules25112717
APA StyleJunghans, U., Bernhardt, J. J., Wollnik, R., Triebert, D., Unkelbach, G., & Pufky-Heinrich, D. (2020). Valorization of Lignin via Oxidative Depolymerization with Hydrogen Peroxide: Towards Carboxyl-Rich Oligomeric Lignin Fragments. Molecules, 25(11), 2717. https://doi.org/10.3390/molecules25112717