
Streptavidin-Hosted Organocatalytic Aldol Addition


Abstract
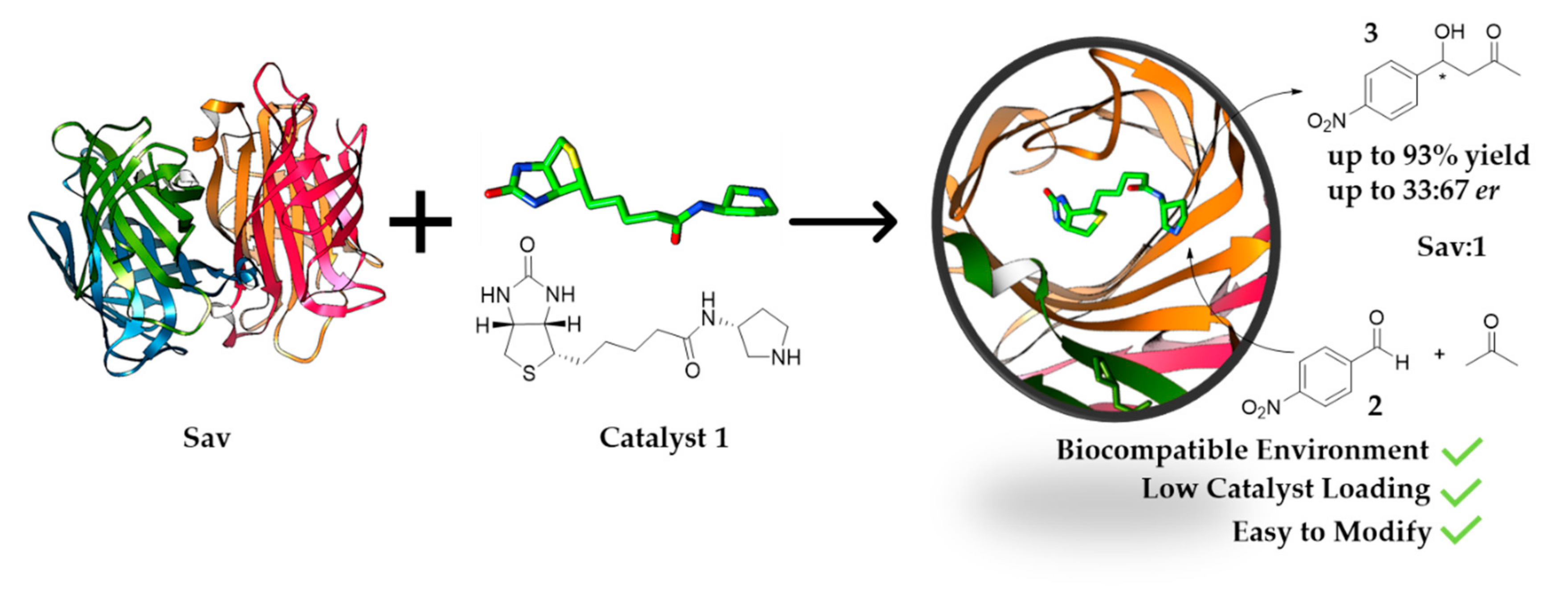
1. Introduction
2. Results and Discussion
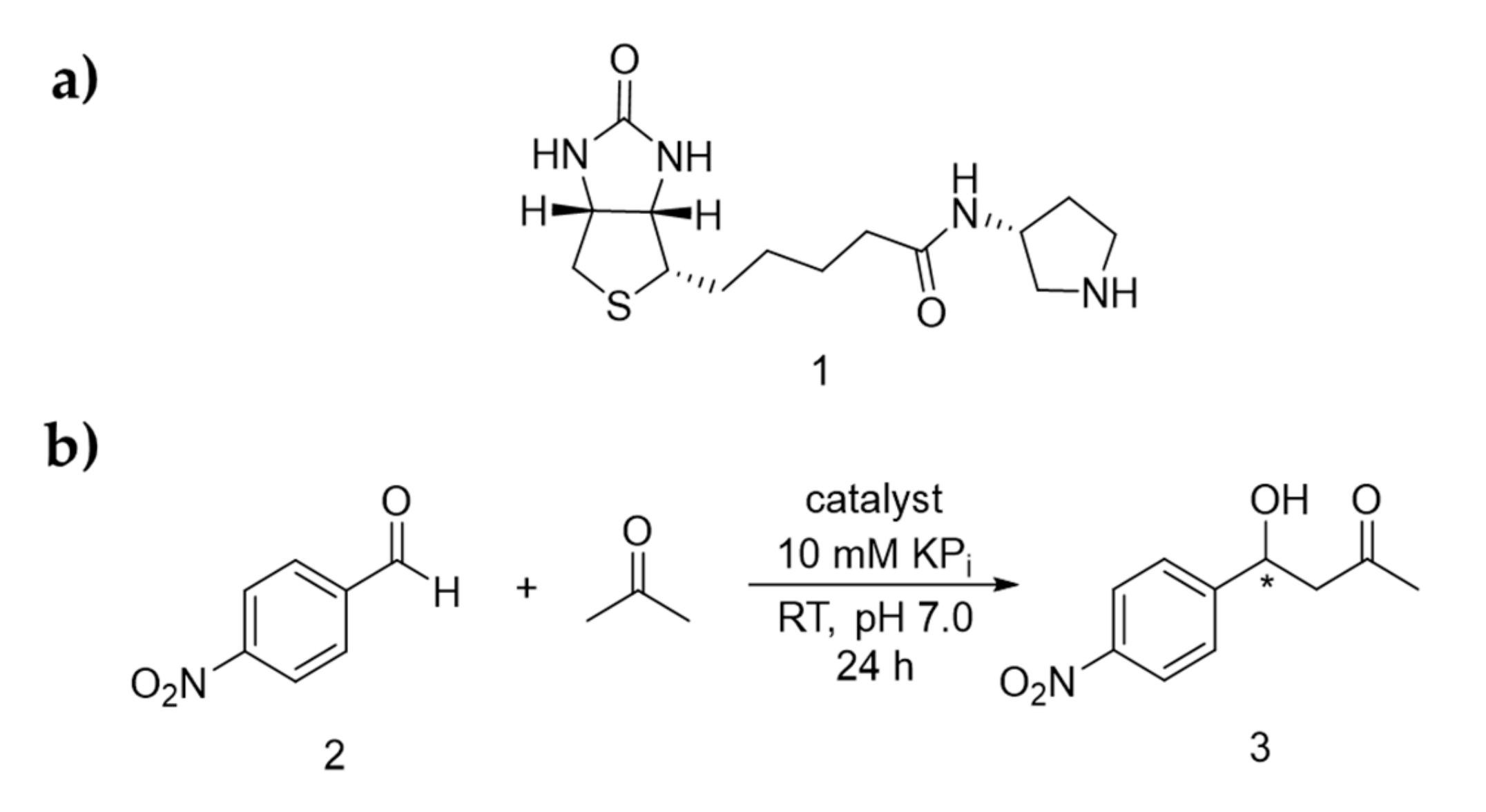
2.1. Screening for Optimised Conditions
2.2. Site-Directed Mutagenesis of the Protein Host
3. Materials and Methods
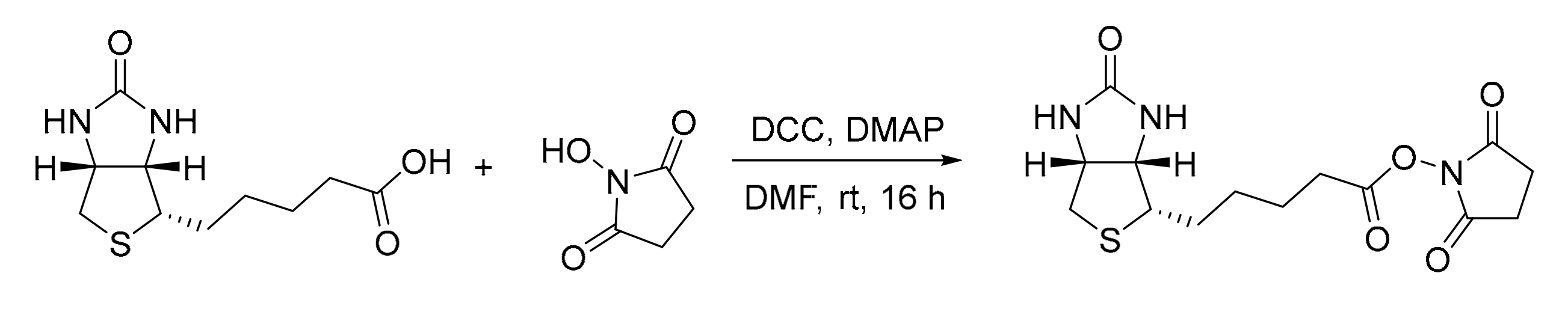
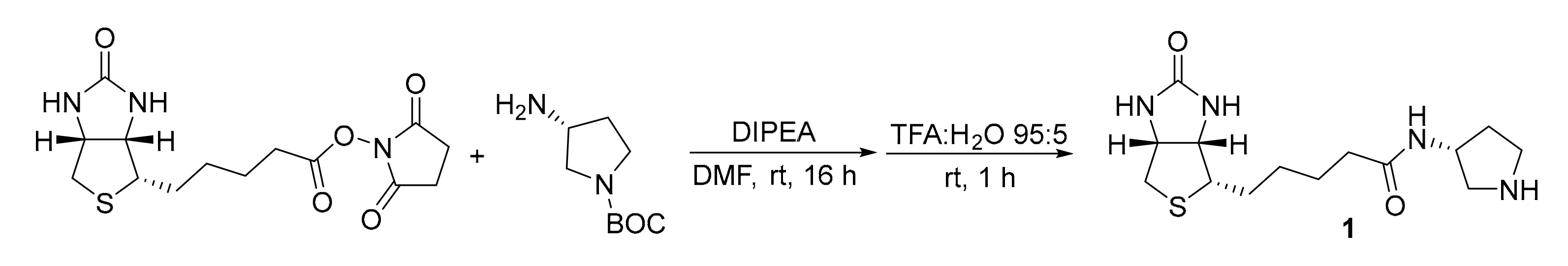
3.1. Experimental Details for the Synthesis of Catalyst 1
3.2. Experimental Details for the Preparation and Purification of T-rSav and Mutants
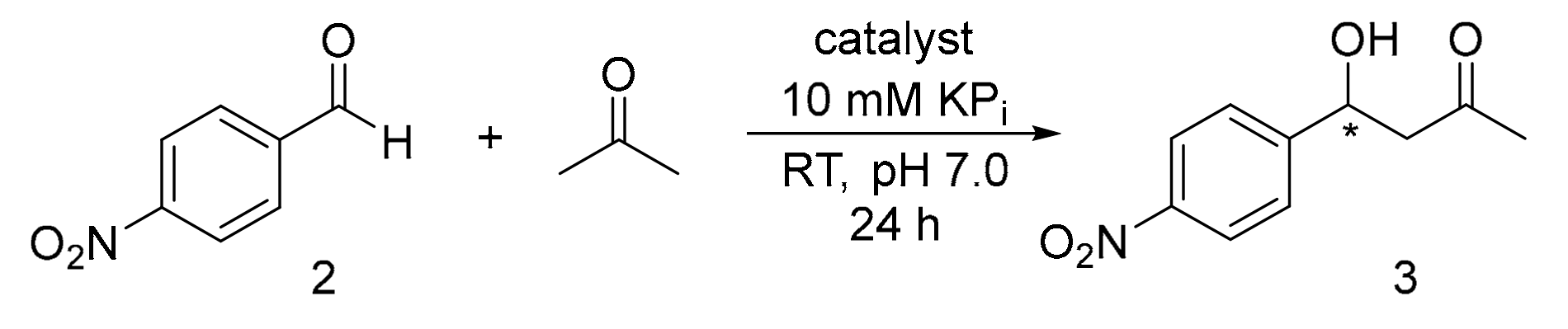
3.3. Experimental Details for the Activity Screening of Catalysts 1 for the Aldol Addition Reaction of Acetone and p-Nitrobenzaldehyde
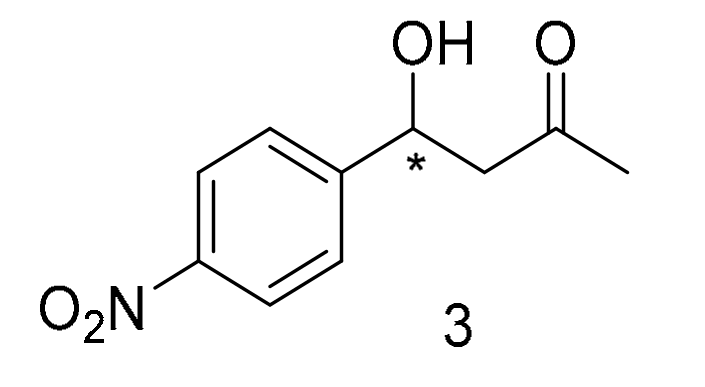
3.4. Synthesis and 1H-NMR Assignment of Aldol Addition Reaction Product
3.5. Chiral HPLC Data of Activity and Selectivity Screening
Screening Reactions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Van der Helm, M.P.; Klemm, B.; Eelkema, R. Organocatalysis in aqueous media. Nat. Rev. Chem. 2019, 3, 491–508. [Google Scholar] [CrossRef]
- Bhowmick, S.; Mondal, A.; Ghosh, A.; Bhowmick, K.C. Water: The most versatile and nature’s friendly media in asymmetric organocatalyzed direct aldol reactions. Tetrahedron Asymmetry 2015, 26, 1215–1244. [Google Scholar] [CrossRef]
- Howard, T.S.; Cohen, R.D.; Nwajiobi, O.; Muneeswaran, Z.P.; Sim, Y.E.; Lahankar, N.N.; Yeh, J.T.H.; Raj, M. Amino-Acid-Catalyzed Direct Aldol Bioconjugation. Org. Lett. 2018, 20, 5344–5347. [Google Scholar] [CrossRef] [PubMed]
- Schober, L.; Ratnam, S.; Yamashita, Y.; Adebar, N.; Pieper, M.; Berkessel, A.; Hessel, V.; Gröger, H. An Asymmetric Organocatalytic Aldol Reaction of a Hydrophobic Aldehyde in Aqueous Medium Running in Flow Mode. Synthesis 2019, 51, 1178–1184. [Google Scholar] [CrossRef]
