
Series of Near-IR-Absorbing Transition Metal Complexes with Redox Active Ligands


 ,
,  ,
,
Abstract

1. Introduction
2. Results and Discussion
2.1. Synthesis of Complexes
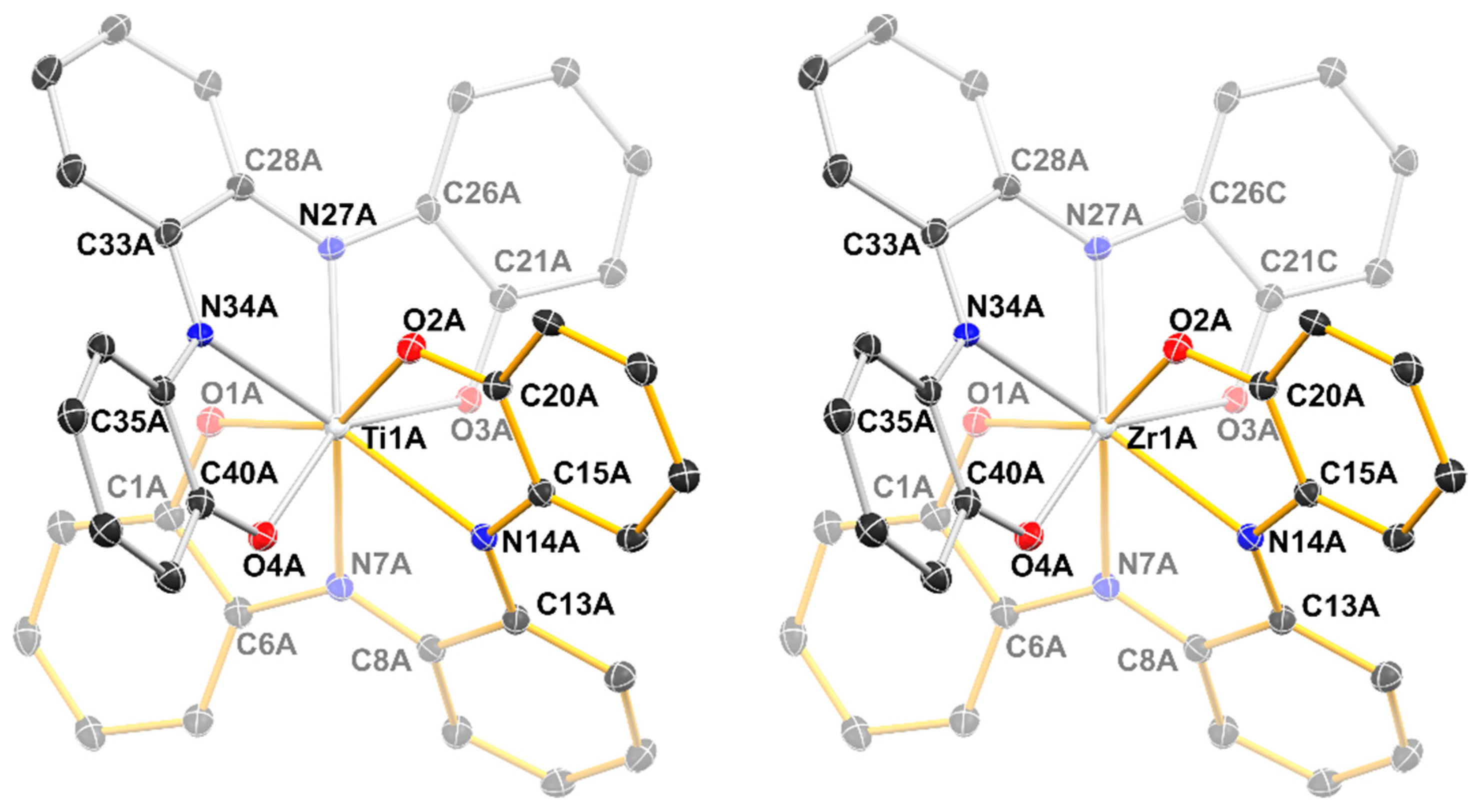
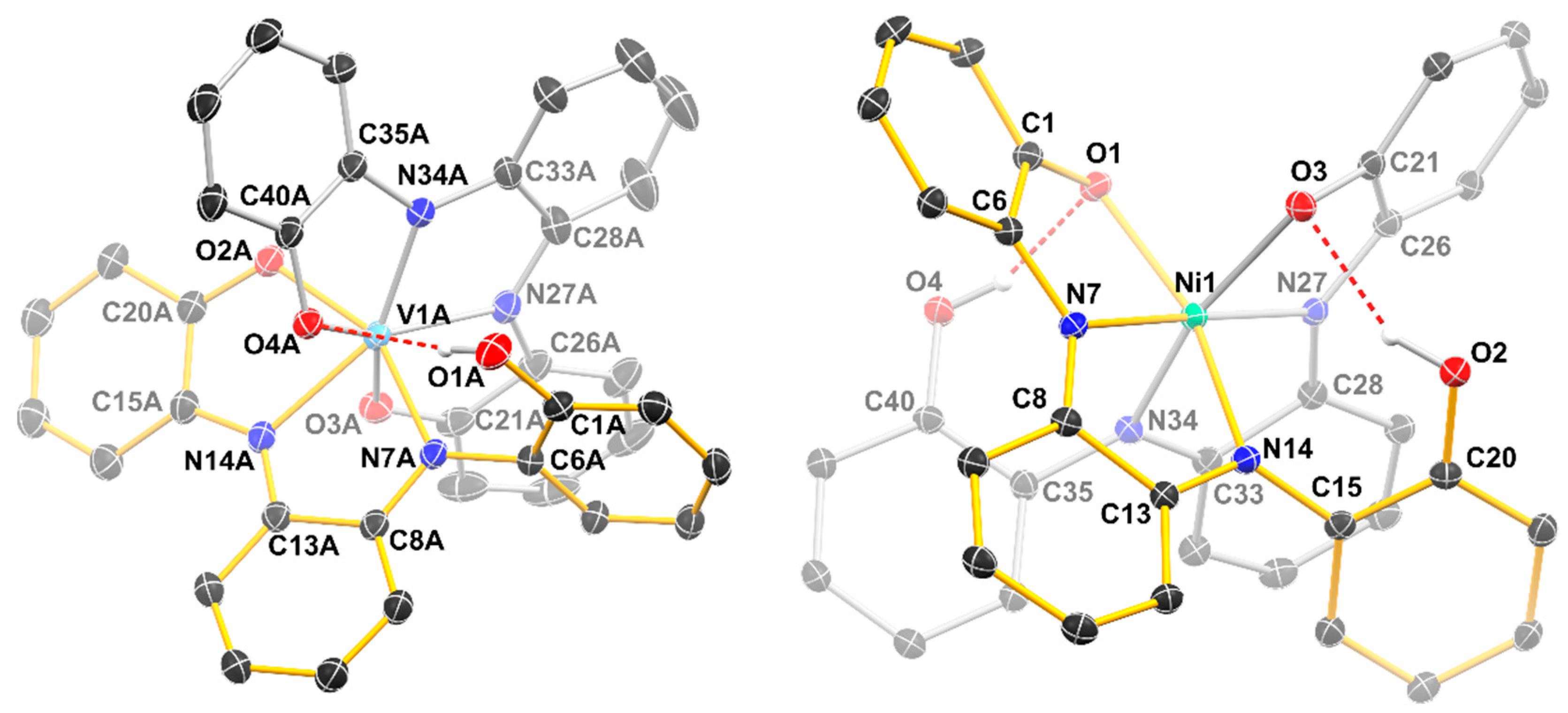
2.2. Structural Studies
2.3. NMR and MS Spectroscopic Studies
2.4. DFT Studies for the Vanadium Complex
2.5. Electrochemical Studies
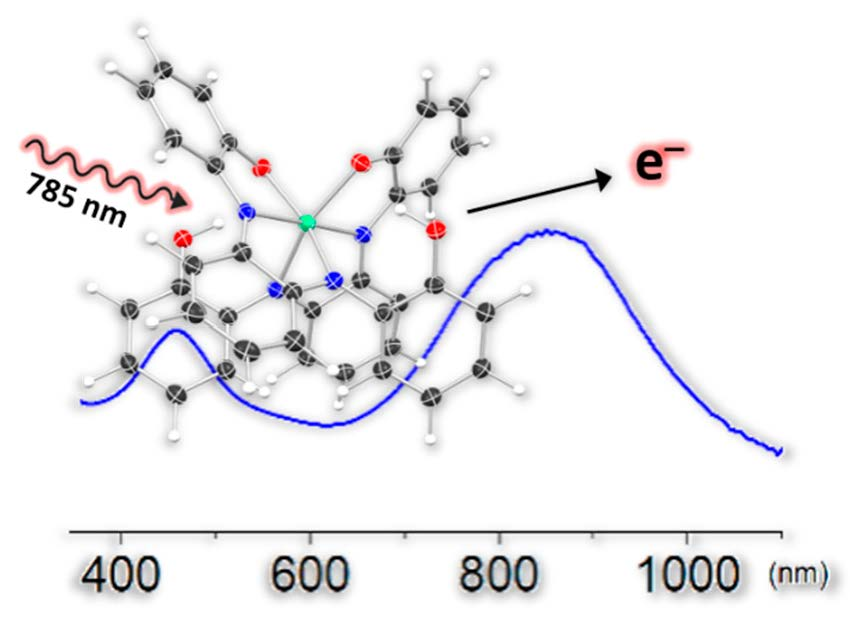
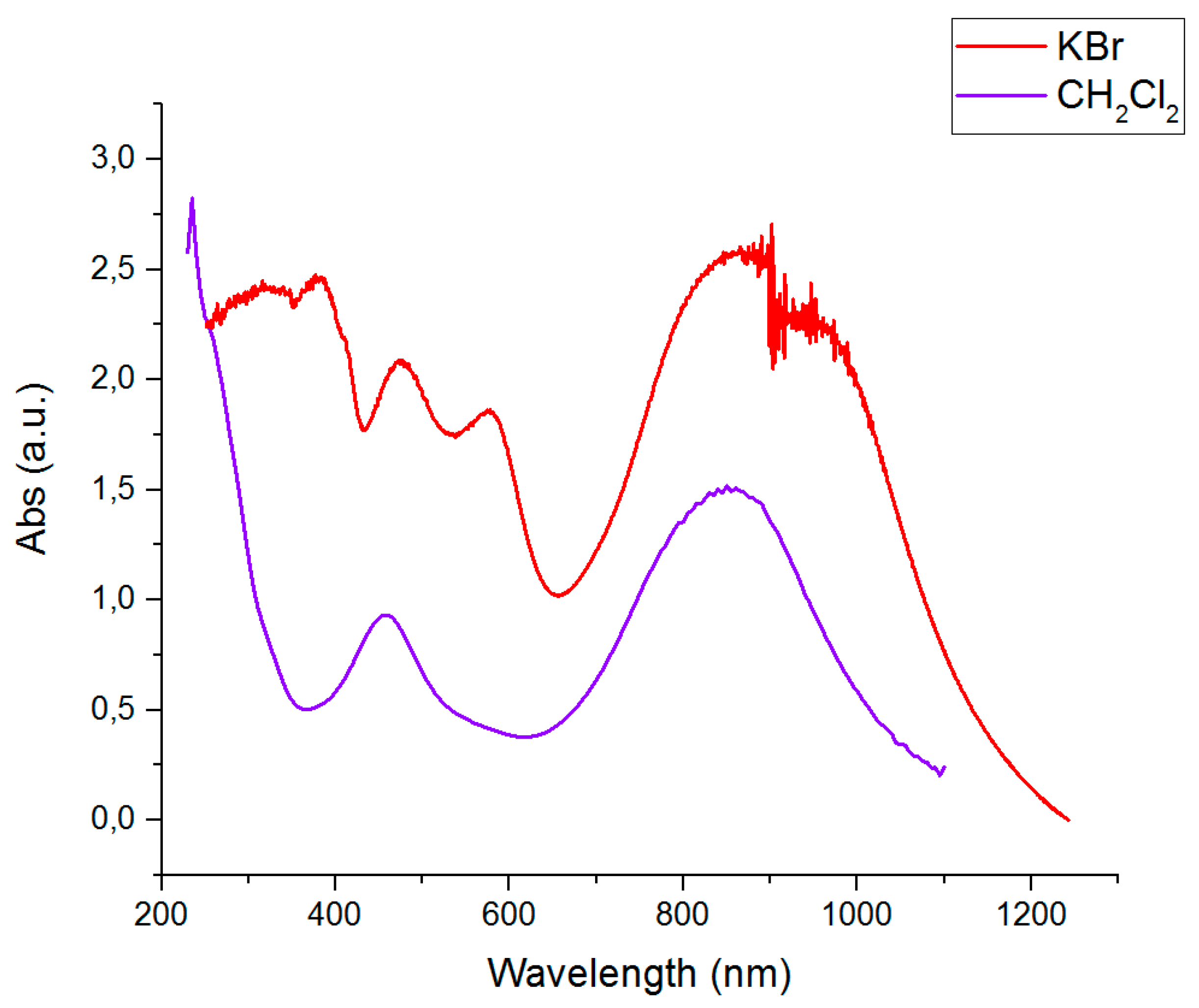
2.6. Optical Absorbtion Studies
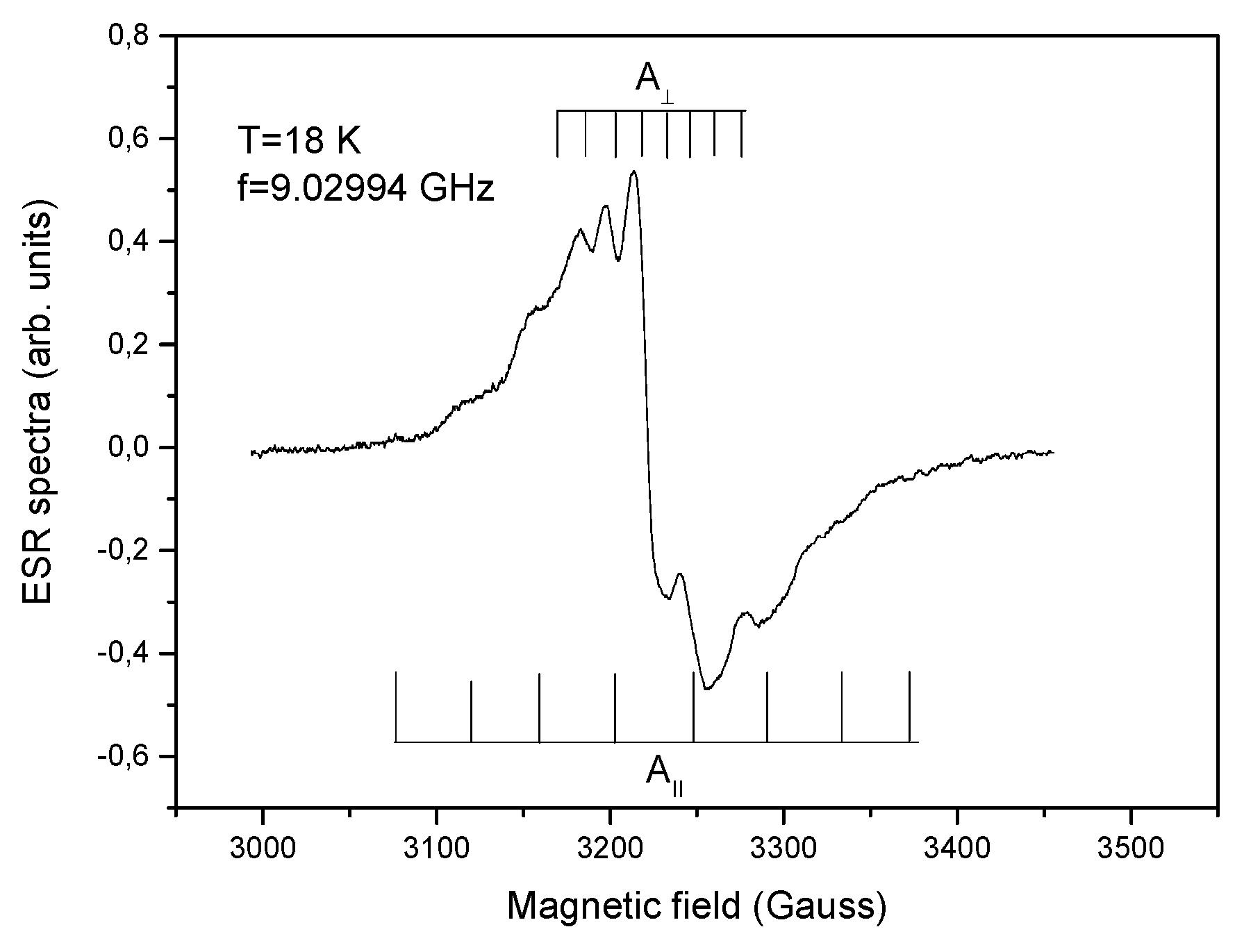
2.7. EPR Studies for the Vanadium and Nickel Complexes
2.8. Magnetic Properties
3. Materials and Methods
3.1. Preparation of [Ti(Lox)2]
3.2. Preparation of [Zr(Lox)2]
3.3. Preparation of [V(Lsq1)(HLox)]
3.4. Preparation of [Ni(HLox)2]
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jørgensen, C.K. Differences between the four halide ligands, and discussion remarks on trigonal-bipyramidal complexes, on oxidation states, and on diagonal elements of one-electron energy. Coord. Chem. Rev. 1966. [Google Scholar] [CrossRef]
- Zelikoff, A.L.; Kopilov, J.; Goldberg, I.; Coates, G.W.; Kol, M. New facets of an old ligand: Titanium and zirconium complexes of phenylenediamine bis(phenolate) in lactide polymerisation catalysisf. Chem. Commun. 2009, 6804–6806. [Google Scholar] [CrossRef]
