Cryptococcus neoformans Capsular GXM Conformation and Epitope Presentation: A Molecular Modelling Study



Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
- What is the secondary structure or conformation of GXM?
- Does the differing xylose substitution pattern in serotypes A and D alter the GXM conformation?
- Does 6-O-acetylation on the mannose backbone alter the conformation of serotype A?
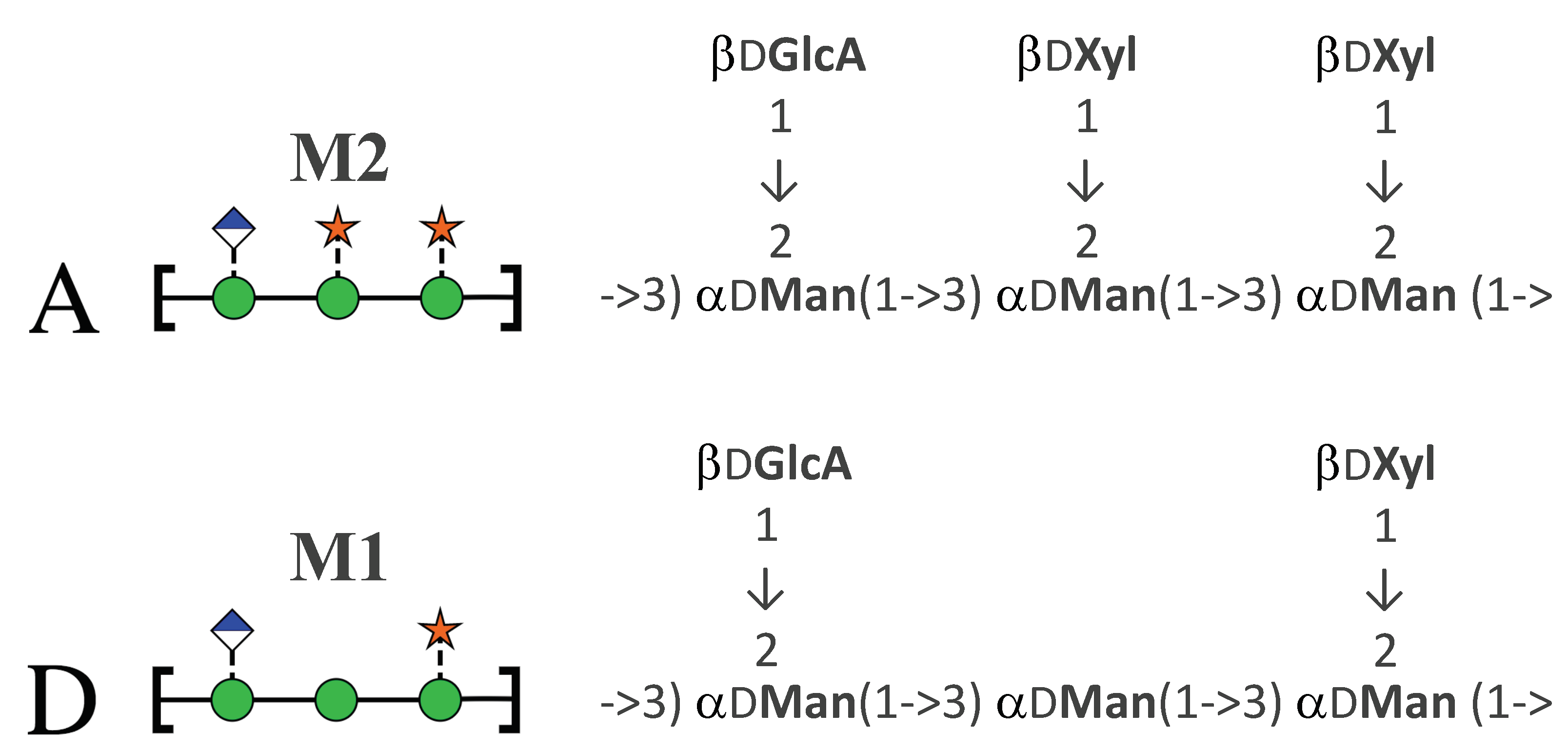
- Do shorter strands with the M2 triad have the same conformational epitope as the GXM serotype A polysaccharide?
- What is mechanism of GXM self-aggregation?
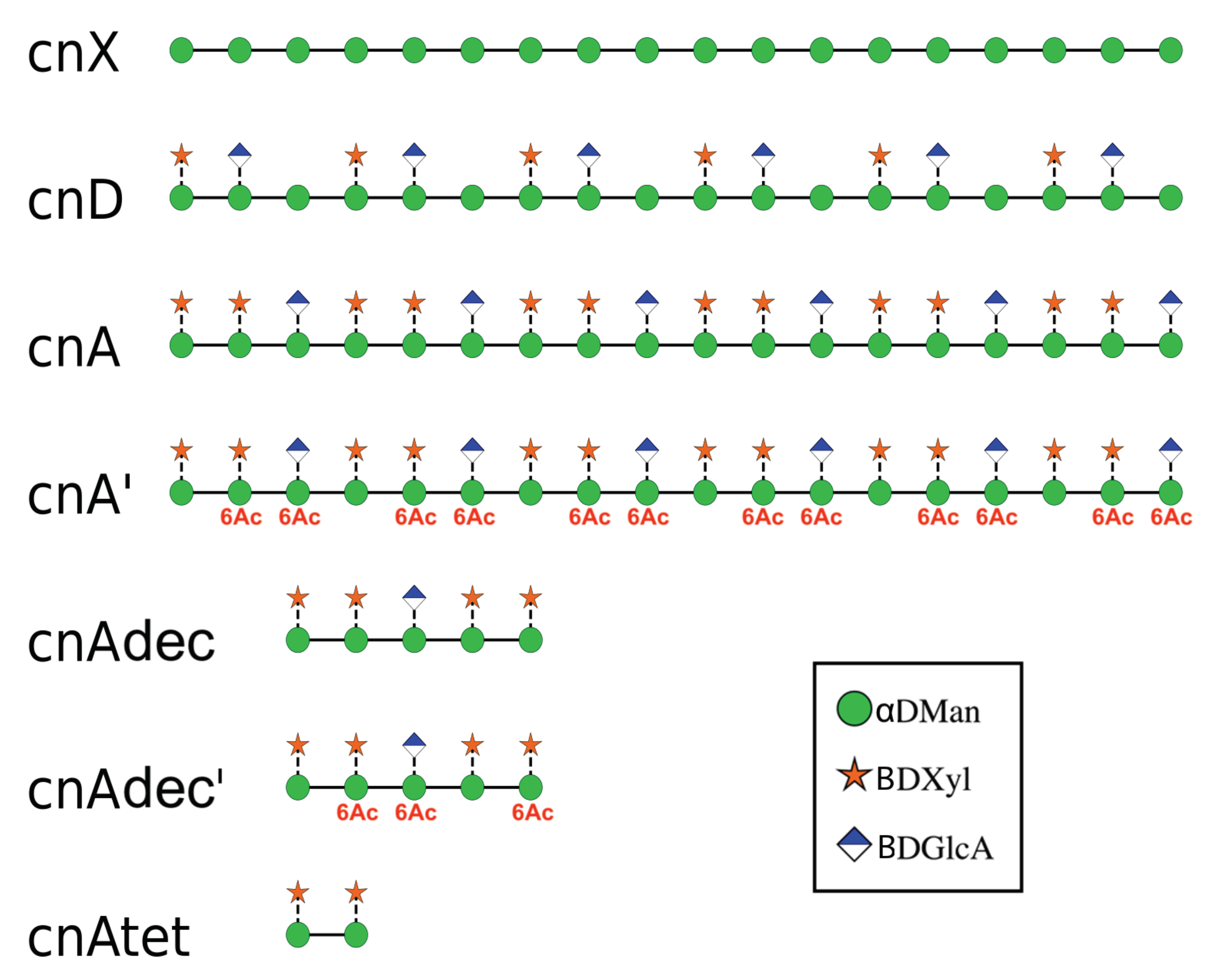
2. Results
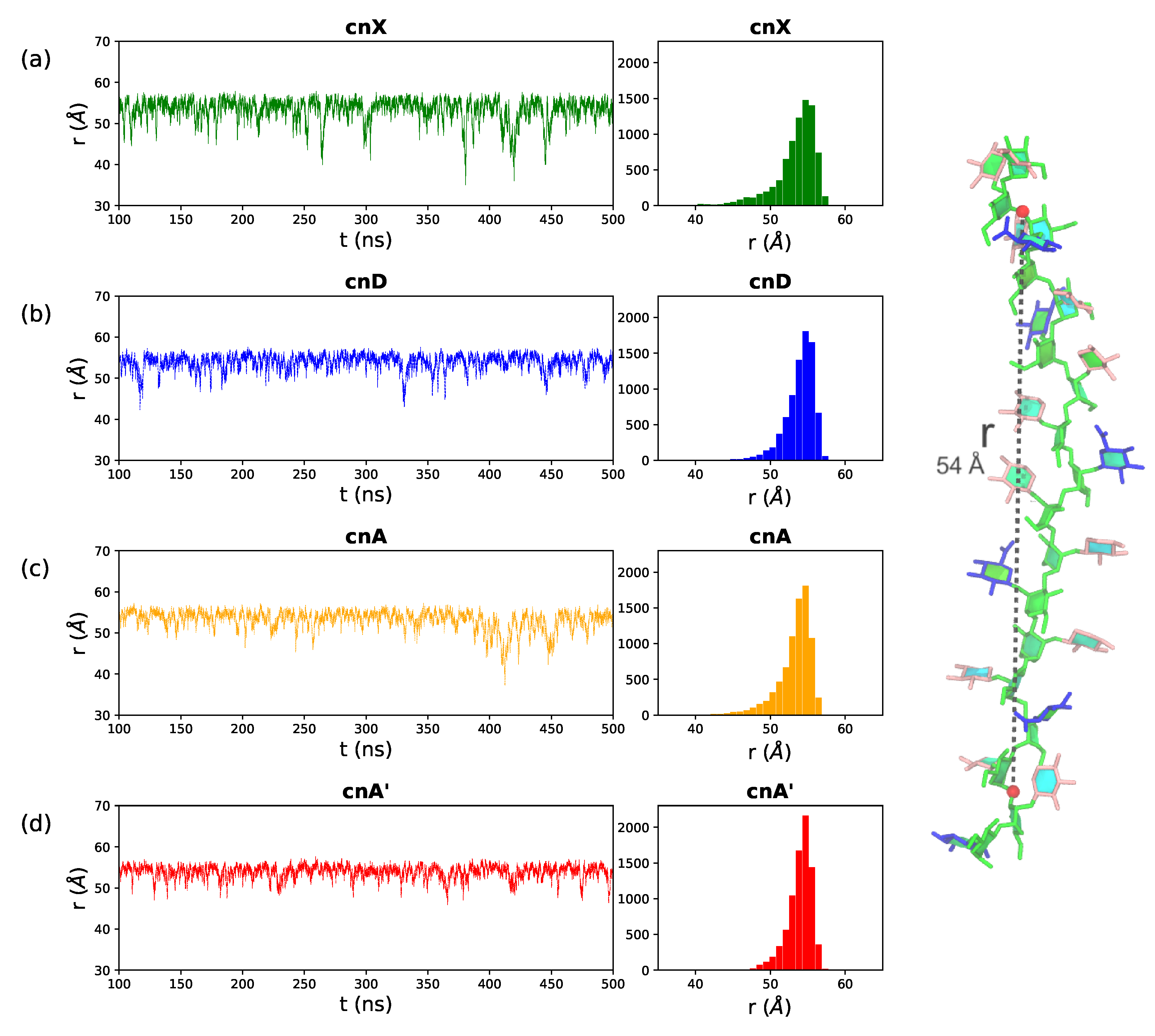
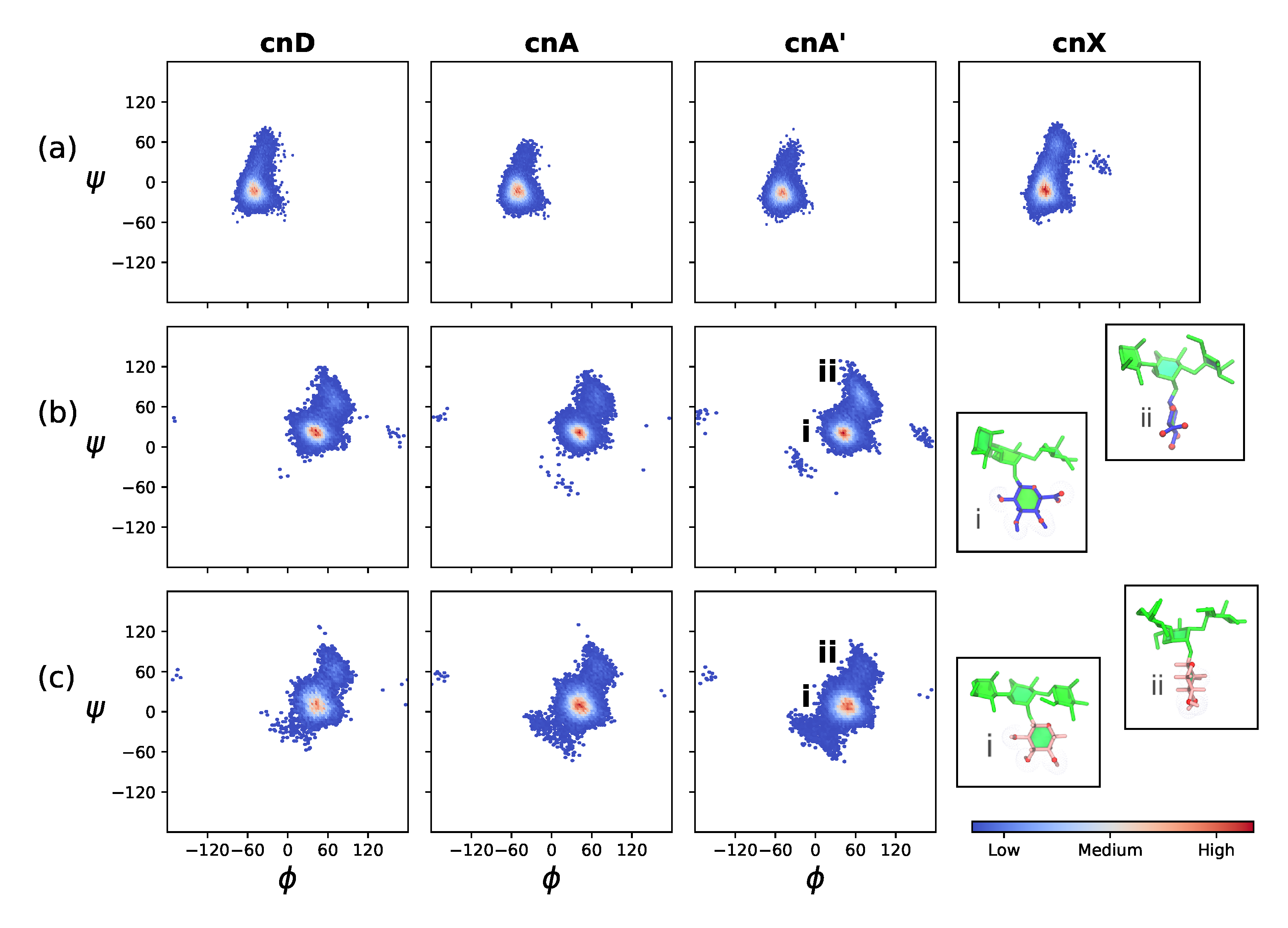
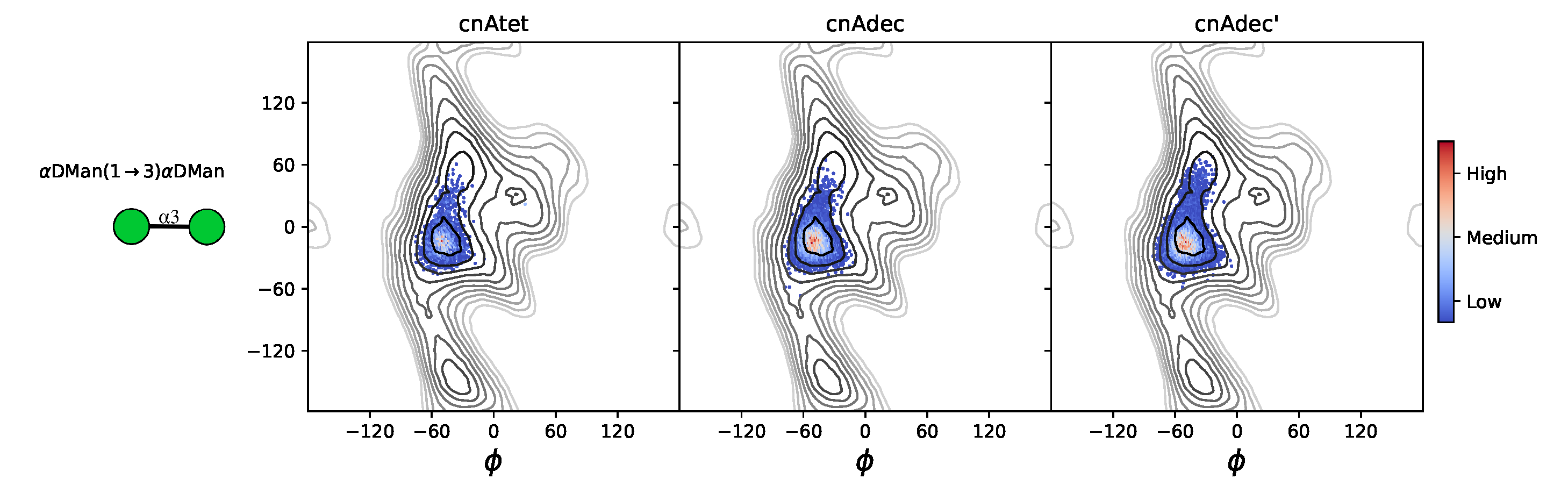
2.1. Comparison of GXM Chain Flexibility
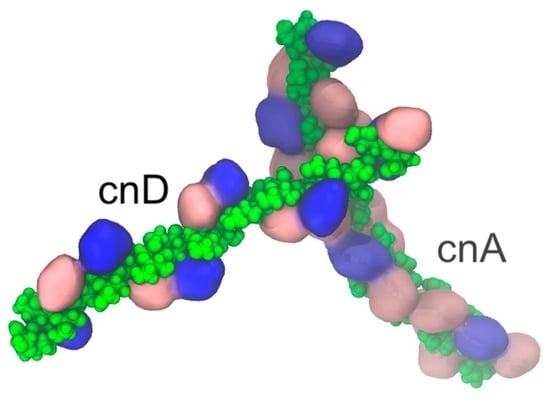
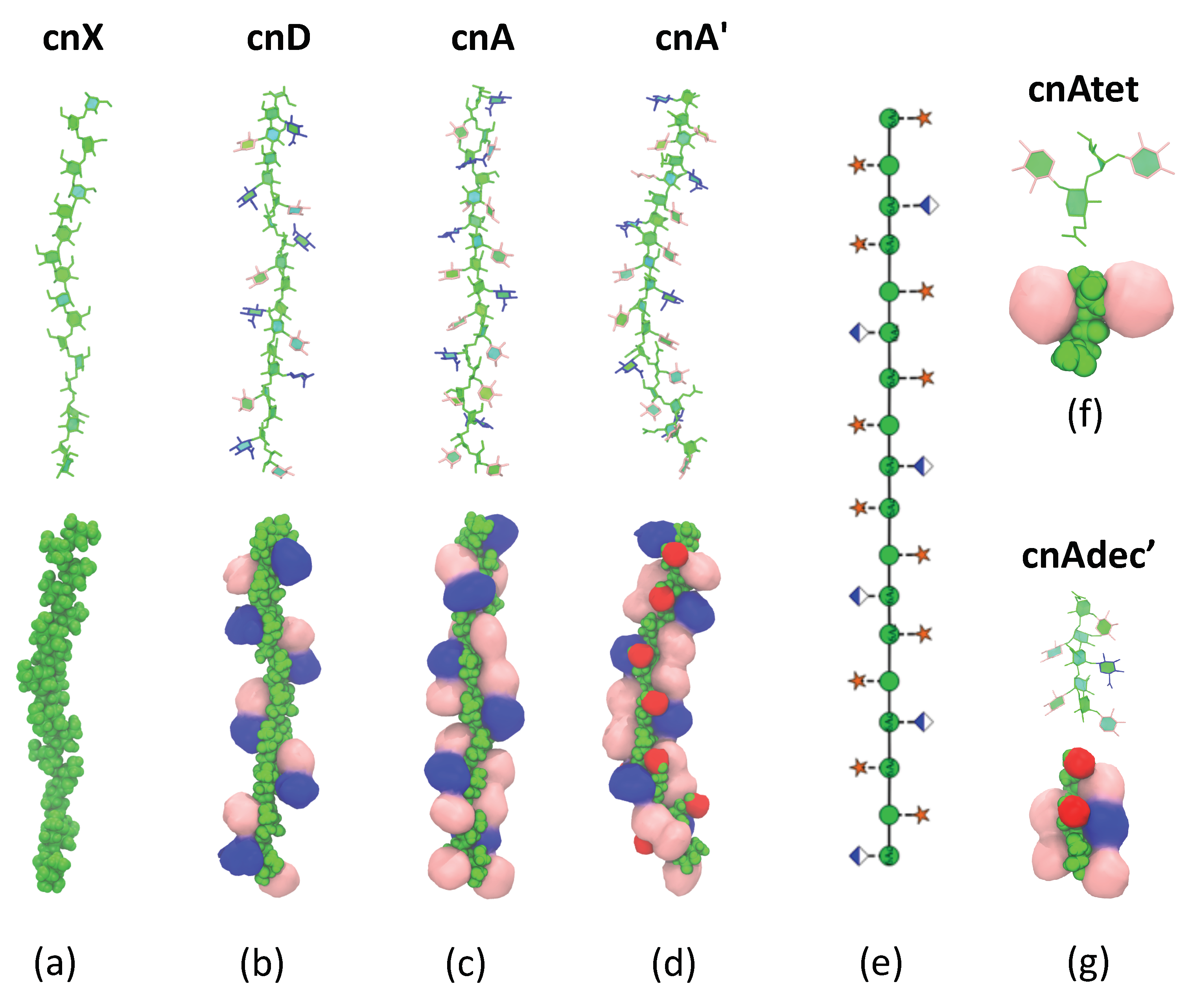
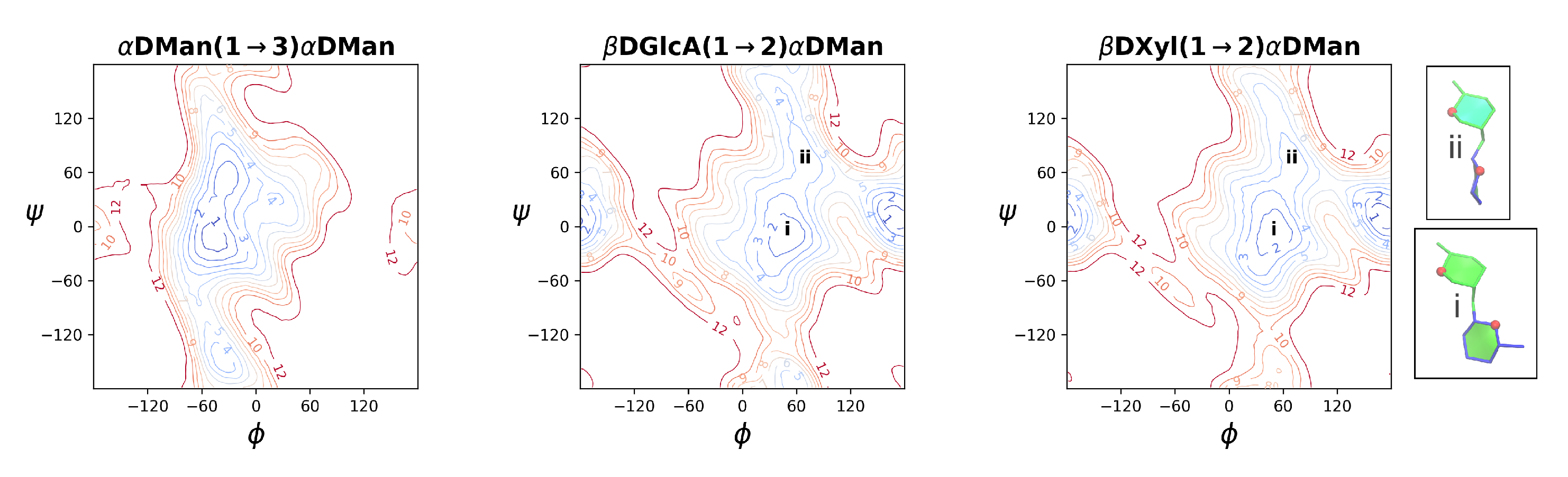
2.2. Comparison of GXM Chain Conformations and Binding Surfaces
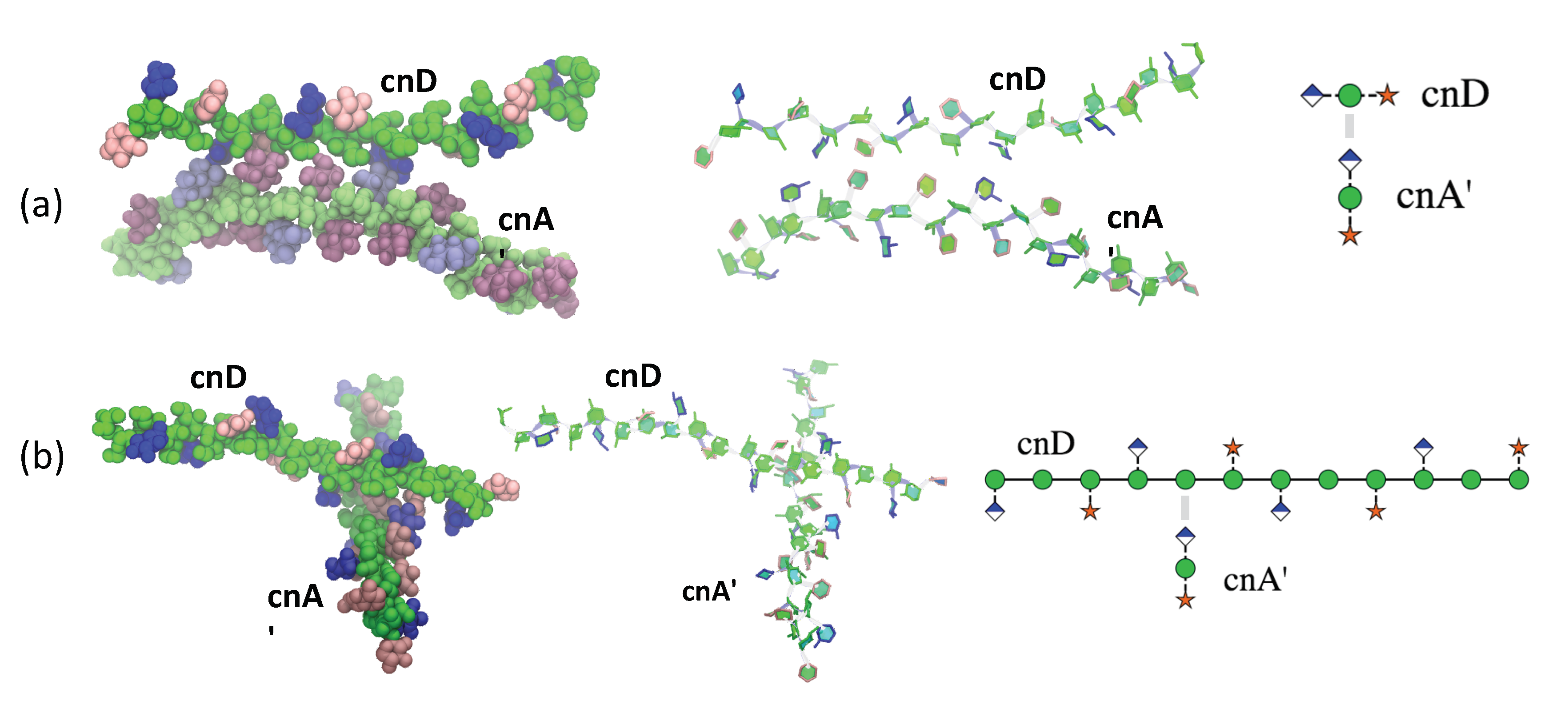
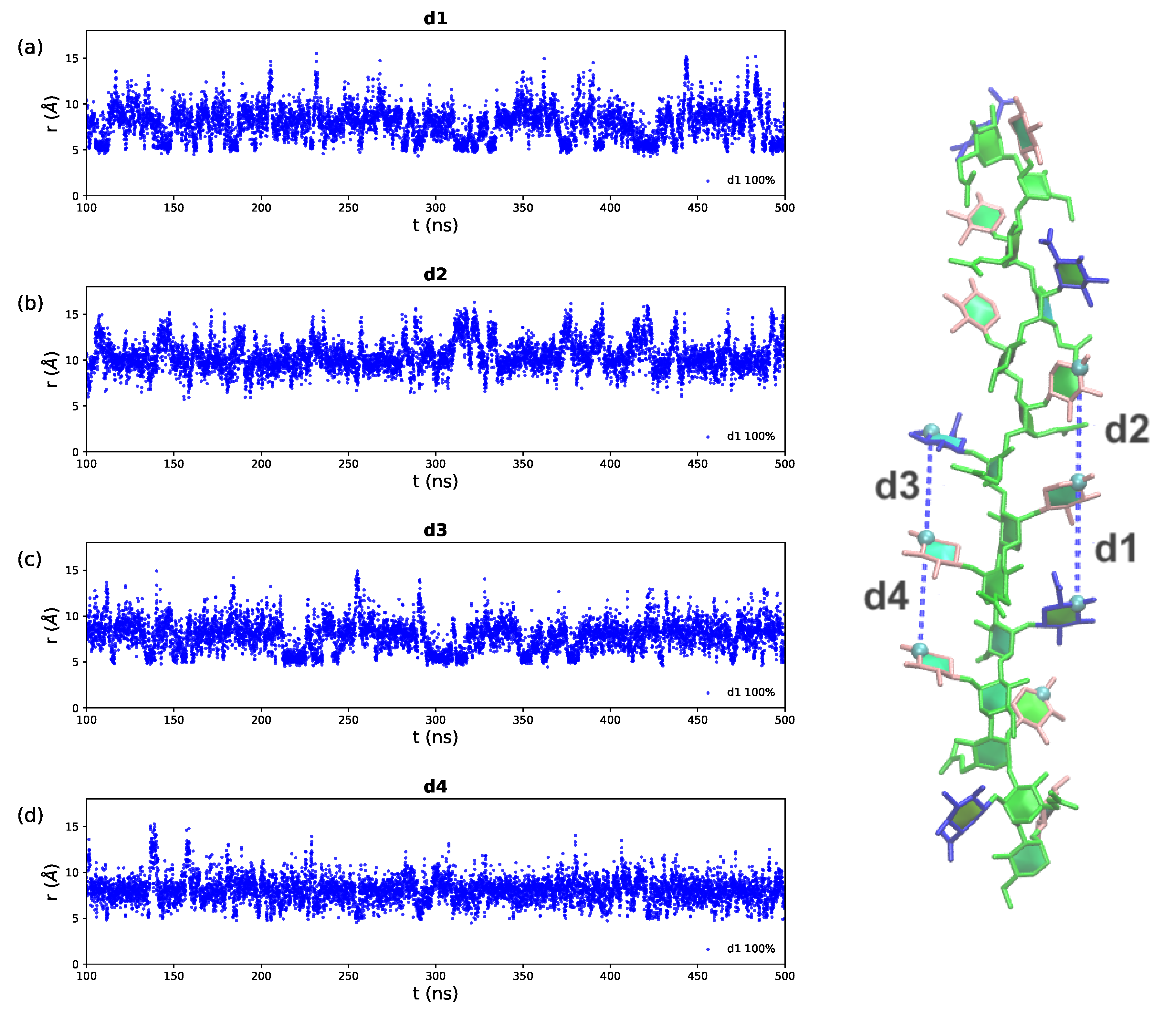
2.3. GXM Self-Aggregation
3. Discussion
4. Materials and Methods
4.1. Disaccharide PMF Calculations
4.2. Molecular Dynamics Simulations
4.2.1. Simulations of Single Saccharide Chains
4.2.2. Simulation of Two Saccharide Chains: cnA’ and cnD
4.3. Data Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Ab | antibody |
| cnA | 6-RU chains of GXM serogroup A primary motif |
| cnA’ | 6-RU chains of GXM serogroup A primary motif, 6-O-acetylated |
| on every second and third mannose in the RU | |
| cnD | 6-RU chains of GXM serogroup D primary motif |
| cnAtet | a tetrasaccharide of GXM serogroup A primary motif |
| cnAdec | a decasaccharide of GXM serogroup A primary motif |
| cnAdec’ | a decasaccharide of GXM Serogroup A primary motif 6-O-acetylated |