- Heidlindemann, M.; Rulli, G.; Berkessel, A.; Hummel, W.; Gröger, H. Combination of Asymmetric Organo- and Biocatalytic Reactions in Organic Media Using Immobilized Catalysts in Different Compartments. ACS Catal. 2014, 4, 1099–1103. [Google Scholar] [CrossRef]
- Rulli, G.; Duangdee, N.; Baer, K.; Hummel, W.; Berkessel, A.; Gröger, H. Direction of Kinetically versus Thermodynamically Controlled Organocatalysis and Its Application in Chemoenzymatic Synthesis. Angew. Chem. Int. Ed. 2011, 50, 7944–7947. [Google Scholar] [CrossRef]
- Baer, K.; Kraußer, M.; Burda, E.; Hummel, W.; Berkessel, A.; Gröger, H. Sequential and Modular Synthesis of Chiral 1,3-Diols with Two Stereogenic Centers: Access to All Four Stereoisomers by Combination of Organo- and Biocatalysis. Angew. Chem. Int. Ed. 2009, 48, 9355–9358. [Google Scholar] [CrossRef]
- Avila-Ortiz, C.G.; Pérez-Venegas, M.; Vargas-Caporali, J.; Juaristi, E. Recent applications of mechanochemistry in enantioselective synthesis. Tetrahedron Lett. 2019, 60, 1749–1757. [Google Scholar] [CrossRef]
- Kamo, S.; Maruo, S.; Kuramochi, K.; Tsubaki, K. Synthesis of enantiomerically pure juglomycin C and NHAB. Tetrahedron 2015, 71, 3478–3484. [Google Scholar] [CrossRef]
- Varun; Sonam; Kakkar, R. Isatin and its derivatives: A survey of recent syntheses, reactions, and applications. Med. Chem. Commun. 2019, 10, 351–368. [Google Scholar] [CrossRef]
- Samanta, B.; Seikowski, J.; Höbartner, C. Fluorogenic Labeling of 5-Formylpyrimidine Nucleotides in DNA and RNA. Angew. Chem. Int. Ed. 2016, 55, 1912–1916. [Google Scholar] [CrossRef] [PubMed]
