
The Positive Side of the Alzheimer’s Disease Amyloid Cross-Interactions: The Case of the Aβ 1-42 Peptide with Tau, TTR, CysC, and ApoA1

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Amyloid Cross-Interactions
3. Amyloid Proteins Displaying Cross-Interaction with Aβ1-42 Peptide
3.1. Tau Protein
3.2. Transthyretin (TTR)
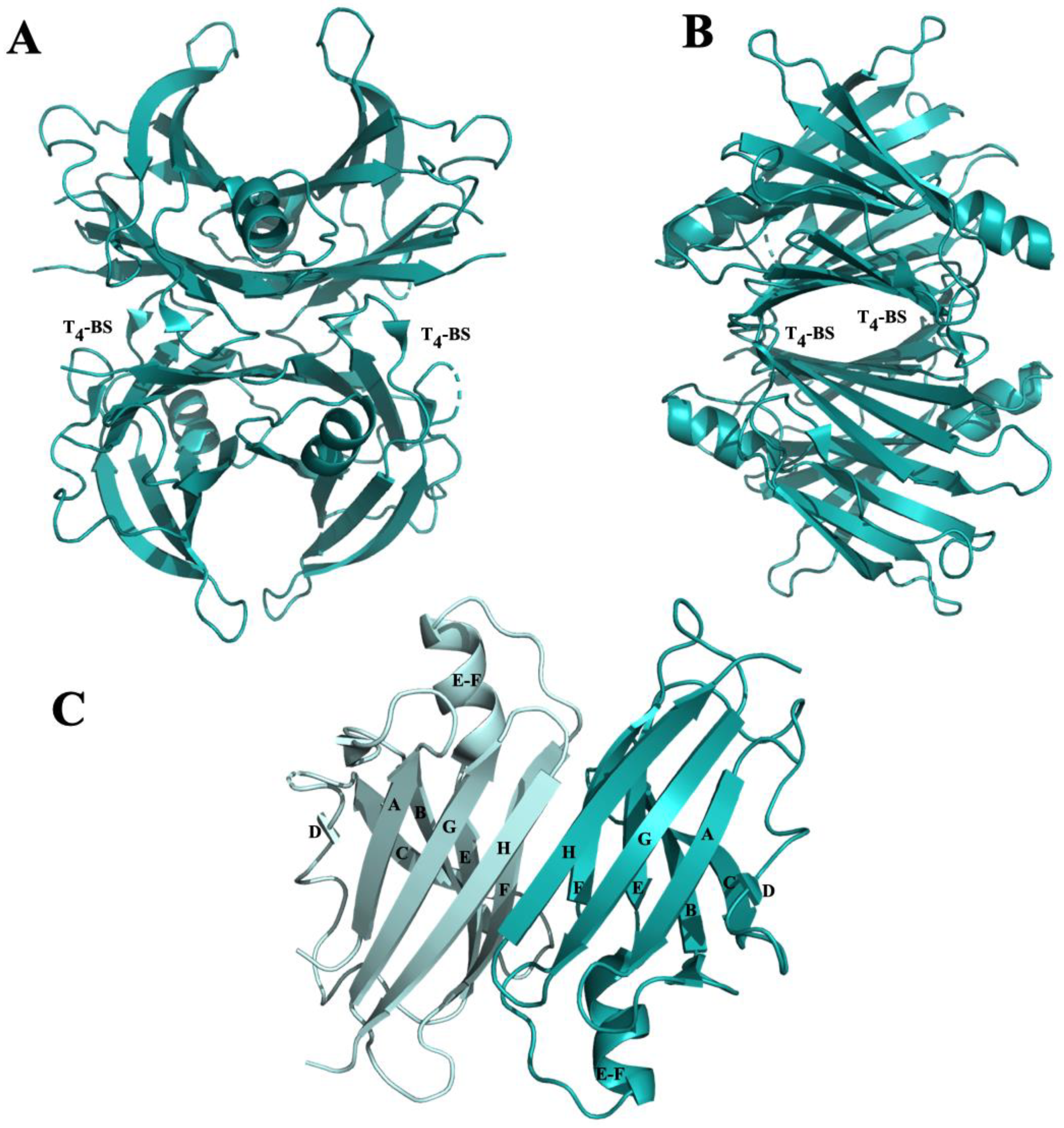
3.2.1. β-Amyloid-Binding Sites on TTR
3.2.2. TTR-Aβ Interaction-Based Strategies to Design Anti-Aβ Agents
3.3. Cystatin C (CysC)
β-Amyloid-Binding Sites on CysC
3.4. Apolipoprotein A1 (ApoA1)
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Ankarcrona, M.; Winblad, B.; Monteiro, C.; Fearns, C.; Powers, E.T.; Johansson, J.; Westermark, G.T.; Presto, J.; Ericzon, B.-G.; Kelly, J.W. Current and future treatment of amyloid diseases. J. Intern. Med. 2016, 280, 177–202. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Abelein, A.; Abrahams, J.P.; Danielsson, J.; Gräslund, A.; Jarvet, J.; Luo, J.; Tiiman, A.; Wärmländer, S.K.T.S. The hairpin conformation of the amyloid β peptide is an important structural motif along the aggregation pathway. J. Biol. Inorg. Chem. 2014, 19, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Peter J Lansbury, J. Alzheimer’s Disease Is the Most Common Neurodegenerative Disorder. Basic Neurochem. Mol. Cell. Med. Asp. 1999, 6, 101–102. [Google Scholar]
- Murphy, M.P.; LeVine, H. Alzheimer’s Disease and the β-Amyloid Peptide. J. Alzheimers Dis. 2010, 19, 311. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Scheltens, P.; Blennow, K.; Breteler, M.M.B.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Nerelius, C.; Sandegren, A.; Sargsyan, H.; Raunak, R.; Leijonmarck, H.; Chatterjee, U.; Fisahn, A.; Imarisio, S.; Lomas, D.A.; Crowther, D.C.; et al. α-Helix targeting reduces amyloid-β peptide toxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 9191–9196. [Google Scholar] [CrossRef]
- Tonali, N.; Dodero, V.I.; Kaffy, J.; Hericks, L.; Ongeri, S.; Sewald, N. Real-Time BODIPY-Binding Assay To Screen Inhibitors of the Early Oligomerization Process of Aβ1–42 Peptide. ChemBioChem 2019, 21, 1129–1135. [Google Scholar] [CrossRef]
- Tonali, N.; Kaffy, J.; Soulier, J.-L.; Gelmi, M.L.; Erba, E.; Taverna, M.; van Heijenoort, C.; Ha-Duong, T.; Ongeri, S. Structure-activity relationships of β-hairpin mimics as modulators of amyloid β-peptide aggregation. Eur. J. Med. Chem. 2018, 154, 280–293. [Google Scholar] [CrossRef]
- Pellegrino, S.; Tonali, N.; Erba, E.; Kaffy, J.; Taverna, M.; Contini, A.; Taylor, M.; Allsop, D.; Gelmi, M.L.; Ongeri, S. β-Hairpin mimics containing a piperidine–pyrrolidine scaffold modulate the β-amyloid aggregation process preserving the monomer species. Chem. Sci. 2017, 8, 1295–1302. [Google Scholar] [CrossRef] [PubMed]
- Kaffy, J.; Brinet, D.; Soulier, J.-L.; Correia, I.; Tonali, N.; Fera, K.F.; Iacone, Y.; Hoffmann, A.R.F.; Khemtémourian, L.; Crousse, B.; et al. Designed Glycopeptidomimetics Disrupt Protein–Protein Interactions Mediating Amyloid β-Peptide Aggregation and Restore Neuroblastoma Cell Viability. J. Med. Chem. 2016, 59, 2025–2040. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.-S.; AhnJo, S.-M. Mechanisms of Amyloid-β Peptide Clearance: Potential Therapeutic Targets for Alzheimer’s Disease. Biomol. Ther. 2012, 20, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Yerbury, J.; Kumita, J. Protein chemistry of amyloid fibrils and chaperones: Implications for amyloid formation and disease. Fac. Sci. Pap. (Arch.) 2010, 89–98. [Google Scholar] [CrossRef]
- Castellano, J.M.; Kim, J.; Stewart, F.R.; Jiang, H.; DeMattos, R.B.; Patterson, B.W.; Fagan, A.M.; Morris, J.C.; Mawuenyega, K.G.; Cruchaga, C.; et al. Human apoE Isoforms Differentially Regulate Brain Amyloid-β Peptide Clearance. Sci. Transl. Med. 2011, 3, 89ra57. [Google Scholar] [CrossRef]
- Kim, J.; Basak, J.M.; Holtzman, D.M. The Role of Apolipoprotein E in Alzheimer’s Disease. Neuron 2009, 63, 287–303. [Google Scholar] [CrossRef]
- Deane, R.; Sagare, A.; Hamm, K.; Parisi, M.; Lane, S.; Finn, M.B.; Holtzman, D.M.; Zlokovic, B.V. apoE isoform–specific disruption of amyloid β peptide clearance from mouse brain. J. Clin. Investig. 2008, 118, 4002–4013. [Google Scholar] [CrossRef]
- Arboleda-Velasquez, J.F.; Lopera, F.; O’Hare, M.; Delgado-Tirado, S.; Marino, C.; Chmielewska, N.; Saez-Torres, K.L.; Amarnani, D.; Schultz, A.P.; Sperling, R.A.; et al. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: A case report. Nat. Med. 2019, 25, 1680–1683. [Google Scholar] [CrossRef]
- Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef]
- Herrup, K. The case for rejecting the amyloid cascade hypothesis. Nat. Neurosci. 2015, 18, 794–799. [Google Scholar] [CrossRef]
- Musiek, E.S.; Holtzman, D.M. Three dimensions of the amyloid hypothesis: Time, space and “wingmen.”. Nat. Neurosci. 2015, 18, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.P.; Matthews, J.M. Protein–protein interactions in human disease. Curr. Opin. Struct. Biol. 2005, 15, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Wärmländer, S.K.T.S.; Gräslund, A.; Abrahams, J.P. Cross-interactions between the Alzheimer Disease Amyloid-β Peptide and Other Amyloid Proteins: A Further Aspect of the Amyloid Cascade Hypothesis. J. Biol. Chem. 2016, 291, 16485–16493. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Cheng, D.; Wang, J.; Duong, D.M.; Losik, T.G.; Gearing, M.; Rees, H.D.; Lah, J.J.; Levey, A.I.; Peng, J. Proteomic Characterization of Postmortem Amyloid Plaques Isolated by Laser Capture Microdissection. J. Biol. Chem. 2004, 279, 37061–37068. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Tanji, K.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease: Molecules implicated in the formation and degradation of α-synuclein aggregates. Neuropathology 2007, 27, 494–506. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Engelender, S.; Yoshimoto, M.; Tsuji, S.; Ross, C.A.; Takahashi, H. Synphilin-1 is present in Lewy bodies in Parkinson’s disease. Ann. Neurol. 2000, 47, 521–523. [Google Scholar] [CrossRef]
- Xia, Q.; Liao, L.; Cheng, D.; Duong, D.M.; Gearing, M.; Lah, J.J.; Levey, A.I.; Peng, J. Proteomic identification of novel proteins associated with Lewy bodies. Front. Biosci. 2008, 13, 3850–3856. [Google Scholar] [CrossRef]
- Horvath, I.; Wittung-Stafshede, P. Cross-talk between amyloidogenic proteins in type-2 diabetes and Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2016, 113, 12473–12477. [Google Scholar] [CrossRef]
- Giasson, B.I. Initiation and Synergistic Fibrillization of Tau and Alpha-Synuclein. Science 2003, 300, 636–640. [Google Scholar] [CrossRef]
- Buxbaum, J.N.; Ye, Z.; Reixach, N.; Friske, L.; Levy, C.; Das, P.; Golde, T.; Masliah, E.; Roberts, A.R.; Bartfai, T. Transthyretin protects Alzheimer’s mice from the behavioral and biochemical effects of Aβ toxicity. Proc. Natl. Acad. Sci. USA 2008, 105, 2681–2686. [Google Scholar] [CrossRef]
- Ono, K.; Takahashi, R.; Ikeda, T.; Yamada, M. Cross-seeding effects of amyloid β-protein and α-synuclein. J. Neurochem. 2012, 122, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Rockenstein, E.; Veinbergs, I.; Sagara, Y.; Mallory, M.; Hashimoto, M.; Mucke, L. β-Amyloid peptides enhance α-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer’s disease and Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 12245–12250. [Google Scholar] [CrossRef] [PubMed]