| on the second and third mannose and last mannose | |
| GXM | glucuronoxylomannan polysaccharide |
| MD | molecular dynamics |
| PMF | potential of mean force |
| RU | repeat unit |
Appendix A
Appendix A.1

Appendix A.2



References
- Park, B.J.; Wannemuehler, K.A.; Marston, B.J.; Govender, N.; Pappas, P.G.; Chiller, T.M. Estimation of the current global burden of cryptococcal meningitis among persons living with HIV/AIDS. Aids 2009, 23, 525–530. [Google Scholar] [CrossRef] [PubMed]
- McClelland, E.E.; Bernhardt, P.; Casadevall, A. Estimating the relative contributions of virulence factors for pathogenic microbes. Infect. Immun. 2006, 74, 1500–1504. [Google Scholar] [CrossRef] [PubMed]
- Zaragoza, O.; Rodrigues, M.L.; Jesus, M.D.; Frases, S.; Dadachova, E.; Casadevall, A. The Capsule of the Fungal Pathogen Cryptococcus neoformans. In Advances in Applied Microbiology; Academic Press: Cambridge, MA, USA, 2009; Volume 68, Chapter 4; pp. 133–216. [Google Scholar]
- Kwon-Chung, K.J.; Varma, A. Do major species concepts support one, two or more species within Cryptococcus neoformans? FEMS Yeast Res. 2006, 6, 574–587. [Google Scholar] [CrossRef] [PubMed]
- Kwon-Chung, K.J.; Bennett, J.E.; Wickes, B.L.; Meyer, W.; Cuomo, C.A.; Wollenburg, K.R.; Bicanic, T.A.; Castañeda, E.; Chang, Y.C.; Chen, J.; et al. The case for adopting the “species complex” nomenclature for the etiologic agents of cryptococcosis. MSphere 2017, 2, e00357-16. [Google Scholar] [CrossRef]
- Cherniak, R.; Sundstrom, J. Polysaccharide antigens of the capsule of Cryptococcus neoformans. Infect. Immun. 1994, 62, 1507–1512. [Google Scholar] [CrossRef]
- Merrifield, E.H.; Stephen, A.M. Structural investigations of two capsular polysaccharides from Cryptococcus neoformans. Carbohydr. Res. 1980, 86, 69–76. [Google Scholar] [CrossRef]