- Blackmore, K.J.; Lal, N.; Ziller, J.W.; Heyduk, A.F. Catalytic reactivity of a zirconium(IV) redox-active ligand complex with 1,2-diphenylhydrazine. J. Am. Chem. Soc. 2008, 130, 2728–2729. [Google Scholar] [CrossRef] [PubMed]
- Blackmore, K.J.; Lal, N.; Ziller, J.W.; Heyduk, A.F. Group IV coordination chemistry of a tetradentate redox-active ligand in two oxidation states. Eur. J. Inorg. Chem. 2009, 735–743. [Google Scholar] [CrossRef]
- Hänninen, M.M.; Paturi, P.; Tuononen, H.M.; Sillanpää, R.; Lehtonen, A. Heptacoordinated molybdenum(VI) complexes of phenylenediamine Bis(phenolate): A stable molybdenum amidophenoxide radical. Inorg. Chem. 2013, 52, 5714–5721. [Google Scholar] [CrossRef]
- Hossain, M.K.; Haukka, M.; Hänninen, M.M.; Lisensky, G.C.; Paturi, P.; Nordlander, E.; Lehtonen, A. An experimental and theoretical study of a heptacoordinated tungsten(VI) complex of a noninnocent phenylenediamine bis(phenolate) ligand. Inorg. Chem. Commun. 2018, 93, 149–152. [Google Scholar] [CrossRef]
- Lesh, F.D.; Lord, R.L.; Heeg, M.J.; Schlegel, H.B.; Verani, C.N. Unexpected formation of a cobalt(III) phenoxazinylate electron reservoir. Eur. J. Inorg. Chem. 2012, 463–466. [Google Scholar] [CrossRef]