- Clinton, L.K.; Blurton-Jones, M.; Myczek, K.; Trojanowski, J.Q.; LaFerla, F.M. Synergistic Interactions between Aβ, Tau, and α-Synuclein: Acceleration of Neuropathology and Cognitive Decline. J. Neurosci. 2010, 30, 7281–7289. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.-P.; Arai, T.; Miklossy, J.; McGeer, P.L. Aβ and tau form soluble complexes that may promote self aggregation of both into the insoluble forms observed in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 1953. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G.; Wong, C.W.; Quaranta, V.; Eanes, E.D. The amyloid deposits in Alzheimer’s disease: Their nature and pathogenesis. Appl. Pathol. 1984, 2, 357–369. [Google Scholar] [PubMed]
- Ittner, L.M.; Götz, J. Amyloid-β and tau—A toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci 2011, 12, 67–72. [Google Scholar] [CrossRef]
- Goedert, M.; Eisenberg, D.S.; Crowther, R.A. Propagation of Tau Aggregates and Neurodegeneration. Annu. Rev. Neurosci. 2017, 40, 189–210. [Google Scholar] [CrossRef]
- Tatarnikova, O.G.; Orlov, M.A.; Bobkova, N.V. Beta-Amyloid and Tau-Protein: Structure, Interaction, and Prion-Like Properties. Biochem. Mosc. 2015, 80, 1800–1819. [Google Scholar] [CrossRef]
- Brier, M.R.; Gordon, B.; Friedrichsen, K.; McCarthy, J.; Stern, A.; Christensen, J.; Owen, C.; Aldea, P.; Su, Y.; Hassenstab, J.; et al. Tau and Ab imaging, CSF measures, and cognition in Alzheimer’s disease. Sci. Transl. Med. 2016, 338, 338ra66. [Google Scholar] [CrossRef]
- Schwarz, A.J.; Yu, P.; Miller, B.B.; Shcherbinin, S.; Dickson, J.; Navitsky, M.; Joshi, A.D.; Devous, M.D.; Mintun, M.S. Regional profiles of the candidate tau PET ligand 18 F-AV-1451 recapitulate key features of Braak histopathological stages. Brain 2016, 139, 1539–1550. [Google Scholar] [CrossRef]
- Neve, R.L.; Harris, P.; Kosik, K.S.; Kurnit, D.M.; Donlon, T.A. Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Brain Res. 1986, 387, 271–280. [Google Scholar] [CrossRef]
- Avila, J.; Lucas, J.J.; Pérez, M.; Hernández, F. Role of Tau Protein in Both Physiological and Pathological Conditions. Physiol. Rev. 2004, 84, 361–384. [Google Scholar] [CrossRef] [PubMed]
- Andreadis, A. Misregulation of tau alternative splicing in neurodegeneration and dementia. Prog. Mol. Subcell. Biol. 2006, 44, 89–107. [Google Scholar] [CrossRef] [PubMed]
- Avila, J.; Jiménez, J.S.; Sayas, C.L.; Bolós, M.; Zabala, J.C.; Rivas, G.; Hernández, F. Tau Structures. Front. Aging Neurosci. 2016, 8, 262. [Google Scholar] [CrossRef]
- Schweers, O.; Schönbrunn-Hanebeck, E.; Marx, A.; Mandelkow, E. Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta-structure. J. Biol. Chem. 1994, 269, 24290–24297. [Google Scholar]
- Mylonas, E.; Hascher, A.; Bernadó, P.; Blackledge, M.; Mandelkow, E.; Svergun, D.I. Domain conformation of tau protein studied by solution small-angle X-ray scattering. Biochemistry 2008, 47, 10345–10353. [Google Scholar] [CrossRef]
- Fontaine, S.N.; Sabbagh, J.J.; Baker, J.; Martinez-Licha, C.R.; Darling, A.; Dickey, C.A. Cellular factors modulating the mechanism of tau protein aggregation. Cell. Mol. Life Sci. 2015, 72, 1863–1879. [Google Scholar] [CrossRef]
- Barbier, P.; Zejneli, O.; Martinho, M.; Lasorsa, A.; Belle, V.; Smet-Nocca, C.; Tsvetkov, P.O.; Devred, F.; Landrieu, I. Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects. Front. Aging Neurosci. 2019, 11, 204. [Google Scholar] [CrossRef]
- Lippens, G.; Landrieu, I.; Smet, C.; Huvent, I.; Gandhi, N.; Gigant, B.; Despres, C.; Qi, H.; Lopez, J. NMR Meets Tau: Insights into Its Function and Pathology. Biomolecules 2016, 6, 28. [Google Scholar] [CrossRef]
- Jouanne, M.; Rault, S.; Voisin-Chiret, A.-S. Tau protein aggregation in Alzheimer’s disease: An attractive target for the development of novel therapeutic agents. Eur. J. Med. Chem. 2017, 139, 153–167. [Google Scholar] [CrossRef]
- Fichou, Y.; Al-Hilaly, Y.K.; Devred, F.; Smet-Nocca, C.; Tsvetkov, P.O.; Verelst, J.; Winderickx, J.; Geukens, N.; Vanmechelen, E.; Perrotin, A.; et al. The elusive tau molecular structures: Can we translate the recent breakthroughs into new targets for intervention? Acta Neuropathol. Commun. 2019, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, A.; Falcon, B.; He, S.; Murzin, A.; Murshudov, G.; Garringer, H.; Crowther, A.; Ghetti, B.F.; Goedert, M.; Scheres, S. CRYO-EM STRUCTURES OF TAU FILAMENTS FROM ALZHEIMER’S DISEASE BRAIN. Alzheimer’s Dement. 2017, 13, P892. [Google Scholar] [CrossRef]
- Selkoe, D.J. Resolving controversies on the path to Alzheimer’s therapeutics. Nat. Med. 2011, 17, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Van der Kant, R.; Goldstein, L.S.B.; Ossenkoppele, R. Amyloid-β-independent regulators of tau pathology in Alzheimer disease. Nat. Rev. Neurosci 2020, 21, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. Embo Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Zempel, H.; Thies, E.; Mandelkow, E.; Mandelkow, E.-M. A Oligomers Cause Localized Ca2+ Elevation, Missorting of Endogenous Tau into Dendrites, Tau Phosphorylation, and Destruction of Microtubules and Spines. J. Neurosci. 2010, 30, 11938–11950. [Google Scholar] [CrossRef]
- Paula, V.D.J.R.D.; Guimarães, F.M.; Diniz, B.S.; Forlenza, O.V. Neurobiological pathways to Alzheimer’s disease: Amyloid-beta, TAU protein or both? Dement. Neuropsychol. 2009, 3, 188–194. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K. The preclinical phase of the pathological process underlying sporadic Alzheimer’s disease. Brain 2015, 138, 2814–2833. [Google Scholar] [CrossRef]
- Manczak, M.; Reddy, P.H. Abnormal Interaction of Oligomeric Amyloid-β with Phosphorylated Tau: Implications to Synaptic Dysfunction and Neuronal Damage. JAD 2013, 36, 285–295. [Google Scholar] [CrossRef]
- Ren, B.; Zhang, Y.; Zhang, M.; Liu, Y.; Zhang, D.; Gong, X.; Feng, Z.; Tang, J.; Chang, Y.; Zheng, J. Fundamentals of cross-seeding of amyloid proteins: An introduction. J. Mater. Chem. B 2019, 7, 7267–7282. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Busche, M.A.; Wegmann, S.; Dujardin, S.; Commins, C.; Schiantarelli, J.; Klickstein, N.; Kamath, T.V.; Carlson, G.A.; Nelken, I.; Hyman, B.T. Tau impairs neural circuits, dominating amyloid-β effects, in Alzheimer models in vivo. Nat. Neurosci. 2019, 22, 57–64. [Google Scholar] [CrossRef]
- Terwel, D.; Muyllaert, D.; Dewachter, I.; Borghgraef, P.; Croes, S.; Devijver, H.; Van Leuven, F. Amyloid Activates GSK-3β to Aggravate Neuronal Tauopathy in Bigenic Mice. Am. J. Pathol. 2008, 172, 786–798. [Google Scholar] [CrossRef]
- De Felice, F.G.; Wu, D.; Lambert, M.P.; Fernandez, S.J.; Velasco, P.T.; Lacor, P.N.; Bigio, E.H.; Jerecic, J.; Acton, P.J.; Shughrue, P.J.; et al. Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by Aβ oligomers. Neurobiol. Aging 2008, 29, 1334–1347. [Google Scholar] [CrossRef]
- Hernández, F.; Gómez de Barreda, E.; Fuster-Matanzo, A.; Lucas, J.J.; Avila, J. GSK3: A possible link between beta amyloid peptide and tau protein. Exp. Neurol. 2010, 223, 322–325. [Google Scholar] [CrossRef] [PubMed]
- Imamura, T.; Yanagihara, Y.T.; Ohyagi, Y.; Nakamura, N.; Iinuma, K.M.; Yamasaki, R.; Asai, H.; Maeda, M.; Murakami, K.; Irie, K.; et al. Insulin deficiency promotes formation of toxic amyloid-β42 conformer co-aggregating with hyper-phosphorylated tau oligomer in an Alzheimer’s disease model. Neurobiol. Dis. 2020, 137, 104739. [Google Scholar] [CrossRef] [PubMed]
- Ikezu, S.; Ingraham Dixie, K.L.; Koro, L.; Watanabe, T.; Kaibuchi, K.; Ikezu, T. Tau-tubulin kinase 1 and amyloid-β peptide induce phosphorylation of collapsin response mediator protein-2 and enhance neurite degeneration in Alzheimer disease mouse models. Acta Neuropathol. Commun. 2020, 8, 12. [Google Scholar] [CrossRef]
- Garwood, C.J.; Pooler, A.M.; Atherton, J.; Hanger, D.P.; Noble, W. Astrocytes are important mediators of Aβ-induced neurotoxicity and tau phosphorylation in primary culture. Cell Death Dis. 2011, 2, e167. [Google Scholar] [CrossRef]