- Cherniak, R.; Reiss, E.; Slodki, M.; Plattner, R.; Blumer, S. Structure and antigenic activity of the capsular polysaccharide of Cryptococcus neoformans serotype A. Mol. Immunol. 1980, 17, 1025–1032. [Google Scholar] [CrossRef]
- Urai, M.; Kaneko, Y.; Ueno, K.; Okubo, Y.; Aizawa, T.; Fukazawa, H.; Sugita, T.; Ohno, H.; Shibuya, K.; Kinjo, Y.; et al. Evasion of Innate Immune Responses by the Highly Virulent Cryptococcus gattii by Altering Capsule Glucuronoxylomannan Structure. Front. Cell. Infect. Microbiol. 2016, 5, 101. [Google Scholar] [CrossRef]
- McFadden, D.C.; Fries, B.C.; Wang, F.; Casadevall, A. Capsule structural heterogeneity and antigenic variation in Cryptococcus neoformans. Eukaryot. Cell 2007, 6, 1464–1473. [Google Scholar] [CrossRef]
- Cherniak, R.; Valafar, H.; Morris, L.C.; Valafar, F. Cryptococcus neoformans chemotyping by quantitative analysis of 1H nuclear magnetic resonance spectra of glucuronoxylomannans with a computer-simulated artificial neural network. Clin. Diagn. Lab. Immunol. 1998, 5, 146–159. [Google Scholar] [CrossRef]
- Ellerbroek, P.M.; Lefeber, D.J.; van Veghel, R.; Scharringa, J.; Brouwer, E.; Gerwig, G.J.; Janbon, G.; Hoepelman, A.I.; Coenjaerts, F.E. O-acetylation of cryptococcal capsular glucuronoxylomannan is essential for interference with neutrophil migration. J. Immunol. 2004, 173, 7513–7520. [Google Scholar] [CrossRef] [PubMed]
- Varki, A.; Cummings, R.D.; Aebi, M.; Packer, N.H.; Seeberger, P.H.; Esko, J.D.; Stanley, P.; Hart, G.; Darvill, A.; Kinoshita, T.; et al. Symbol Nomenclature for Graphical Representations of Glycans. Glycobiology 2015, 25, 1323–1324. [Google Scholar] [CrossRef] [PubMed]
- Neelamegham, S.; Aoki-Kinoshita, K.; Bolton, E.; Frank, M.; Lisacek, F.; Lütteke, T.; O’Boyle, N.; Packer, N.H.; Stanley, P.; Toukach, P.; et al. Updates to the Symbol Nomenclature for Glycans guidelines. Glycobiology 2019, 29, 620–624. [Google Scholar] [CrossRef] [PubMed]