- Chaudhuri, P.; Hess, M.; Müller, J.; Hildenbrand, K.; Bill, E.; Weyhermüller, T.; Wieghardt, K. Aerobic Oxidation of Primary Alcohols (Including Methanol) by Copper(II)− and Zinc(II)−Phenoxyl Radical Catalysts. J. Am. Chem. Soc. 1999, 121, 9599–9610. [Google Scholar] [CrossRef]
- Amb, C.M.; Heth, C.L.; Evenson, S.J.; Pokhodnya, K.I.; Rasmussen, S.C. Thiophene-Fused Nickel Dithiolenes: A Synthetic Scaffold for Highly Delocalized π-Electron Systems. Inorg. Chem. 2016, 55, 10978–10989. [Google Scholar] [CrossRef]
- Aragoni, M.C.; Arca, M.; Devillanova, F.A.; Isaia, F.; Lippolis, V.; Mancini, A.; Pala, L.; Verani, G.; Agostinelli, T.; Caironi, M.; et al. First example of a near-IR photodetector based on neutral [M(R-dmet)2] bis(1,2-dithiolene) metal complexes. Inorg. Chem. Commun. 2007, 10, 191–194. [Google Scholar] [CrossRef]
- Mueller-Westerhoff, U.T.; Vance, B.; Ihl Yoon, D. The synthesis of dithiolene dyes with strong near-IR absorption. Tetrahedron 1991, 47, 909–932. [Google Scholar] [CrossRef]
- Bai, J.F.; Zuo, J.L.; Tan, W.L.; Ji, W.; Shen, Z.; Fun, H.K.; Chinnakali, K.; Razak, I.A.; You, X.Z.; Che, C.M. Synthesis, structure and optical limiting effect of two new nickel complexes containing strongly bound geometrically fixed multisulfur 1,2-dithiolene ligands showing remarkable near-IR absorption. J. Mater. Chem. 1999, 9, 2419–2423. [Google Scholar] [CrossRef]