- Stancu, I.-C.; Vasconcelos, B.; Terwel, D.; Dewachter, I. Models of β-amyloid induced Tau-pathology: The long and “folded” road to understand the mechanism. Mol. Neurodegener. 2014, 9, 51. [Google Scholar] [CrossRef]
- Qi, R.; Luo, Y.; Wei, G.; Nussinov, R.; Ma, B. Aβ “Stretching-and-Packing” Cross-Seeding Mechanism Can Trigger Tau Protein Aggregation. J. Phys. Chem. Lett. 2015, 6, 3276–3282. [Google Scholar] [CrossRef]
- McAllister, B.B.; Lacoursiere, S.G.; Sutherland, R.J.; Mohajerani, M.H. Intracerebral seeding of amyloid-β and tau pathology in mice: Factors underlying prion-like spreading and comparisons with α-synuclein. Neurosci. Biobehav. Rev. 2020, 112, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J. Enhanced Neurofibrillary Degeneration in Transgenic Mice Expressing Mutant Tau and APP. Science 2001, 293, 1487–1491. [Google Scholar] [CrossRef] [PubMed]
- Gotz, J. Formation of Neurofibrillary Tangles in P301L Tau Transgenic Mice Induced by Abeta 42 Fibrils. Science 2001, 293, 1491–1495. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic Model of Alzheimer’s Disease With Plaques and Tangles: Intracellular Abeta and Synaptic Dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Bolmont, T.; Clavaguera, F.; Meyer-Luehmann, M.; Herzig, M.C.; Radde, R.; Staufenbiel, M.; Lewis, J.; Hutton, M.; Tolnay, M.; Jucker, M. Induction of Tau Pathology by Intracerebral Infusion of Amyloid-β-Containing Brain Extract and by Amyloid-β Deposition in APP × Tau Transgenic Mice. Am. J. Pathol. 2007, 171, 2012–2020. [Google Scholar] [CrossRef]
- Vasconcelos, B.; Stancu, I.-C.; Buist, A.; Bird, M.; Wang, P.; Vanoosthuyse, A.; Van Kolen, K.; Verheyen, A.; Kienlen-Campard, P.; Octave, J.-N.; et al. Heterotypic seeding of Tau fibrillization by pre-aggregated Abeta provides potent seeds for prion-like seeding and propagation of Tau-pathology in vivo. Acta Neuropathol. 2016, 131, 549–569. [Google Scholar] [CrossRef]
- Bennett, R.E.; DeVos, S.L.; Dujardin, S.; Corjuc, B.; Gor, R.; Gonzalez, J.; Roe, A.D.; Frosch, M.P.; Pitstick, R.; Carlson, G.A.; et al. Enhanced Tau Aggregation in the Presence of Amyloid β. Am. J. Pathol. 2017, 187, 1601–1612. [Google Scholar] [CrossRef]
- He, Z.; Guo, J.L.; McBride, J.D.; Narasimhan, S.; Kim, H.; Changolkar, L.; Zhang, B.; Gathagan, R.J.; Yue, C.; Dengler, C.; et al. Amyloid-β plaques enhance Alzheimer’s brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat. Med. 2018, 24, 29–38. [Google Scholar] [CrossRef]
- Vergara, C.; Houben, S.; Suain, V.; Yilmaz, Z.; De Decker, R.; Vanden Dries, V.; Boom, A.; Mansour, S.; Leroy, K.; Ando, K.; et al. Amyloid-β pathology enhances pathological fibrillary tau seeding induced by Alzheimer PHF in vivo. Acta Neuropathol. 2019, 137, 397–412. [Google Scholar] [CrossRef]
- Saito, T.; Mihira, N.; Matsuba, Y.; Sasaguri, H.; Hashimoto, S.; Narasimhan, S.; Zhang, B.; Murayama, S.; Higuchi, M.; Lee, V.M.Y.; et al. Humanization of the entire murine Mapt gene provides a murine model of pathological human tau propagation. J. Biol. Chem. 2019, 294, 12754–12765. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.S.; Di, J.; Murray, K.A.; Sun, C.; Li, B.; Bitan, G.; Jiang, L. Different Amyloid-β Self-Assemblies Have Distinct Effects on Intracellular Tau Aggregation. Front. Mol. Neurosci. 2019, 12, 268. [Google Scholar] [CrossRef] [PubMed]
- Di Scala, C.; Yahi, N.; Boutemeur, S.; Flores, A.; Rodriguez, L.; Chahinian, H.; Fantini, J. Common molecular mechanism of amyloid pore formation by Alzheimer’s β-amyloid peptide and α-synuclein. Sci Rep. 2016, 6, 28781. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.S.; Di, J.; Cao, Q.; Li, B.; Seidler, P.M.; Murray, K.A.; Bitan, G.; Jiang, L. Amyloid β-protein oligomers promote the uptake of tau fibril seeds potentiating intracellular tau aggregation. Alz. Res. Ther. 2019, 11, 86. [Google Scholar] [CrossRef]
- Wallin, C.; Hiruma, Y.; Wärmländer, S.K.T.S.; Huvent, I.; Jarvet, J.; Abrahams, J.P.; Gräslund, A.; Lippens, G.; Luo, J. The Neuronal Tau Protein Blocks in Vitro Fibrillation of the Amyloid-β (Aβ) Peptide at the Oligomeric Stage. J. Am. Chem. Soc. 2018, 140, 8138–8146. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, Y.H.; Hebisch, M.; Sliwinski, C.; Lee, S.; D’Avanzo, C.; Chen, H.; Hooli, B.; Asselin, C.; Muffat, J.; et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 2014, 515, 274–278. [Google Scholar] [CrossRef]
- Kim, Y.H.; Choi, S.H.; D’Avanzo, C.; Hebisch, M.; Sliwinski, C.; Bylykbashi, E.; Washicosky, K.J.; Klee, J.B.; Brüstle, O.; Tanzi, R.E.; et al. A 3D human neural cell culture system for modeling Alzheimer’s disease. Nat. Protoc. 2015, 10, 985–1006. [Google Scholar] [CrossRef]
- Kwak, S.S.; Washicosky, K.J.; Brand, E.; von Maydell, D.; Aronson, J.; Kim, S.; Capen, D.E.; Cetinbas, M.; Sadreyev, R.; Ning, S.; et al. Amyloid-β42/40 ratio drives tau pathology in 3D human neural cell culture models of Alzheimer’s disease. Nat. Commun. 2020, 11, 1377. [Google Scholar] [CrossRef]
- Bitan, G.; Kirkitadze, M.D.; Lomakin, A.; Vollers, S.S.; Benedek, G.B.; Teplow, D.B. Amyloid -protein (A ) assembly: A 40 and A 42 oligomerize through distinct pathways. Proc. Natl. Acad. Sci. USA 2003, 100, 330–335. [Google Scholar] [CrossRef]
- Johnson, R.D.; Schauerte, J.A.; Chang, C.-C.; Wisser, K.C.; Althaus, J.C.; Carruthers, C.J.L.; Sutton, M.A.; Steel, D.G.; Gafni, A. Single-Molecule Imaging Reveals Aβ42:Aβ40 Ratio-Dependent Oligomer Growth on Neuronal Processes. Biophys. J. 2013, 104, 894–903. [Google Scholar] [CrossRef]
- Chang, Y.-J.; Chen, Y.-R. The coexistence of an equal amount of Alzheimer’s amyloid-β 40 and 42 forms structurally stable and toxic oligomers through a distinct pathway. FEBS J. 2014, 281, 2674–2687. [Google Scholar] [CrossRef] [PubMed]
- McGowan, E.; Pickford, F.; Kim, J.; Onstead, L.; Eriksen, J.; Yu, C.; Skipper, L.; Murphy, M.P.; Beard, J.; Das, P.; et al. Aβ42 Is Essential for Parenchymal and Vascular Amyloid Deposition in Mice. Neuron 2005, 47, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.M.; Bernstein, S.L.; Nyugen, V.; Condron, M.M.; Teplow, D.B.; Bowers, M.T. Amyloid β Protein: Aβ40 Inhibits Aβ42 Oligomerization. J. Am. Chem. Soc. 2009, 131, 6316–6317. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, X.; Zhao, M.; Gottesdiener, A.; Luo, W.; Paul, S. Tau pathogenesis is promoted by Aβ1-42 but not Aβ1-40. Mol. Neurodegener. 2014, 9, 52. [Google Scholar] [CrossRef]
- Rojas, A.V.; Maisuradze, G.G.; Scheraga, H.A. Dependence of the Formation of Tau and Aβ Peptide Mixed Aggregates on the Secondary Structure of the N-Terminal Region of Aβ. J. Phys. Chem. B 2018, 122, 7049–7056. [Google Scholar] [CrossRef]
- Mohamed, T.; Gujral, S.S.; Rao, P.P.N. Tau Derived Hexapeptide AcPHF6 Promotes Beta-Amyloid (Aβ) Fibrillogenesis. Acs Chem. Neurosci. 2018, 9, 773–782. [Google Scholar] [CrossRef]
- Griner, S.L.; Seidler, P.; Bowler, J.; Murray, K.A.; Yang, T.P.; Sahay, S.; Sawaya, M.R.; Cascio, D.; Rodriguez, J.A.; Philipp, S.; et al. Structure-based inhibitors of amyloid beta core suggest a common interface with tau. eLife 2019, 8, e46924. [Google Scholar] [CrossRef]
- Miller, Y.; Ma, B.; Nussinov, R. Synergistic Interactions between Repeats in Tau Protein and Aβ Amyloids May Be Responsible for Accelerated Aggregation via Polymorphic States. Biochemistry 2011, 50, 5172–5181. [Google Scholar] [CrossRef]
- Tripathi, T.; Khan, H. Direct Interaction between the β-Amyloid Core and Tau Facilitates Cross-Seeding: A Novel Target for Therapeutic Intervention. Biochemistry 2020, 59, 341–342. [Google Scholar] [CrossRef]