- Micoli, F.; Costantino, P.; Adamo, R. Potential targets for next generation antimicrobial glycoconjugate vaccines. FEMS Microbiol. Rev. 2018, 42, 388–423. [Google Scholar] [CrossRef]
- Maitta, R.W.; Datta, K.; Chang, Q.; Luo, R.X.; Witover, B.; Subramaniam, K.; Pirofski, L.A. Protective and Nonprotective Human Immunoglobulin M Monoclonal Antibodies to Cryptococcus neoformans Glucuronoxylomannan Manifest Different Specificities and Gene Use Profiles. Infect. Immun. 2004, 72, 4810–4818. [Google Scholar] [CrossRef] [PubMed]
- Nakouzi, A.; Zhang, T.; Oscarson, S.; Casadevall, A. The common Cryptococcus neoformans glucuronoxylomannan M2 motif elicits non-protective antibodies. Vaccine 2009, 27, 3513–3518. [Google Scholar] [CrossRef]
- Guazzelli, L.; Ulc, R.; Bowen, A.; Crawford, C.; McCabe, O.; Jedlicka, A.J.; Wear, M.P.; Casadevall, A.; Oscarson, S. A Synthetic Glycan Array Containing Cryptococcus Neoformans Glucuronoxylomannan Capsular Polysaccharide Fragments Allows the Mapping of Protective Epitopes. ChemRxiv. 2020. [Google Scholar] [CrossRef]
- Casadevall, A.; Mukherjee, J.; Devi, S.J.; Schneerson, R.; Robbins, J.B.; Scharff, M.D. Antibodies elicited by a Cryptococcus neoformans-tetanus toxoid conjugate vaccine have the same specificity as those elicited in infection. J. Infect. Dis. 1992, 165, 1086–1093. [Google Scholar] [CrossRef]
- Casadevall, A.; Coelho, C.; Cordero, R.J.; Dragotakes, Q.; Jung, E.; Vij, R.; Wear, M.P. The capsule of Cryptococcus neoformans. Virulence 2019, 10, 822–831. [Google Scholar] [CrossRef]
- Oscarson, S.; Alpe, M.; Svahnberg, P.; Nakouzi, A.; Casadevall, A. Synthesis and immunological studies of glycoconjugates of Cryptococcus neoformans capsular glucuronoxylomannan oligosaccharide structures. Vaccine 2005, 23, 3961–3972. [Google Scholar] [CrossRef]