- Cho, J.-Y.; Fu, J.; Padilha, L.A.; Barlow, S.; Van Stryland, E.W.; Hagan, D.J.; Bishop, M.; Marder, S.R. Synthesis of a Nickel Bis(dithiolene) Complex with Strong Near-Infrared Two-Photon Absorption. Mol. Cryst. Liq. Cryst. 2008, 485, 915–927. [Google Scholar] [CrossRef]
- Garreau-de Bonneval, B.; Moineau-Chane Ching, K.I.; Alary, F.; Bui, T.T.; Valade, L. Neutral d8 metal bis-dithiolene complexes: Synthesis, electronic properties and applications. Coord. Chem. Rev. 2010, 254, 1457–1467. [Google Scholar] [CrossRef]
- Marshall, K.L.; Painter, G.; Lotito, K.; Noto, A.G.; Chang, P. Transition Metal Dithiolene Near-IR Dyes and Their Applications in Liquid Crystal Devices. Mol. Cryst. Liq. Cryst. 2006, 454, 47–449. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, Z.; Chen, X.; Xu, S.; Cao, S. Near-infrared absorbing dyes at 1064 nm: Soluble dithiolene nickel complexes with alkylated electron-donating groups as Peripheral substituents. Dye. Pigment. 2016, 128, 179–189. [Google Scholar] [CrossRef]
- Miao, Q.; Gao, J.; Wang, Z.; Yu, H.; Luo, Y.; Ma, T. Syntheses and characterization of several nickel bis(dithiolene) complexes with strong and broad Near-IR absorption. Inorganica Chim. Acta 2011, 376, 619–627. [Google Scholar] [CrossRef]