- Gomes, L.A.; Hipp, S.A.; Rijal Upadhaya, A.; Balakrishnan, K.; Ospitalieri, S.; Koper, M.J.; Largo-Barrientos, P.; Uytterhoeven, V.; Reichwald, J.; Rabe, S.; et al. Aβ-induced acceleration of Alzheimer-related τ-pathology spreading and its association with prion protein. Acta Neuropathol. 2019, 138, 913–941. [Google Scholar] [CrossRef]
- Wojtczak, A.; Neumann, P.; Cody, V. Structure of a new polymorphic monoclinic form of human transthyretin at 3 Å resolution reveals a mixed complex between unliganded and T4-bound tetramers of TTR. Acta Crystallogr. Sect. D 2001, 57, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Martone, R.L.; Herbert, J.; Dwork, A.; Schon, E.A. Transthyretin is synthesized in the mammalian eye. Biochem. Biophys. Res. Commun. 1988, 151, 905–912. [Google Scholar] [CrossRef]
- Landers, K.A.; Mortimer, R.H.; Richard, K. Transthyretin and the human placenta. Placenta 2013, 34, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.A.; Benson, M.D. Transthyretin: A review from a structural perspective. Cell. Mol. Life Sci. 2001, 58, 1491–1521. [Google Scholar] [CrossRef] [PubMed]
- Cornwell, G.G.; Sletten, K.; Johansson, B.; Westermark, P. Evidence that the amyloid fibril protein in senile systemic amyloidosis is derived from normal prealbumin. Biochem. Biophys. Res. Commun. 1988, 154, 648–653. [Google Scholar] [CrossRef]
- Brito, R.M.M.; Damas, A.M.; Saraiva, M.J. Amyloid Formation by Transthyretin: From Protein Stability to Protein Aggregation. Available online: http://www.eurekaselect.com/91733/article (accessed on 20 April 2020).
- Westermark, P.; Sletten, K.; Johansson, B.; Cornwell, G.G. Fibril in senile systemic amyloidosis is derived from normal transthyretin. Proc. Natl. Acad. Sci. USA 1990, 87, 2843–2845. [Google Scholar] [CrossRef]
- Tantau, A.; Laszlo, M.; Laszlo, I. Transthyretin amyloidosis: An over review. Cardiovasc. Regen. Med. 2015, 9, 387–404. [Google Scholar]
- Nencetti, S.; Orlandini, E. TTR Fibril Formation Inhibitors: Is there a SAR? Curr. Med. Chem. 2012, 19, 2356–2379. [Google Scholar]
- Guo, X.; Liu, Z.; Zheng, Y.; Li, Y.; Li, L.; Liu, H.; Chen, Z.; Wu, L. Review on the Structures and Activities of Transthyretin Amyloidogenesis Inhibitors. Available online: https://www.dovepress.com/review-on-the-structures-and-activities-of-transthyretin-amyloidogenes-peer-reviewed-fulltext-article-DDDT (accessed on 22 April 2020).
- Ciccone, L.; Nencetti, S.; Rossello, A.; Tepshi, L.; Stura, E.A.; Orlandini, E. X-ray crystal structure and activity of fluorenyl-based compounds as transthyretin fibrillogenesis inhibitors. J. Enzym. Inhib. Med. Chem. 2016, 31, 824–833. [Google Scholar] [CrossRef]
- Ciccone, L.; Nencetti, S.; Rossello, A.; Stura, E.A.; Orlandini, E. Synthesis and structural analysis of halogen substituted fibril formation inhibitors of Human Transthyretin (TTR). J. Enzym. Inhib. Med. Chem. 2016, 31, 40–51. [Google Scholar] [CrossRef][Green Version]
- Palaninathan, S.K.; Mohamedmohaideen, N.N.; Orlandini, E.; Ortore, G.; Nencetti, S.; Lapucci, A.; Rossello, A.; Freundlich, J.S.; Sacchettini, J.C. Novel Transthyretin Amyloid Fibril Formation Inhibitors: Synthesis, Biological Evaluation, and X-Ray Structural Analysis. PLoS ONE 2009, 4, e6290. [Google Scholar] [CrossRef] [PubMed]
- Ortore, G.; Orlandini, E.; Braca, A.; Ciccone, L.; Rossello, A.; Martinelli, A.; Nencetti, S. Targeting Different Transthyretin Binding Sites with Unusual Natural Compounds. ChemMedChem 2016, 11, 1865–1874. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, L.; Tonali, N.; Nencetti, S.; Orlandini, E. Natural compounds as inhibitors of transthyretin amyloidosis and neuroprotective agents: Analysis of structural data for future drug design. J. Enzym. Inhib. Med. Chem. 2020, 35, 1145–1162. [Google Scholar] [CrossRef] [PubMed]
- Schwarzman, A.L.; Gregori, L.; Vitek, M.P.; Lyubski, S.; Strittmatter, W.J.; Enghilde, J.J.; Bhasin, R.; Silverman, J.; Weisgraber, K.H.; Coyle, P.K. Transthyretin sequesters amyloid ft protein and prevents amyloid formation. Proc. Natl. Acad. Sci. USA 1994, 91, 8368–8372. [Google Scholar] [CrossRef]
- Han, S.-H.; Jung, E.S.; Sohn, J.-H.; Hong, H.J.; Hong, H.S.; Kim, J.W.; Na, D.L.; Kim, M.; Kim, H.; Ha, H.J.; et al. Human Serum Transthyretin Levels Correlate Inversely with Alzheimer’s Disease. JAD 2011, 25, 77–84. [Google Scholar] [CrossRef]
- Serot, J.-M.; Christmann, D.; Dubost, T.; Couturier, M. Cerebrospinal fluid transthyretin: Aging and late onset Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 1997, 63, 506–508. [Google Scholar] [CrossRef]
- Li, X.; Buxbaum, J.N. Transthyretin and the brain re-visited: Is neuronal synthesis of transthyretin protective in Alzheimer’s disease? Mol. Neurodegener. 2011, 6, 79. [Google Scholar] [CrossRef]
- Oliveira, S.M.; Ribeiro, C.A.; Cardoso, I.; Saraiva, M.J. Gender-Dependent Transthyretin Modulation of Brain Amyloid-β Levels: Evidence from a Mouse Model of Alzheimer’s Disease. JAD 2011, 27, 429–439. [Google Scholar] [CrossRef]
- Li, X.; Zhang, X.; Ladiwala, A.R.A.; Du, D.; Yadav, J.K.; Tessier, P.M.; Wright, P.E.; Kelly, J.W.; Buxbaum, J.N. Mechanisms of Transthyretin Inhibition of -Amyloid Aggregation In Vitro. J. Neurosci. 2013, 33, 19423–19433. [Google Scholar] [CrossRef]
- Ribeiro, C.A.; Oliveira, S.M.; Guido, L.F.; Magalhães, A.; Valencia, G.; Arsequell, G.; Saraiva, M.J.; Cardoso, I. Transthyretin stabilization by iododiflunisal promotes amyloid-β peptide clearance, decreases its deposition, and ameliorates cognitive deficits in an Alzheimer’s disease mouse model. J. Alzheimers Dis. 2014, 39, 357–370. [Google Scholar] [CrossRef]
- Ribeiro, C.A.; Saraiva, M.J.; Cardoso, I. Stability of the Transthyretin Molecule as a Key Factor in the Interaction with A-Beta Peptide—Relevance in Alzheimer’s Disease. PLoS ONE 2012, 7, e45368. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Cho, P.Y.; Yang, D.T.; Murphy, R.M. Identification of beta-amyloid-binding sites on transthyretin. Protein Eng. Des. Sel. 2012, 25, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Murphy, R.M. Characterization of the Interaction of β-Amyloid with Transthyretin Monomers and Tetramers. Biochemistry 2010, 49, 8276–8289. [Google Scholar] [CrossRef] [PubMed]
- Ghadami, S.A.; Chia, S.; Ruggeri, F.S.; Meisl, G.; Bemporad, F.; Habchi, J.; Cascella, R.; Dobson, C.M.; Vendruscolo, M.; Knowles, T.P.J.; et al. Transthyretin Inhibits Primary and Secondary Nucleations of Amyloid-β Peptide Aggregation and Reduces the Toxicity of Its Oligomers. Biomacromolecules 2020, 21, 1112–1125. [Google Scholar] [CrossRef] [PubMed]
- Alemi, M.; Gaiteiro, C.; Ribeiro, C.A.; Santos, L.M.; Gomes, J.R.; Oliveira, S.M.; Couraud, P.-O.; Weksler, B.; Romero, I.; Saraiva, M.J.; et al. Transthyretin participates in beta-amyloid transport from the brain to the liver-involvement of the low-density lipoprotein receptor-related protein 1? Sci. Rep. 2016, 6, 1–15. [Google Scholar] [CrossRef]
- Alemi, M.; Silva, S.C.; Santana, I.; Cardoso, I. Transthyretin stability is critical in assisting beta amyloid clearance—Relevance of transthyretin stabilization in Alzheimer’s disease. Cns Neurosci. 2017, 23, 605–619. [Google Scholar] [CrossRef]
- Yang, D.T.; Joshi, G.; Cho, P.Y.; Johnson, J.A.; Murphy, R.M. Transthyretin as both Sensor and Scavenger of Aβ Oligomers. Biochemistry 2013, 52, 2849–2861. [Google Scholar] [CrossRef]
- Gimeno, A.; Santos, L.M.; Alemi, M.; Rivas, J.; Blasi, D.; Cotrina, E.Y.; Llop, J.; Valencia, G.; Cardoso, I.; Quintana, J.; et al. Insights on the Interaction between Transthyretin and Aβ in Solution. A Saturation Transfer Difference (STD) NMR Analysis of the Role of Iododiflunisal. J. Med. Chem. 2017, 60, 5749–5758. [Google Scholar] [CrossRef]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef]