- McFadden, D.C.; De Jesus, M.; Casadevall, A. The physical properties of the capsular polysaccharides from Cryptococcus neoformans suggest features for capsule construction. J. Biol. Chem. 2006, 281, 1868–1875. [Google Scholar] [CrossRef] [PubMed]
- Nimrichter, L.; Frases, S.; Cinelli, L.P.; Viana, N.B.; Nakouzi, A.; Travassos, L.R.; Casadevall, A.; Rodrigues, M.L. Self-aggregation of Cryptococcus neoformans capsular glucuronoxylomannan is dependent on divalent cations. Eukaryot. Cell 2007, 6, 1400–1410. [Google Scholar] [CrossRef]
- Frases, S.; Nimrichter, L.; Viana, N.B.; Nakouzi, A.; Casadevall, A. Cryptococcus neoformans capsular polysaccharide and exopolysaccharide fractions manifest physical, chemical, and antigenic differences. Eukaryot. Cell 2008, 7, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Kuttel, M.M.; Ravenscroft, N. The Role of Molecular Modeling in Predicting Carbohydrate Antigen Conformation and Understanding Vaccine Immunogenicity. In Carbohydrate-Based Vaccines: From Concept to Clinic; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2018; Volume 1290, Chapter 7; pp. 139–173. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Cross, S.; Kuttel, M.M.; Stone, J.E.; Gain, J.E. Visualisation of cyclic and multi-branched molecules with VMD. J. Mol. Graph. Modell. 2009, 28, 131–139. [Google Scholar] [CrossRef]
- Frases, S.; Pontes, B.; Nimrichter, L.; Rodrigues, M.L.; Viana, N.B.; Casadevall, A. The elastic properties of the Cryptococcus neoformans capsule. Biophys. J. 2009, 97, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Maxson, M.E.; Cook, E.; Casadevall, A.; Zaragoza, O. The volume and hydration of the Cryptococcus neoformans polysaccharide capsule. Fungal Genet. Biol. 2007, 44, 180–186. [Google Scholar] [CrossRef]