- Saifuddin, M.; Guo, C.; Biewenga, L.; Saravanan, T.; Charnock, S.J.; Poelarends, G.J. Enantioselective Aldol Addition of Acetaldehyde to Aromatic Aldehydes Catalyzed by Proline-Based Carboligases. ACS Catal. 2020, 10, 2522–2527. [Google Scholar] [CrossRef] [PubMed]
- Spears, R.J.; Brabham, R.L.; Budhadev, D.; Keenan, T.; McKenna, S.; Walton, J.; Brannigan, J.A.; Brzozowski, A.M.; Wilkinson, A.J.; Plevin, M.; et al. Site-selective C–C modification of proteins at neutral pH using organocatalyst-mediated cross aldol ligations. Chem. Sci. 2018, 9, 5585–5593. [Google Scholar] [CrossRef] [PubMed]
- Roldán, R.; Hernandez, K.; Joglar, J.; Bujons, J.; Parella, T.; Sánchez-Moreno, I.; Hélaine, V.; Lemaire, M.; Guérard-Hélaine, C.; Fessner, W.-D.; et al. Biocatalytic Aldol Addition of Simple Aliphatic Nucleophiles to Hydroxyaldehydes. ACS Catal. 2018, 8, 8804–8809. [Google Scholar] [CrossRef]
- Rahimi, M.; Geertsema, E.M.; Miao, Y.; van der Meer, J.Y.; van den Bosch, T.; de Haan, P.; Zandvoort, E.; Poelarends, G.J. Inter- and intramolecular aldol reactions promiscuously catalyzed by a proline-based tautomerase. Org. Biomol. Chem. 2017, 15, 2809–2816. [Google Scholar] [CrossRef]
- Yamashita, Y.; Yasukawa, T.; Yoo, W.-J.; Kitanosono, T.; Kobayashi, S. Catalytic enantioselective aldol reactions. Chem. Soc. Rev. 2018, 47, 4388–4480. [Google Scholar] [CrossRef]
- Zeymer, C.; Hilvert, D. Directed Evolution of Protein Catalysts. Annu. Rev. Biochem. 2018, 87, 131–157. [Google Scholar] [CrossRef]
- Obexer, R.; Godina, A.; Garrabou, X.; Mittl, P.R.E.; Baker, D.; Griffiths, A.D.; Hilvert, D. Emergence of a catalytic tetrad during evolution of a highly active artificial aldolase. Nat. Chem. 2016, 9, 50. [Google Scholar] [CrossRef]
- Mase, N.; Nakai, Y.; Ohara, N.; Yoda, H.; Takabe, K.; Tanaka, F.; Barbas, C.F. Organocatalytic Direct Asymmetric Aldol Reactions in Water. J. Am. Chem. Soc. 2006, 128, 734–735. [Google Scholar] [CrossRef]