- Brzyska, M.; Trzesniewska, K.; Wieckowska, A.; Szczepankiewicz, A.; Elbaum, D. Electrochemical and Conformational Consequences of Copper (Cu I and Cu II ) Binding to β-Amyloid(1-40). ChemBioChem 2009, 10, 1045–1055. [Google Scholar] [CrossRef]
- Pietropaolo, A.; Satriano, C.; Strano, G.; La Mendola, D.; Rizzarelli, E. Different zinc(II) complex species and binding modes at Aβ N-terminus drive distinct long range cross-talks in the Aβ monomers. J. Inorg. Biochem. 2015, 153, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-T.; Liao, Y.-H.; Yu, H.-M.; Cheng, I.H.; Chen, Y.-R. Distinct Effects of Zn2+, Cu2+, Fe3+, and Al3+ on Amyloid-β Stability, Oligomerization, and Aggregation: AMYLOID-β DESTABILIZATION PROMOTES ANNULAR PROTOFIBRIL FORMATION. J. Biol. Chem. 2011, 286, 9646–9656. [Google Scholar] [CrossRef] [PubMed]
- Leal, S.S.; Botelho, H.M.; Gomes, C.M. Metal ions as modulators of protein conformation and misfolding in neurodegeneration. Coord. Chem. Rev. 2012, 256, 2253–2270. [Google Scholar] [CrossRef]
- Castro-Rodrigues, A.F.; Gales, L.; Saraiva, M.J.; Damas, A.M. Structural insights into a zinc-dependent pathway leading to Leu55Pro transthyretin amyloid fibrils. Acta Cryst. D Biol. Cryst. 2011, 67, 1035–1044. [Google Scholar] [CrossRef]
- Liz, M.A.; Leite, S.C.; Juliano, L.; Saraiva, M.J.; Damas, A.M.; Bur, D.; Sousa, M.M. Transthyretin is a metallopeptidase with an inducible active site. Biochem. J. 2012, 443, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, L.; Fruchart-Gaillard, C.; Mourier, G.; Savko, M.; Nencetti, S.; Orlandini, E.; Servent, D.; Stura, E.A.; Shepard, W. Copper mediated amyloid-β binding to Transthyretin. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Ciccone, L.; Policar, C.; Stura, E.A.; Shepard, W. Human TTR conformation altered by rhenium tris-carbonyl derivatives. J. Struct. Biol. 2016, 195, 353–364. [Google Scholar] [CrossRef]
- Quintela, T.; Alves, C.H.; Gonçalves, I.; Baltazar, G.; Saraiva, M.J.; Santos, C.R.A. 5Alpha-dihydrotestosterone up-regulates transthyretin levels in mice and rat choroid plexus via an androgen receptor independent pathway. Brain Res. 2008, 1229, 18–26. [Google Scholar] [CrossRef]
- Quintela, T.; Gonçalves, I.; Baltazar, G.; Alves, C.H.; Saraiva, M.J.; Santos, C.R.A. 17beta-estradiol induces transthyretin expression in murine choroid plexus via an oestrogen receptor dependent pathway. Cell. Mol. Neurobiol. 2009, 29, 475–483. [Google Scholar] [CrossRef]
- Quintela, T.; Gonçalves, I.; Martinho, A.; Alves, C.H.; Saraiva, M.J.; Rocha, P.; Santos, C.R.A. Progesterone enhances transthyretin expression in the rat choroid plexus in vitro and in vivo via progesterone receptor. J. Mol. Neurosci. 2011, 44, 152–158. [Google Scholar] [CrossRef]
- Nalivaeva, N.N.; Belyaev, N.D.; Kerridge, C.; Turner, A.J. Amyloid-clearing proteins and their epigenetic regulation as a therapeutic target in Alzheimer’s disease. Front. Aging Neurosci 2014, 6. [Google Scholar] [CrossRef]
- Rios, X.; Gómez-Vallejo, V.; Martín, A.; Cossío, U.; Morcillo, M.Á.; Alemi, M.; Cardoso, I.; Quintana, J.; Jiménez-Barbero, J.; Cotrina, E.Y.; et al. Radiochemical examination of transthyretin (TTR) brain penetration assisted by iododiflunisal, a TTR tetramer stabilizer and a new candidate drug for AD. Sci. Rep. 2019, 9, 13672. [Google Scholar] [CrossRef] [PubMed]
- Cotrina, E.Y.; Gimeno, A.; Llop, J.; Jiménez-Barbero, J.; Quintana, J.; Valencia, G.; Cardoso, I.; Prohens, R.; Arsequell, G. Calorimetric Studies of Binary and Ternary Molecular Interactions between Transthyretin, Aβ Peptides, and Small-Molecule Chaperones toward an Alternative Strategy for Alzheimer’s Disease Drug Discovery. J. Med. Chem. 2020, 63, 3205–3214. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.M.; Rodrigues, D.; Alemi, M.; Silva, S.C.; Ribeiro, C.A.; Cardoso, I. Resveratrol Administration Increases Transthyretin Protein Levels, Ameliorating AD Features: The Importance of Transthyretin Tetrameric Stability. Mol Med. 2016, 22, 597–607. [Google Scholar] [CrossRef]
- Velayudhan, L.; Killick, R.; Hye, A.; Kinsey, A.; Güntert, A.; Lynham, S.; Ward, M.; Leung, R.; Lourdusamy, A.; To, A.W.M.; et al. Plasma transthyretin as a candidate marker for Alzheimer’s disease. J. Alzheimers Dis. 2012, 28, 369–375. [Google Scholar] [CrossRef]
- Robertson, N.S.; Spring, D.R. Using Peptidomimetics and Constrained Peptides as Valuable Tools for Inhibiting Protein−Protein Interactions. Molecules 2018, 23, 959. [Google Scholar] [CrossRef]
- Ran, X.; Gestwicki, J.E. Inhibitors of protein-protein interactions (PPIs): An analysis of scaffold choices and buried surface area. Curr. Opin. Chem. Biol. 2018, 44, 75–86. [Google Scholar] [CrossRef]
- Bruzzoni-Giovanelli, H.; Alezra, V.; Wolff, N.; Dong, C.-Z.; Tuffery, P.; Rebollo, A. Interfering peptides targeting protein-protein interactions: The next generation of drugs? Drug Discov. Today 2018, 23, 272–285. [Google Scholar] [CrossRef]
- Laxio Arenas, J.; Kaffy, J.; Ongeri, S. Peptides and peptidomimetics as inhibitors of protein-protein interactions involving β-sheet secondary structures. Curr. Opin. Chem. Biol. 2019, 52, 157–167. [Google Scholar] [CrossRef]
- Cho, P.Y.; Joshi, G.; Johnson, J.A.; Murphy, R.M. Transthyretin-Derived Peptides as β-Amyloid Inhibitors. Acs Chem. Neurosci. 2014, 5, 542–551. [Google Scholar] [CrossRef]
- Cho, P.Y.; Joshi, G.; Boersma, M.D.; Johnson, J.A.; Murphy, R.M. A Cyclic Peptide Mimic of the β-Amyloid Binding Domain on Transthyretin. Acs Chem. Neurosci. 2015, 6, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Brickson, C.R.; Murphy, R.M. TANGO-Inspired Design of Anti-Amyloid Cyclic Peptides. Acs Chem. Neurosci. 2016, 7, 1264–1274. [Google Scholar] [CrossRef] [PubMed]
- Pate, K.M.; Kim, B.J.; Shusta, E.V.; Murphy, R.M. Transthyretin Mimetics as Anti-β-Amyloid Agents: A Comparison of Peptide and Protein Approaches. ChemMedChem 2018, 13, 968–979. [Google Scholar] [CrossRef] [PubMed]
- Tonali, N.; Correia, I.; Lesma, J.; Bernadat, G.; Ongeri, S.; Lequin, O. Introducing sequential aza-amino acids units induces repeated β-turns and helical conformations in peptides. Org. Biomol. Chem. 2020, 18, 3452–3458. [Google Scholar] [CrossRef]
- Turk, V.; Bode, W. The cystatins: Protein inhibitors of cysteine proteinases. Febs Lett. 1991, 285, 213–219. [Google Scholar] [CrossRef]
- Abrahamson, M.; Barrett, A.J.; Salvesen, G.; Grubb, A. Isolation of six cysteine proteinase inhibitors from human urine. Their physicochemical and enzyme kinetic properties and concentrations in biological fluids. J. Biol. Chem. 1986, 261, 11282–11289. [Google Scholar]
- Håkansson, K.; Huh, C.; Grubb, A.; Karlsson, S.; Abrahamson, M. Mouse and rat cystatin C: Escherichia coli production, characterization and tissue distribution. Comp. Biochem. Physiol. Part. B Biochem. Mol. Biol. 1996, 114, 303–311. [Google Scholar] [CrossRef]
- Bernstein, H.-G.; Kirschke, H.; Wiederanders, B.; Pollak, K.-H.; Zipress, A.; Rinne, A. The possible place of cathepsins and cystatins in the puzzle of Alzheimer disease: A review. Mol. Chem. Neuropathol. 1996, 27, 225–247. [Google Scholar] [CrossRef]
- Lenarčič, B.; Krašovec, M.; Ritonja, A.; Olafsson, I.; Turk, V. Inactivation of human cystatin C and kininogen by human cathepsin D. Febs Lett. 1991, 280, 211–215. [Google Scholar] [CrossRef]
- Ekiel, I.; Abrahamson, M. Folding-related Dimerization of Human Cystatin C. J. Biol. Chem. 1996, 271, 1314–1321. [Google Scholar] [CrossRef]
- Wahlbom, M.; Wang, X.; Lindström, V.; Carlemalm, E.; Jaskolski, M.; Grubb, A. Fibrillogenic Oligomers of Human Cystatin C Are Formed by Propagated Domain Swapping. J. Biol. Chem. 2007, 282, 18318–18326. [Google Scholar] [CrossRef] [PubMed]