- Aiken, D.C.; Ramsey, S.; Mayo, T.; Bellemare, J.; Lambrakos, S.G.; Peak, J. Inverse analysis of near infrared transmission spectra for triarylamine, tetraaryldiamine, nickel dithiolene and indolium-iodide dyes. J. Near Infrared Spectrosc. 2015, 23, 123–132. [Google Scholar] [CrossRef]
- Thorley, K.J.; Hales, J.M.; Anderson, H.L.; Perry, J.W. Porphyrin dimer carbocations with strong near infrared absorption and third-order optical nonlinearity. Angew. Chemie - Int. Ed. 2008, 47, 7095–7098. [Google Scholar] [CrossRef]
- Demir, S.; Jeon, I.R.; Long, J.R.; Harris, T.D. Radical ligand-containing single-molecule magnets. Coord. Chem. Rev. 2015, 289–290, 149–176. [Google Scholar] [CrossRef]
- Atanasov, M.; Aravena, D.; Suturina, E.; Bill, E.; Maganas, D.; Neese, F. First principles approach to the electronic structure, magnetic anisotropy and spin relaxation in mononuclear 3d-transition metal single molecule magnets. Coord. Chem. Rev. 2015, 289–290, 177–214. [Google Scholar] [CrossRef]
- Gómez-Coca, S.; Aravena, D.; Morales, R.; Ruiz, E. Large magnetic anisotropy in mononuclear metal complexes. Coord. Chem. Rev. 2015, 289–290, 379–392. [Google Scholar] [CrossRef]
- Liu, K.; Shi, W.; Cheng, P. Toward heterometallic single-molecule magnets: Synthetic strategy, structures and properties of 3d-4f discrete complexes. Coord. Chem. Rev. 2015, 289–290, 74–122. [Google Scholar] [CrossRef]