- Raj, M.; Vishnumaya; Ginotra, S.K.; Singh, V.K. Highly Enantioselective Direct Aldol Reaction Catalyzed by Organic Molecules. Org. Lett. 2006, 8, 4097–4099. [Google Scholar] [CrossRef]
- List, B.; Lerner, R.A.; Barbas, C.F. Proline-Catalyzed Direct Asymmetric Aldol Reactions. J. Am. Chem. Soc. 2000, 122, 2395–2396. [Google Scholar] [CrossRef]
- Oliveira, V.G.; Cardoso, M.F.C.; Forezi, L.S.M. Organocatalysis: A Brief Overview on Its Evolution and Applications. Catalysts 2018, 8, 605. [Google Scholar] [CrossRef]
- Nödling, A.R.; Santi, N.; Williams, T.L.; Tsai, Y.-H.; Luk, L.Y.P. Enabling protein-hosted organocatalytic transformations. RSC Adv. 2020, 10, 16147–16161. [Google Scholar] [CrossRef]
- Nödling, A.R.; Świderek, K.; Castillo, R.; Hall, J.W.; Angelastro, A.; Morrill, L.C.; Jin, Y.; Tsai, Y.-H.; Moliner, V.; Luk, L.Y.P. Reactivity and Selectivity of Iminium Organocatalysis Improved by a Protein Host. Angew. Chem. Int. Ed. 2018, 57, 12478–12482. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Cotelle, Y.; Liu, L.; Lopez-Andarias, J.; Bornhof, A.B.; Akamatsu, M.; Sakai, N.; Matile, S. The Emergence of Anion-pi Catalysis. Acc. Chem. Res. 2018, 51, 2255–2263. [Google Scholar] [CrossRef]
- Guo, C.; Saifuddin, M.; Saravanan, T.; Sharifi, M.; Poelarends, G.J. Biocatalytic Asymmetric Michael Additions of Nitromethane to α,β-Unsaturated Aldehydes via Enzyme-bound Iminium Ion Intermediates. ACS Catal. 2019, 4369–4373. [Google Scholar] [CrossRef]
- Leveson-Gower, R.B.; Mayer, C.; Roelfes, G. The importance of catalytic promiscuity for enzyme design and evolution. Nat. Rev. Chem. 2019, 3, 687–705. [Google Scholar] [CrossRef]