- Sastre, M.; Calero, M.; Pawlik, M.; Mathews, P.M.; Kumar, A.; Danilov, V.; Schmidt, S.D.; Nixon, R.A.; Frangione, B.; Levy, E. Binding of cystatin C to Alzheimer’s amyloid β inhibits in vitro amyloid fibril formation. Neurobiol. Aging 2004, 25, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Palsdottir, A.; Snorradottir, A.O.; Thorsteinsson, L. Hereditary Cystatin C Amyloid Angiopathy: Genetic, Clinical, and Pathological Aspects. Brain Pathol. 2006, 16, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Levy, E.; Jaskolski, M.; Grubb, A. The Role of Cystatin C in Cerebral Amyloid Angiopathy and Stroke: Cell Biology and Animal Models. Brain Pathol. 2006, 16, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Lundgren, E.; Persson, H.; Andersson, K.; Olofsson, A.; Dacklin, I.; Goldsteins, G. Mapping protein conformations in fibril structures using monoclonal antibodies. Methods Enzymol. 1999, 309, 591–605. [Google Scholar]
- Kolodziejczyk, R.; Michalska, K.; Hernandez-Santoyo, A.; Wahlbom, M.; Grubb, A.; Jaskolski, M. Crystal structure of human cystatin C stabilized against amyloid formation: Structure of monomeric cystatin C. FEBS J. 2010, 277, 1726–1737. [Google Scholar] [CrossRef]
- Janowski, R.; Kozak, M.; Jankowska, E.; Grzonka, Z.; Grubb, A.; Abrahamson, M.; Jaskolski, M. Human cystatin C, an amyloidogenic protein, dimerizes through three-dimensional domain swapping. Nat. Struct. Biol. 2001, 8, 316–320. [Google Scholar] [CrossRef][Green Version]
- Bennett, M.J.; Choe, S.; Eisenberg, D. Domain swapping: Entangling alliances between proteins. Proc. Natl. Acad. Sci. USA 1994, 91, 3127–3131. [Google Scholar] [CrossRef]
- Nilsson, M.; Wang, X.; Rodziewicz-Motowidlo, S.; Janowski, R.; Lindström, V.; Önnerfjord, P.; Westermark, G.; Grzonka, Z.; Jaskolski, M.; Grubb, A. Prevention of Domain Swapping Inhibits Dimerization and Amyloid Fibril Formation of Cystatin C: USE OF ENGINEERED DISULFIDE BRIDGES, ANTIBODIES, AND CARBOXYMETHYLPAPAIN TO STABILIZE THE MONOMERIC FORM OF CYSTATIN C. J. Biol. Chem. 2004, 279, 24236–24245. [Google Scholar] [CrossRef]
- Orlikowska, M.; Jankowska, E.; Kołodziejczyk, R.; Jaskólski, M.; Szymańska, A. Hinge-loop mutation can be used to control 3D domain swapping and amyloidogenesis of human cystatin C. J. Struct. Biol. 2011, 173, 406–413. [Google Scholar] [CrossRef]
- Mathews, P.M.; Levy, E. Cystatin C in aging and in Alzheimer’s disease. Ageing Res. Rev. 2016, 32, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Haan, J.; Maat-Schieman, M.L.; van Duinen, S.G.; Jensson, O.; Thorsteinsson, L.; Roos, R.A. Co-localization of beta/A4 and cystatin C in cortical blood vessels in Dutch, but not in Icelandic hereditary cerebral hemorrhage with amyloidosis. Acta Neurol. Scand. 1994, 89, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Levy, E.; Sastre, M.; Kumar, A.; Gallo, G.; Piccardo, P.; Ghetti, B.; Tagliavini, F. Codeposition of Cystatin C with Amyloid-β Protein in the Brain of Alzheimer Disease Patients. J. Neuropathol. Exp. Neurol. 2001, 60, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Vattemi, G.; King Engel, W.; McFerrin, J.; Askanas, V. Cystatin C colocalizes with amyloid-β and coimmunoprecipitates with amyloid-β precursor protein in sporadic inclusion-body myositis muscles: Cystatin C and amyloid-β in inclusion-body myositis. J. Neurochem. 2003, 85, 1539–1546. [Google Scholar] [CrossRef]
- Wang, B.; Xie, Y.; Yang, Z.; Peng, D.; Wang, J.; Zhou, S.; Li, S.; Ma, X. Lack of an Association between Alzheimer’s Disease and the Cystatin C (CST3) Gene G73A Polymorphism in Mainland Chinese. Dement. Geriatr. Cogn. Disord. 2008, 25, 461–464. [Google Scholar] [CrossRef]
- Hua, Y.; Zhao, H.; Lu, X.; Kong, Y.; Jin, H. Meta-Analysis of the Cystatin C( CST3 ) Gene G73A Polymorphism and Susceptibility to Alzheimer’s Disease. Int. J. Neurosci. 2012, 122, 431–438. [Google Scholar] [CrossRef]
- Beyer, K.; Lao, J.I.; Gómez, M.; Riutort, N.; Latorre, P.; Mate, J.L.; Ariza, A. Alzheimer’s disease and the cystatin C gene polymorphism: An association study. Neurosci. Lett. 2001, 315, 17–20. [Google Scholar] [CrossRef]
- Cathcart, H.M.; Huang, R.; Lanham, I.S.; Corder, E.H.; Poduslo, S.E. Cystatin C as a risk factor for Alzheimer disease. Neurology 2005, 64, 755–757. [Google Scholar] [CrossRef]
- Mueller-Steiner, S.; Zhou, Y.; Arai, H.; Roberson, E.D.; Sun, B.; Chen, J.; Wang, X.; Yu, G.; Esposito, L.; Mucke, L.; et al. Antiamyloidogenic and Neuroprotective Functions of Cathepsin B: Implications for Alzheimer’s Disease. Neuron 2006, 51, 703–714. [Google Scholar] [CrossRef]
- Sun, B.; Zhou, Y.; Halabisky, B.; Lo, I.; Cho, S.-H.; Mueller-Steiner, S.; Devidze, N.; Wang, X.; Grubb, A.; Gan, L. Cystatin C-Cathepsin B Axis Regulates Amyloid Beta Levels and Associated Neuronal Deficits in an Animal Model of Alzheimer’s Disease. Neuron 2008, 60, 247–257. [Google Scholar] [CrossRef]
- Wang, C.; Sun, B.; Zhou, Y.; Grubb, A.; Gan, L. Cathepsin B Degrades Amyloid-β in Mice Expressing Wild-type Human Amyloid Precursor Protein. J. Biol. Chem. 2012, 287, 39834–39841. [Google Scholar] [CrossRef] [PubMed]
- Mi, W.; Jung, S.S.; Yu, H.; Schmidt, S.D.; Nixon, R.A.; Mathews, P.M.; Tagliavini, F.; Levy, E. Complexes of Amyloid-β and Cystatin C in the Human Central Nervous System. JAD 2009, 18, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Kaeser, S.A.; Herzig, M.C.; Coomaraswamy, J.; Kilger, E.; Selenica, M.-L.; Winkler, D.T.; Staufenbiel, M.; Levy, E.; Grubb, A.; Jucker, M. Cystatin C modulates cerebral β-amyloidosis. Nat. Genet. 2007, 39, 1437–1439. [Google Scholar] [CrossRef] [PubMed]
- Juszczyk, P.; Paraschiv, G.; Szymanska, A.; Kolodziejczyk, A.S.; Rodziewicz-Motowidlo, S.; Grzonka, Z.; Przybylski, M. Binding Epitopes and Interaction Structure of the Neuroprotective Protease Inhibitor Cystatin C with β-Amyloid Revealed by Proteolytic Excision Mass Spectrometry and Molecular Docking Simulation. J. Med. Chem. 2009, 52, 2420–2428. [Google Scholar] [CrossRef] [PubMed]
- Spodzieja, M.; Kalejta, K.; Kołodziejczyk, A.S.; Maszota-Zieleniak, M.; Rodziewicz-Motowidło, S.; Żmudzińska, W.; Czaplewska, P. Characteristics of C-terminal, β-amyloid peptide binding fragment of neuroprotective protease inhibitor, cystatin C. J. Mol. Recognit. 2017, 30. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.K.; Persichetti, J.; Tale, E.; Prelvukaj, G.; Cropley, T.; Choudhury, R. A computational examination of the binding interactions of amyloidβ and human cystatin C. J. Comput. Chem. 2017, 17, 1850001. [Google Scholar] [CrossRef]
- Iłowska, E.; Sawicka, J.; Szymańska, A. Synthesis and physicochemical studies of amyloidogenic hexapeptides derived from human cystatin C. J. Pept. Sci. 2018, 24, e3073. [Google Scholar] [CrossRef]
- Sorci-Thomas, M.G.; Thomas, M.J. The Effects of Altered Apolipoprotein A-I Structure on Plasma HDL Concentration. Trends Cardiovasc. Med. 2002, 12, 121–128. [Google Scholar] [CrossRef]
- Wang, X.; Rader, D.J. Molecular regulation of macrophage reverse cholesterol transport. Curr. Opin. Cardiol. 2007, 22, 368–372. [Google Scholar] [CrossRef]
- Fielding, C.J.; Fielding, P.E. Molecular physiology of reverse cholesterol transport. J. Lipid Res. 1995, 36, 211–228. [Google Scholar]
- Obici, L.; Franceschini, G.; Calabresi, L.; Giorgetti, S.; Stoppini, M.; Merlini, G.; Bellotti, V. Structure, function and amyloidogenic propensity of apolipoprotein A-I. Amyloid 2006, 13, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Stoffel, W. Synthesis, transport, and processing of apolipoproteins of high density lipoproteins. J. Lipid Res. 1984, 25, 1586–1592. [Google Scholar] [PubMed]