- Fabian, J.; Nakazumi, H.; Matsuoka, M. Near-Infrared Absorbing Dyes. Chem. Rev. 1992. [Google Scholar] [CrossRef]
- Brown, S.N. Metrical oxidation states of 2-amidophenoxide and catecholate ligands: Structural signatures of metal-Ligand π bonding in potentially noninnocent ligands. Inorg. Chem. 2012, 51, 1251–1260. [Google Scholar] [CrossRef]
- Evans, D.F. Paramagnetic Susceptibility, etc. 2003 400. The Determination of the Pararnagnetic Susceptibility. J. Chem. Soc. 1959, 2003–2005. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ernzerhof, M.; Burke, K. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982–9985. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Ernzerhof, M.; Scuseria, G.E. Assessment of the Perdew-Burke-Ernzerhof exchange-correlation functional. J. Chem. Phys. 1999, 110, 5029–5036. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Nazeeruddin, M.K.; Zakeeruddin, S.M.; Humphry-Baker, R.; Jirousek, M.; Liska, P.; Vlachopoulos, N.; Shklover, V.; Fischer, C.-H.; Grätzel, M. Acid−Base Equilibria of (2,2‘-Bipyridyl-4,4‘-dicarboxylic acid)ruthenium(II) Complexes and the Effect of Protonation on Charge-Transfer Sensitization of Nanocrystalline Titania. Inorg. Chem. 1999, 38, 6298–6305. [Google Scholar] [CrossRef] [PubMed]