- Frases, S.; Pontes, B.; Nimrichter, L.; Viana, N.B.; Rodrigues, M.L.; Casadevall, A. Capsule of Cryptococcus neoformans grows by enlargement of polysaccharide molecules. Proc. Natl. Acad. Sci. USA 2009, 106, 1228–1233. [Google Scholar] [CrossRef]
- Vij, R.; Crawford, C.J.; Casadevall, A. Variation in cell surface hydrophobicity among Cryptococcus neoformans strains influences interactions with amoeba. mSphere 2020, 5, e00310-20. [Google Scholar] [CrossRef]
- Szu, S.C.; Li, X.; Stone, A.L.; Robbins, J.B. Relation between structure and immunologic properties of the Vi capsular polysaccharide. Infect. Immun. 1991, 59, 4555–4561. [Google Scholar] [CrossRef]
- Hitri, K.; Kuttel, M.M.; De Benedetto, G.; Lockyer, K.; Gao, F.; Hansal, P.; Rudd, T.R.; Beamish, E.; Rijpkema, S.; Ravenscroft, N.; et al. O-acetylation of typhoid capsular polysaccharide confers polysaccharide rigidity and immunodominance by masking additional epitopes. Vaccine 2019, 37, 3866–3875. [Google Scholar] [CrossRef] [PubMed]
- Probert, M.; Zhou, X.; Goodall, M.; Johnston, S.A.; Bielska, E.; Ballou, E.R.; May, R.C. A glucuronoxylomannan epitope exhibits serotype-specific accessibility and redistributes towards the capsule surface during titanization of the fungal pathogen Cryptococcus neoformans. Infect. Immun. 2019, 87, e00731-18. [Google Scholar] [CrossRef] [PubMed]
- Cordero, R.J.; Frases, S.; Guimaräes, A.J.; Rivera, J.; Casadevall, A. Evidence for branching in cryptococcal capsular polysaccharides and consequences on its biological activity. Mol. Microbiol. 2011, 79, 1101–1117. [Google Scholar] [CrossRef] [PubMed]
- Kuttel, M.M.; Jackson, G.E.; Mafata, M.; Ravenscroft, N. Capsular polysaccharide conformations in pneumococcal serotypes 19F and 19A. Carbohydr. Res. 2015, 406, 27–33. [Google Scholar] [CrossRef]