- Brewer, H.B.; Fairwell, T.; Kay, L.; Meng, M.; Ronan, R.; Law, S.; Light, J.A. Human plasma proapoA-I: Isolation and amino-terminal sequence. Biochem. Biophys. Res. Commun. 1983, 113, 626–632. [Google Scholar] [CrossRef]
- Gordon, J.I.; Sims, H.F.; Lentz, S.R.; Edelstein, C.; Scanu, A.M.; Strauss, A.W. Proteolytic processing of human preproapolipoprotein A-I. A proposed defect in the conversion of pro A-I to A-I in Tangier’s disease. J. Biol. Chem. 1993, 258, 4037–4044. [Google Scholar]
- Karathanasis, S.K.; Zannis, V.I.; Breslow, J.L. Isolation and characterization of the human apolipoprotein A-I gene. Proc. Natl. Acad. Sci. USA 1983, 80, 6147–6151. [Google Scholar] [CrossRef] [PubMed]
- Rye, K.-A.; Barter, P.J. Formation and metabolism of prebeta-migrating, lipid-poor apolipoprotein A-I. Arter. Thromb. Vasc. Biol. 2004, 24, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P.; Benson, M.D.; Buxbaum, J.N.; Cohen, A.S.; Frangione, B.; Ikeda, S.-I.; Masters, C.L.; Merlini, G.; Saraiva, M.J.; Sipe, J.D.; et al. Amyloid: Toward terminology clarification. Report from the Nomenclature Committee of the International Society of Amyloidosis. Amyloid 2005, 12, 1–4. [Google Scholar] [CrossRef]
- Borhani, D.W.; Rogers, D.P.; Engler, J.A.; Brouillette, C.G. Crystal structure of truncated human apolipoprotein A-I suggests a lipid-bound conformation. Proc. Natl. Acad. Sci. USA 1997, 94, 12291–12296. [Google Scholar] [CrossRef]
- Paula-Lima, A.C.; Tricerri, M.A.; Brito-Moreira, J.; Bomfim, T.R.; Oliveira, F.F.; Magdesian, M.H.; Grinberg, L.T.; Panizzutti, R.; Ferreira, S.T. Human apolipoprotein A–I binds amyloid-β and prevents Aβ-induced neurotoxicity. Int. J. Biochem. Cell Biol. 2009, 41, 1361–1370. [Google Scholar] [CrossRef]
- Frank, P.G.; Marcel, Y.L. Apolipoprotein A-I: Structure–function relationships. J. Lipid Res. 2000, 41, 853–872. [Google Scholar]
- Westermark, P.; Mucchiano, G.; Marthin, T.; Johnson, K.H.; Sletten, K. Apolipoprotein A1-derived amyloid in human aortic atherosclerotic plaques. Am. J. Pathol. 1995, 147, 1186–1192. [Google Scholar] [PubMed]
- Girych, M.; Gorbenko, G.; Trusova, V.; Adachi, E.; Mizuguchi, C.; Nagao, K.; Kawashima, H.; Akaji, K.; Lund-Katz, S.; Phillips, M.C.; et al. Interaction of Thioflavin T with amyloid fibrils of apolipoprotein A-I N-terminal fragment: Resonance energy transfer study. J. Struct. Biol. 2014, 185, 116–124. [Google Scholar] [CrossRef]
- Röcken, C.; Shakespeare, A. Pathology, diagnosis and pathogenesis of AA amyloidosis. Virchows Arch. 2002, 440, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.H.; Lau, H.S.H.; Kezdy, F.J.; Kaiser, E.T. The use of polymer-bound oximes for the synthesis of large peptides usable in segment condensation: Synthesis of a 44 amino acid amphiphilic peptide model of apolipoprotein A-1. J. Am. Chem. Soc. 1985, 107, 7087–7092. [Google Scholar] [CrossRef]
- Kanellis, P.; Romans, A.Y.; Johnson, B.J.; Kercret, H.; Chiovetti, R.; Allen, T.M.; Segrest, J.P. Studies of synthetic peptide analogs of the amphipathic helix. Effect of charged amino acid residue topography on lipid affinity. J. Biol. Chem. 1980, 255, 11464–11472. [Google Scholar]
- Segrest, J.P.; Jones, M.K.; De Loof, H.; Brouillette, C.G.; Venkatachalapathi, Y.V.; Anantharamaiah, G.M. The amphipathic helix in the exchangeable apolipoproteins: A review of secondary structure and function. J. Lipid Res. 1992, 33, 141–166. [Google Scholar]
- Leman, L.J.; Maryanoff, B.E.; Ghadiri, M.R. Molecules That Mimic Apolipoprotein A-I: Potential Agents for Treating Atherosclerosis. J. Med. Chem. 2014, 57, 2169–2196. [Google Scholar] [CrossRef] [PubMed]
- Mei, X.; Atkinson, D. Crystal Structure of C-terminal Truncated Apolipoprotein A-I Reveals the Assembly of High Density Lipoprotein (HDL) by Dimerization. J. Biol. Chem. 2011, 286, 38570–38582. [Google Scholar] [CrossRef]
- Gregorini, G.; Izzi, C.; Obici, L.; Tardanico, R.; Röcken, C.; Viola, B.F.; Capistrano, M.; Donadei, S.; Biasi, L.; Scalvini, T.; et al. Renal apolipoprotein A-I amyloidosis: A rare and usually ignored cause of hereditary tubulointerstitial nephritis. J. Am. Soc. Nephrol. 2005, 16, 3680–3686. [Google Scholar] [CrossRef]
- Soutar, A.K.; Hawkins, P.N.; Vigushin, D.M.; Tennent, G.A.; Booth, S.E.; Hutton, T.; Nguyen, O.; Totty, N.F.; Feest, T.G.; Hsuan, J.J. Apolipoprotein AI mutation Arg-60 causes autosomal dominant amyloidosis. Proc. Natl. Acad. Sci. USA 1992, 89, 7389–7393. [Google Scholar] [CrossRef]
- Nichols, W.C.; Dwulet, F.E.; Liepnieks, J.; Benson, M.D. Variant apolipoprotein AI as a major constituent of a human hereditary amyloid. Biochem. Biophys. Res. Commun. 1988, 156, 762–768. [Google Scholar] [CrossRef]
- Mucchiano, G.I.; Jonasson, L.; Häggqvist, B.; Einarsson, E.; Westermark, P. Apolipoprotein A-I-derived amyloid in atherosclerosis. Its association with plasma levels of apolipoprotein A-I and cholesterol. Am. J. Clin. Pathol. 2001, 115, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.; Schönland, S.; Yumlu, S.; Hegenbart, U.; von Hutten, H.; Gioeva, Z.; Lohse, P.; Büttner, J.; Schmidt, H.; Röcken, C. Hereditary Apolipoprotein AI-Associated Amyloidosis in Surgical Pathology Specimens. J. Mol. Diagn. 2009, 11, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Mizuguchi, C.; Nakagawa, M.; Namba, N.; Sakai, M.; Kurimitsu, N.; Suzuki, A.; Fujita, K.; Horiuchi, S.; Baba, T.; Ohgita, T.; et al. Mechanisms of aggregation and fibril formation of the amyloidogenic N-terminal fragment of apolipoprotein A-I. J. Biol. Chem. 2019, 294, 13515–13524. [Google Scholar] [CrossRef]
- Benson, M.D. The hereditary amyloidoses. Best Pr. Res. Clin. Rheumatol. 2003, 17, 909–927. [Google Scholar] [CrossRef]
- Nichols, W.C.; Gregg, R.E.; Brewer, H.B.; Benson, M.D. A mutation in apolipoprotein A-I in the Iowa type of familial amyloidotic polyneuropathy. Genomics 1990, 8, 318–323. [Google Scholar] [CrossRef]
- Lagerstedt, J.O.; Cavigiolio, G.; Roberts, L.M.; Hong, H.-S.; Jin, L.-W.; Fitzgerald, P.G.; Oda, M.N.; Voss, J.C. Mapping the structural transition in an amyloidogenic apolipoprotein A-I. Biochemistry 2007, 46, 9693–9699. [Google Scholar] [CrossRef]
- Gursky, O.; Mei, X.; Atkinson, D. The Crystal Structure of the C-Terminal Truncated Apolipoprotein A-I Sheds New Light on Amyloid Formation by the N-Terminal Fragment. Biochemistry 2012, 51, 10–18. [Google Scholar] [CrossRef]
- Chetty, P.S.; Ohshiro, M.; Saito, H.; Dhanasekaran, P.; Lund-Katz, S.; Mayne, L.; Englander, W.; Phillips, M.C. Effects of the Iowa and Milano Mutations on Apolipoprotein A-I Structure and Dynamics Determined by Hydrogen Exchange and Mass Spectrometry. Biochemistry 2012, 51, 8993–9001. [Google Scholar] [CrossRef]
- Wisniewski, T.; Golabek, A.A.; Kida, E.; Wisniewski, K.E.; Frangione, B. Conformational mimicry in Alzheimer’s disease. Role of apolipoproteins in amyloidogenesis. Am. J. Pathol. 1995, 147, 238–244. [Google Scholar]
- Koudinov, A.R.; Berezov, T.T.; Kumar, A.; Koudinova, N.V. Alzheimer’s amyloid β interaction with normal human plasma high density lipoprotein: Association with apolipoprotein and lipids. Clin. Chim. Acta 1998, 270, 75–84. [Google Scholar] [CrossRef]
- Koudinov, A.; Matsubara, E.; Frangione, B.; Ghiso, J. The soluble form of Alzheimer’s amyloid beta protein is complexed to high density lipoprotein 3 and very high density lipoprotein in normal human plasma. Biochem. Biophys. Res. Commun. 1994, 205, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Kawano, M.; Kawakami, M.; Otsuka, M.; Yashima, H.; Yaginuma, T.; Ueki, A. Marked decrease of plasma apolipoprotein AI and AII in Japanese patients with late-onset non-familial Alzheimer’s disease. Clin. Chim. Acta 1995, 239, 209–211. [Google Scholar] [CrossRef]