- Vogler, A.; Kunkely, H. Ligand-to-ligand and intraligand charge transfer and their relation to charge transfer interactions in organic zwitterions. Coord. Chem. Rev. 2007, 251, 577–583. [Google Scholar] [CrossRef]
- Paul, A.; Assabghy, F. Optical and esr spectra of vanadium (IV) in different simple germanate, phosphate and borate glasses. J. Mater. Sci. 1975, 10, 613–620. [Google Scholar] [CrossRef]
- Gallay, R.; Van Der Klink, J.J.; Moser, J. EPR study of vanadium (4+) in the anatase and rutile phases of TiO2. Phys. Rev. B 1986, 34, 3060–3068. [Google Scholar] [CrossRef]
- Smith, T.S.; LoBrutto, R.; Pecoraro, V.L. Paramagnetic spectroscopy of vanadyl complexes and its applications to biological systems. Coord. Chem. Rev. 2002, 228, 1–18. [Google Scholar] [CrossRef]
- Gilinskaya, L.G. EPR spectra of V(IV) complexes and the structure of oil porphyrins. J. Struct. Chem. 2008, 49, 245–254. [Google Scholar] [CrossRef]
- Magon, C.J.; Lima, J.F.; Donoso, J.P.; Lavayen, V.; Benavente, E.; Navas, D.; Gonzalez, G. Deconvolution of the EPR spectra of vanadium oxide nanotubes. J. Magn. Reson. 2012, 222, 26–33. [Google Scholar] [CrossRef]
- CrysAlisPro program, version 1.171.38.43 2015. Rigaku Oxford Diffraction. 2018. Available online: https://www.rigaku.com/products/smc/crysalis (accessed on 28 April 2020).