- Kuttel, M.M.; Timol, Z.; Ravenscroft, N. Cross-protection in Neisseria meningitidis serogroups Y and W polysaccharides: A comparative conformational analysis. Carbohydr. Res. 2017, 446-447, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Hlozek, J.; Kuttel, M.M.; Ravenscroft, N. Conformations of Neisseria meningitidis serogroup A and X polysaccharides: The effects of chain length and O-acetylation. Carbohyd. Res. 2018, 465, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Kuttel, M.M.; Ravenscroft, N. Conformation and cross-protection in Group B Streptococcus serotype III and Streptococcus pneumoniae serotype 14: A molecular modeling study. Pharmaceuticals 2019, 12, 28. [Google Scholar] [CrossRef]
- Hlozek, J.; Ravenscroft, N.; Kuttel, M.M. The Effects of Glucosylation and O-Acetylation on the Conformation of Shigella Flexneri Serogroup 2 O-Antigen Vaccine Targets. J. Phys. Chem. B 2020, 124, 2806–2814. [Google Scholar] [CrossRef]
- Kuttel, M.M.; Stähle, J.; Widmalm, G. CarbBuilder: Software for Building Molecular Models of Complex Oligo- and Polysaccharide Structures. J. Comput. Chem. 2016, 37, 2098–2105. [Google Scholar] [CrossRef]
- Laio, A.; Parrinello, M. Escaping free energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12565. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Guvench, O.; Greene, S.N.; Kamath, G.; Brady, J.W.; Venable, R.M.; Pastor, R.W.; MacKerell, A.D. Additive Empirical Force Field for Hexopyranose Monosaccharides. J. Comput. Chem. 2008, 29, 2543–2564. [Google Scholar] [CrossRef] [PubMed]
- Guvench, O.; Hatcher, E.; Venable, R.M.; Pastor, R.W.; Alexander, D.; MacKerell, J. CHARMM Additive All-Atom Force Field for Glycosidic Linkages between Hexopyranoses. J. Chem. Theory Comput. 2009, 5, 2353–2370. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; MacKerell, A.D. Conformational Sampling of Oligosaccharides Using Hamiltonian Replica Exchange with Two-Dimensional Dihedral Biasing Potentials and the Weighted Histogram Analysis Method (WHAM). J. Chem. Theory Comput. 2015, 11, 788–799. [Google Scholar] [CrossRef]