- Jacobsen, E.N.; MacMillan, D.W.C. Organocatalysis. Proc. Natl. Acad. Sci. USA 2010, 107, 20618. [Google Scholar] [CrossRef]
- MacMillan, D.W.C. The advent and development of organocatalysis. Nature 2008, 455, 304–308. [Google Scholar] [CrossRef]
- Donslund, B.S.; Johansen, T.K.; Poulsen, P.H.; Halskov, K.S.; Jorgensen, K.A. The Diarylprolinol Silyl Ethers: Ten Years After. Angew. Chem. Int. Ed. 2015, 54, 13860–13874. [Google Scholar] [CrossRef]
- Jensen, K.L.; Dickmeiss, G.; Jiang, H.; Albrecht, L.; Jorgensen, K.A. The diarylprolinol silyl ether system: A general organocatalyst. Acc. Chem. Res. 2012, 45, 248–264. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Yang, J.W.; Hoffmann, S.; List, B. Asymmetric Enamine Catalysis. Chem. Rev. 2007, 107, 5471–5569. [Google Scholar] [CrossRef] [PubMed]
- Anebouselvy, K.; Shruthi, K.S.; Ramachary, D.B. Asymmetric Supramolecular Organocatalysis: A Complementary Upgrade to Organocatalysis. Eur. J. Org. Chem. 2017, 2017, 5460–5483. [Google Scholar] [CrossRef]
- Bertelsen, S.; Jorgensen, K.A. Organocatalysis--after the gold rush. Chem. Soc. Rev. 2009, 38, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.-Q.; Hörmann, F.M.; Bach, T. Iminium and enamine catalysis in enantioselective photochemical reactions. Chem. Soc. Rev. 2018, 47, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Erkkila, A.; Majander, I.; Pihko, P.M. Iminium catalysis. Chem. Rev. 2007, 107, 5416–5470. [Google Scholar] [CrossRef]
- Davis, H.J.; Ward, T.R. Artificial Metalloenzymes: Challenges and Opportunities. ACS Cent. Sci. 2019, 5, 1120–1136. [Google Scholar] [CrossRef]
- Pellizzoni, M.M.; Schwizer, F.; Wood, C.W.; Sabatino, V.; Cotelle, Y.; Matile, S.; Woolfson, D.N.; Ward, T.R. Chimeric Streptavidins as Host Proteins for Artificial Metalloenzymes. ACS Catal. 2018, 8, 1476–1484. [Google Scholar] [CrossRef]