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ti1A-O1A | 1.977(2) | Zr1A-O1A | 2.113(4) |
| Ti1A-O2A | 2.019(2) | Zr1A-O2A | 2.131(4) |
| Ti1A-O3A | 2.016(2) | Zr1A-O3A | 2.096(3) |
| Ti1A-O4A | 2.022(2) | Zr1A-O4A | 2.134(3) |
| Ti1A-N7A | 2.318(2) | Zr1A-N7A | 2.382(4) |
| Ti1A-N14A | 2.198(3) | Zr1A-N14A | 2.325(4) |
| Ti1A-N27A | 2.282(3) | Zr1A-N27A | 2.367(4) |
| Ti1A-N34A | 2.228(3) | Zr1A-N34A | 2.319(4) |
| N7A-C6A | 1.379(4) | N7A-C6A | 1.377(7) |
| N7A-C8A | 1.333(4) | N7A-C8A | 1.333(6) |
| N14A-C13A | 1.327(4) | N14A-C13A | 1.346(6) |
| N14A-C15A | 1.377(4) | N14A-C15A | 1.379(6) |
| N27A-C26A | 1.361(4) | N27A-C26C | 1.399(13) |
| N27A-C28A | 1.350(4) | N27A-C28A | 1.342(6) |
| N34A-C33A | 1.340(4) | N34A-C33A | 1.340(6) |
| N34A-C35A | 1.365(4) | N34A-C35A | 1.387(6) |
| V1A-O1A | 3.571(2) | Ni1-O1 | 2.0616(16) |
| V1A-O2A | 1.9712(19) | Ni1-O2 | 3.548(2) |
| V1A-O3A | 1.9496(19) | Ni1-O3 | 2.0315(14) |
| V1A-O4A | 2.0544(19) | Ni1-O4 | 3.476(2) |
| V1A-N7A | 2.071(2) | Ni1-N7 | 1.9924(16) |
| V1A-N14A | 2.155(2) | Ni1-N14 | 2.1021(17) |
| V1A-N27A | 2.052(2) | Ni1-N27 | 1.9882(16) |
| V1A-N34A | 1.989(2) | Ni1-N34 | 2.0824(16) |
| N7A-C6A | 1.425(3) | N7-C6 | 1.375(3) |
| N7A-C8A | 1.322(4) | N7-C8 | 1.335(3) |
| N14A-C13A | 1.323(4) | N14-C13 | 1.323(2) |
| N14A-C15A | 1.362(4) | N14-C15 | 1.414(3) |
| N27A-C26A | 1.397(4) | N27-C26 | 1.363(3) |
| N27A-C28A | 1.392(4) | N27-C28 | 1.308(3) |
| N34A-C33A | 1.371(4) | N34-C33 | 1.310(2) |
| N34A-C35A | 1.402(3) | N34-C35 | 1.415(2) |
| Complex | Reversible One-Electron Redox Waves (V) | |||||
|---|---|---|---|---|---|---|
| [Ti(Lox)2] | −1.64 | −1.19 | −0.17 | +0.24 | +1.06 | |
| [Zr(Lox)2] | −1.95 | −1.49 | −1.09 | −0.02 | +0.41 | +0.74 |
| [V(Lsq1)(HLox)] | +0.01 | +0.54 | ||||
| [Ni(HLox)2] | −1.47 | −1.07 | −0.92 | +0.04 | +0.36 | |
| Complex | λmax (nm) | ε (103 × L mol−1 cm−1) |
|---|---|---|
| [Ti(Lox)2] | 390 | 4.3 |
| 570 (sh) | 2.6 | |
| 710 | 5.3 | |
| 1050 | 2.2 | |
| [Zr(Lox)2] | 285 | 19 |
| 370 | 14 | |
| 760 | 13 | |
| 1020 | 8.5 | |
| [V(Lsq1)(HLox)] | 315 | 16 |
| 430 | 7.8 | |
| 540 (sh) | 8.1 | |
| 680 | 13 | |
| [Ni(HLox)2] | 460 | 14 |
| 850 | 23 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salojärvi, E.; Peuronen, A.; Lahtinen, M.; Huhtinen, H.; Vlasenko, L.S.; Lastusaari, M.; Lehtonen, A. Series of Near-IR-Absorbing Transition Metal Complexes with Redox Active Ligands. Molecules 2020, 25, 2531. https://doi.org/10.3390/molecules25112531
Salojärvi E, Peuronen A, Lahtinen M, Huhtinen H, Vlasenko LS, Lastusaari M, Lehtonen A. Series of Near-IR-Absorbing Transition Metal Complexes with Redox Active Ligands. Molecules. 2020; 25(11):2531. https://doi.org/10.3390/molecules25112531
Chicago/Turabian StyleSalojärvi, Esko, Anssi Peuronen, Manu Lahtinen, Hannu Huhtinen, Leonid S. Vlasenko, Mika Lastusaari, and Ari Lehtonen. 2020. "Series of Near-IR-Absorbing Transition Metal Complexes with Redox Active Ligands" Molecules 25, no. 11: 2531. https://doi.org/10.3390/molecules25112531
APA StyleSalojärvi, E., Peuronen, A., Lahtinen, M., Huhtinen, H., Vlasenko, L. S., Lastusaari, M., & Lehtonen, A. (2020). Series of Near-IR-Absorbing Transition Metal Complexes with Redox Active Ligands. Molecules, 25(11), 2531. https://doi.org/10.3390/molecules25112531