- Kuttel, M.M. Conformational free energy maps for globobiose (α-D-Gal-(1-4)-β-D-Gal) in implicit and explict aqueous solution. Carbohydr. Res. 2008, 343, 1091–1098. [Google Scholar] [CrossRef]
- Stone, J.E.; Phillips, J.C.; Freddolino, P.L.; Hardy, J.; Trabuco, L.G.; Schulten, K. Accelerating Molecular Modeling Applications with Graphics Processors. J. Comput. Chem. 2007, 28, 2618–2639. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulations of liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Kuttel, M.; Ravenscroft, N.; Foschiatti, M.; Cescutti, P.; Rizzo, R. Conformational properties of two exopolysaccharides produced by Inquilinus limosus, a cystic fibrosis lung pathogen. Carbohydr. Res. 2012, 350, 40–48. [Google Scholar] [CrossRef]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Nose, S.; Lein, M.L. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuttel, M.M.; Casadevall, A.; Oscarson, S. Cryptococcus neoformans Capsular GXM Conformation and Epitope Presentation: A Molecular Modelling Study. Molecules 2020, 25, 2651. https://doi.org/10.3390/molecules25112651
Kuttel MM, Casadevall A, Oscarson S. Cryptococcus neoformans Capsular GXM Conformation and Epitope Presentation: A Molecular Modelling Study. Molecules. 2020; 25(11):2651. https://doi.org/10.3390/molecules25112651
Chicago/Turabian StyleKuttel, Michelle M., Arturo Casadevall, and Stefan Oscarson. 2020. "Cryptococcus neoformans Capsular GXM Conformation and Epitope Presentation: A Molecular Modelling Study" Molecules 25, no. 11: 2651. https://doi.org/10.3390/molecules25112651
APA StyleKuttel, M. M., Casadevall, A., & Oscarson, S. (2020). Cryptococcus neoformans Capsular GXM Conformation and Epitope Presentation: A Molecular Modelling Study. Molecules, 25(11), 2651. https://doi.org/10.3390/molecules25112651