- Schwizer, F.; Okamoto, Y.; Heinisch, T.; Gu, Y.; Pellizzoni, M.M.; Lebrun, V.; Reuter, R.; Kohler, V.; Lewis, J.C.; Ward, T.R. Artificial Metalloenzymes: Reaction Scope and Optimization Strategies. Chem. Rev. 2018, 118, 142–231. [Google Scholar] [CrossRef]
- Hassan, I.S.; Ta, A.N.; Danneman, M.W.; Semakul, N.; Burns, M.; Basch, C.H.; Dippon, V.N.; McNaughton, B.R.; Rovis, T. Asymmetric δ-Lactam Synthesis with a Monomeric Streptavidin Artificial Metalloenzyme. J. Am. Chem. Soc. 2019, 141, 4815–4819. [Google Scholar] [CrossRef]
- Lechner, H.; Emann, V.R.; Breuning, M.; Höcker, B. An Artificial Cofactor catalyzing the Baylis-Hillman Reaction using Designed Streptavidin as Protein Host. bioRxiv 2020. [Google Scholar] [CrossRef]
- Cotelle, Y.; Lebrun, V.; Sakai, N.; Ward, T.R.; Matile, S. Anion-π Enzymes. ACS Cent. Sci. 2016, 2, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Le, Q.; Nguyen, V.; Park, S. Recent advances in the engineering and application of streptavidin-like molecules. Appl. Microbio. Biotech. 2019, 103, 7355–7365. [Google Scholar] [CrossRef]
- Okamoto, Y.; Kojima, R.; Schwizer, F.; Bartolami, E.; Heinisch, T.; Matile, S.; Fussenegger, M.; Ward, T.R. A cell-penetrating artificial metalloenzyme regulates a gene switch in a designer mammalian cell. Nat. Commun. 2018, 9, 1943. [Google Scholar] [CrossRef] [PubMed]
- Raines, D.J.; Clarke, J.E.; Blagova, E.V.; Dodson, E.J.; Wilson, K.S.; Duhme-Klair, A.-K. Redox-switchable siderophore anchor enables reversible artificial metalloenzyme assembly. Nat. Catal. 2018, 1, 680–688. [Google Scholar] [CrossRef]