- Vollbach, H.; Heun, R.; Morris, C.M.; Edwardson, J.A.; McKeith, I.G.; Jessen, F.; Schulz, A.; Maier, W.; Kölsch, H. APOA1 polymorphism influences risk for early-onset nonfamiliar AD. Ann. Neurol. 2005, 58, 436–441. [Google Scholar] [CrossRef]
- Weisgraber, K.H.; Bersot, T.P.; Mahley, R.W.; Franceschini, G.; Sirtori, C.R. A-Imilano apoprotein. Isolation and characterization of a cysteine-containing variant of the A-I apoprotein from human high density lipoproteins. J. Clin. Investig. 1980, 66, 901–907. [Google Scholar] [CrossRef]
- Weisgraber, K.H.; Rall, S.C.J.; Bersot, T.P.; Mahley, R.W.; Franceschini, G.; Sirtori, C.R. Apolipoprotein A-IMilano. Detection of normal A-I in affected subjects and evidence for a cysteine for arginine substitution in the variant A-I. J. Biol. Chem. 1983, 258, 2508–2513. [Google Scholar]
- Nissen, S.E.; Tsunoda, T.; Tuzcu, E.M.; Schoenhagen, P.; Cooper, C.J.; Yasin, M.; Eaton, G.M.; Lauer, M.A.; Sheldon, W.S.; Grines, C.L.; et al. Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: A randomized controlled trial. JAMA 2003, 290, 2292–2300. [Google Scholar] [CrossRef]
- Fernández-de Retana, S.; Montañola, A.; Marazuela, P.; De La Cuesta, M.; Batlle, A.; Fatar, M.; Grudzenski, S.; Montaner, J.; Hernández-Guillamon, M. Intravenous treatment with human recombinant ApoA-I Milano reduces beta amyloid cerebral deposition in the APP23-transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 2017, 60, 116–128. [Google Scholar] [CrossRef]
- Koldamova, R.P.; Lefterov, I.M.; Lefterova, M.I.; Lazo, J.S. Apolipoprotein A-I Directly Interacts with Amyloid Precursor Protein and Inhibits Aβ Aggregation and Toxicity. Biochemistry 2001, 40, 3553–3560. [Google Scholar] [CrossRef]
- Saczynski, J.S.; White, L.; Peila, R.L.; Rodriguez, B.L.; Launer, L.J. The Relation between Apolipoprotein A-I and DementiaThe Honolulu-Asia Aging Study. Am. J. Epidemiol 2007, 165, 985–992. [Google Scholar] [CrossRef]
- Merched, A.; Xia, Y.; Visvikis, S.; Serot, J.M.; Siest, G. Decreased high-density lipoprotein cholesterol and serum apolipoprotein AI concentrations are highly correlated with the severity of Alzheimer’s disease☆. Neurobiol. Aging 2000, 21, 27–30. [Google Scholar] [CrossRef]
- Kivipelto, M.; Helkala, E.L.; Laakso, M.P.; Hänninen, T.; Hallikainen, M.; Alhainen, K.; Soininen, H.; Tuomilehto, J.; Nissinen, A. Midlife vascular risk factors and Alzheimer’s disease in later life: Longitudinal, population based study. BMJ 2001, 322, 1447–1451. [Google Scholar] [CrossRef] [PubMed]
- Fitz, N.F.; Tapias, V.; Cronican, A.A.; Castranio, E.L.; Saleem, M.; Carter, A.Y.; Lefterova, M.; Lefterov, I.; Koldamova, R. Opposing effects of Apoe/Apoa1 double deletion on amyloid-β pathology and cognitive performance in APP mice. Brain 2015, 138, 3699–3715. [Google Scholar] [CrossRef] [PubMed]
- Dal Magro, R.; Simonelli, S.; Cox, A.; Formicola, B.; Corti, R.; Cassina, V.; Nardo, L.; Mantegazza, F.; Salerno, D.; Grasso, G.; et al. The Extent of Human Apolipoprotein A-I Lipidation Strongly Affects the β-Amyloid Efflux Across the Blood-Brain Barrier in vitro. Front. Neurosci. 2019, 13, 419. [Google Scholar] [CrossRef] [PubMed]
- Lewis, T.L.; Cao, D.; Lu, H.; Mans, R.A.; Su, Y.R.; Jungbauer, L.; Linton, M.F.; Fazio, S.; LaDu, M.J.; Li, L. Overexpression of Human Apolipoprotein A-I Preserves Cognitive Function and Attenuates Neuroinflammation and Cerebral Amyloid Angiopathy in a Mouse Model of Alzheimer Disease. J. Biol. Chem. 2010, 285, 36958–36968. [Google Scholar] [CrossRef] [PubMed]
- Lefterov, I.; Fitz, N.F.; Cronican, A.A.; Fogg, A.; Lefterov, P.; Kodali, R.; Wetzel, R.; Koldamova, R. Apolipoprotein A-I Deficiency Increases Cerebral Amyloid Angiopathy and Cognitive Deficits in APP/PS1ΔE9 Mice. J. Biol. Chem. 2010, 285, 36945–36957. [Google Scholar] [CrossRef] [PubMed]
- Ibanez, B.; Giannarelli, C.; Cimmino, G.; Santos-Gallego, C.G.; Alique, M.; Pinero, A.; Vilahur, G.; Fuster, V.; Badimon, L.; Badimon, J.J. Recombinant HDLMilano exerts greater anti-inflammatory and plaque stabilizing properties than HDLwild-type. Atherosclerosis 2012, 220, 72–77. [Google Scholar] [CrossRef]
- Badimon, J.J.; Badimon, L.; Galvez, A.; Dische, R.; Fuster, V. High density lipoprotein plasma fractions inhibit aortic fatty streaks in cholesterol-fed rabbits. Lab. Investig. 1989, 60, 455–461. [Google Scholar]
- Stoekenbroek, R.M.; Stroes, E.S.; Hovingh, G.K. ApoA-I Mimetics. Handb. Exp. Pharm. 2015, 224, 631–648. [Google Scholar] [CrossRef]
- Bailey, D.; Jahagirdar, R.; Gordon, A.; Hafiane, A.; Campbell, S.; Chatur, S.; Wagner, G.S.; Hansen, H.C.; Chiacchia, F.S.; Johansson, J.; et al. RVX-208: A small molecule that increases apolipoprotein A-I and high-density lipoprotein cholesterol in vitro and in vivo. J. Am. Coll. Cardiol. 2010, 55, 2580–2589. [Google Scholar] [CrossRef]
- Kingwell, B.A.; Chapman, M.J.; Kontush, A.; Miller, N.E. HDL-targeted therapies: Progress, failures and future. Nat. Rev. Drug Discov. 2014, 13, 445–464. [Google Scholar] [CrossRef] [PubMed]
- RVX 208. Drugs R D 2011, 11, 207–213. [CrossRef] [PubMed][Green Version]
- Lerch, P.G.; Förtsch, V.; Hodler, G.; Bolli, R. Production and Characterization of a Reconstituted High Density Lipoprotein for Therapeutic Applications. Vox Sang. 1996, 71, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Robert, J.; Stukas, S.; Button, E.; Cheng, W.H.; Lee, M.; Fan, J.; Wilkinson, A.; Kulic, I.; Wright, S.D.; Wellington, C.L. Reconstituted high-density lipoproteins acutely reduce soluble brain Aβ levels in symptomatic APP/PS1 mice. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2016, 1862, 1027–1036. [Google Scholar] [CrossRef]
- Bloedon, L.T.; Dunbar, R.; Duffy, D.; Pinell-Salles, P.; Norris, R.; DeGroot, B.J.; Movva, R.; Navab, M.; Fogelman, A.M.; Rader, D.J. Safety, pharmacokinetics, and pharmacodynamics of oral apoA-I mimetic peptide D-4F in high-risk cardiovascular patients. J. Lipid Res. 2008, 49, 1344–1352. [Google Scholar] [CrossRef] [PubMed]
- An, S.; Fu, L. Small-molecule PROTACs: An emerging and promising approach for the development of targeted therapy drugs. EBioMedicine 2018, 36, 553–562. [Google Scholar] [CrossRef]
- Xi, M.; Chen, Y.; Yang, H.; Xu, H.; Du, K.; Wu, C.; Xu, Y.; Deng, L.; Luo, X.; Yu, L.; et al. Small molecule PROTACs in targeted therapy: An emerging strategy to induce protein degradation. Eur. J. Med. Chem. 2019, 174, 159–180. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciccone, L.; Shi, C.; di Lorenzo, D.; Van Baelen, A.-C.; Tonali, N. The Positive Side of the Alzheimer’s Disease Amyloid Cross-Interactions: The Case of the Aβ 1-42 Peptide with Tau, TTR, CysC, and ApoA1. Molecules 2020, 25, 2439. https://doi.org/10.3390/molecules25102439
Ciccone L, Shi C, di Lorenzo D, Van Baelen A-C, Tonali N. The Positive Side of the Alzheimer’s Disease Amyloid Cross-Interactions: The Case of the Aβ 1-42 Peptide with Tau, TTR, CysC, and ApoA1. Molecules. 2020; 25(10):2439. https://doi.org/10.3390/molecules25102439
Chicago/Turabian StyleCiccone, Lidia, Chenghui Shi, Davide di Lorenzo, Anne-Cécile Van Baelen, and Nicolo Tonali. 2020. "The Positive Side of the Alzheimer’s Disease Amyloid Cross-Interactions: The Case of the Aβ 1-42 Peptide with Tau, TTR, CysC, and ApoA1" Molecules 25, no. 10: 2439. https://doi.org/10.3390/molecules25102439
APA StyleCiccone, L., Shi, C., di Lorenzo, D., Van Baelen, A.-C., & Tonali, N. (2020). The Positive Side of the Alzheimer’s Disease Amyloid Cross-Interactions: The Case of the Aβ 1-42 Peptide with Tau, TTR, CysC, and ApoA1. Molecules, 25(10), 2439. https://doi.org/10.3390/molecules25102439