- Heinisch, T.; Schwizer, F.; Garabedian, B.; Csibra, E.; Jeschek, M.; Vallapurackal, J.; Pinheiro, V.B.; Marliere, P.; Panke, S.; Ward, T.R.E. coli surface display of streptavidin for directed evolution of an allylic deallylase. Chem. Sci. 2018, 9, 5383–5388. [Google Scholar] [CrossRef]
- Sano, T.; Pandori, W.M.; Chen, X.; Smith, L.C.; Cantor, R.C. A minimum-sized core streptavidin has enhanced structural stability and higher accessibility to biotinylated macromolecules. J. Biol. Chem. 1995, 270, 28204–28209. [Google Scholar] [CrossRef]
- Hosseini, M.; Stiasni, N.; Barbieri, V.; Kappe, C.O. Microwave-Assisted Asymmetric Organocatalysis. A Probe for Nonthermal Microwave Effects and the Concept of Simultaneous Cooling. J. Org. Chem. 2007, 72, 1417–1424. [Google Scholar] [CrossRef]
- Gao, J.; Bai, S.; Gao, Q.; Liu, Y.; Yang, Q. Acid controlled diastereoselectivity in asymmetric aldol reaction of cycloketones with aldehydes using enamine-based organocatalysts. Chem. Commun. 2011, 47, 6716–6718. [Google Scholar] [CrossRef]
- Gryko, D.; Zimnicka, M.; Lipiński, R. Brønsted Acids as Additives for the Direct Asymmetric Aldol Reaction Catalyzed by l-Prolinethioamides. Direct Evidence for Enamine-Iminium Catalysis. J. Org. Chem. 2007, 72, 964–970. [Google Scholar] [CrossRef]
- Sutar, R.L.; Joshi, N.N. Role of Additives in Chiral Amine-Catalyzed Direct Aldol Reaction. Synth. Commun. 2014, 44, 352–360. [Google Scholar] [CrossRef]
- Zhong, G.; Lerner, R.A.; Barbas, I.C. Broadening the Aldolase Catalytic Antibody Repertoire by Combining Reactive Immunization and Transition State Theory: New Enantio- and Diastereoselectivities. Angew. Chem. Int. Ed. 1999, 38, 3738–3741. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compound 3 are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | Eqv. Acetone | Co-Solvent | Loading (mol%) | Estimated Conversion a/% | er (R:S) |
|---|---|---|---|---|---|---|
| 1 | None | 20 vol% | NA | None | 17 | 50:50 |
| 2 | 1 | 1 | 36 | 50:50 | ||
| 3 | Sav | 0.1 | 16 | 50:50 | ||
| 4 | Sav | 0.5 | 23 | 50:50 | ||
| 5 | Sav | 1 | 33 | 50:50 | ||
| 6 | Sav:1 | 0.1 | 47 | 38:62 | ||
| 7 | Sav:1 | 0.5 | 79 | 33:67 | ||
| 8 | Sav:1 | 1 | 93 | 33:67 | ||
| 9 b | Sav:1 | 1 | 92 | 33:67 | ||
| 10 c | Sav:1 | 1 | 37 | 34:66 | ||
| 11 | Sav:1 | 5 | 25% MeOH | 1 | 20 | 33:67 |
| 12 | Sav:1 | 5 | 25% ACN | 1 | 12 | 33:67 |
| 13 | Sav:1 | 5 | 25% i-PrOH | 1 | 1 | NA |
| 14 | Sav:1 | 10 | 1 | 5 | NA | |
| 15 | Sav:1 | 20 | 1 | 9 | 36:64 | |
| 16 | Sav:1 | 50 | 1 | 18 | 33:67 |
| Catalyst | Estimated Conversion a/% | er (R:S) |
|---|---|---|
| Sav:1 | 93 | 33:67 |
| T-rSav:1 | 92 | 35:65 |
| S112E:1 | 14 | 39:61 |
| K121A:1 | 89 | 44:56 |
| Mutation | Primer (5’ to 3’) |
|---|---|
| S112E | Forward GGCTGCTGACCGAAGGCACCACCGAGG Reverse CCTCGGTGGTGCCTTCGGTCAGCAGCC |
| K121A | Forward ACCGAGGCCAACGCCTGGGCGTCCACGCTGGTCGGC Reverse GGCGTTGGCCTCGGTGGTGCCGGA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santi, N.; Morrill, L.C.; Luk, L.Y.P. Streptavidin-Hosted Organocatalytic Aldol Addition. Molecules 2020, 25, 2457. https://doi.org/10.3390/molecules25102457
Santi N, Morrill LC, Luk LYP. Streptavidin-Hosted Organocatalytic Aldol Addition. Molecules. 2020; 25(10):2457. https://doi.org/10.3390/molecules25102457
Chicago/Turabian StyleSanti, Nicolò, Louis C. Morrill, and Louis Y. P. Luk. 2020. "Streptavidin-Hosted Organocatalytic Aldol Addition" Molecules 25, no. 10: 2457. https://doi.org/10.3390/molecules25102457
APA StyleSanti, N., Morrill, L. C., & Luk, L. Y. P. (2020). Streptavidin-Hosted Organocatalytic Aldol Addition. Molecules, 25(10), 2457. https://doi.org/10.3390/molecules25102457