
Sphingosine 1-Phosphate Signaling and Metabolism in Chemoprevention and Chemoresistance in Colon Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
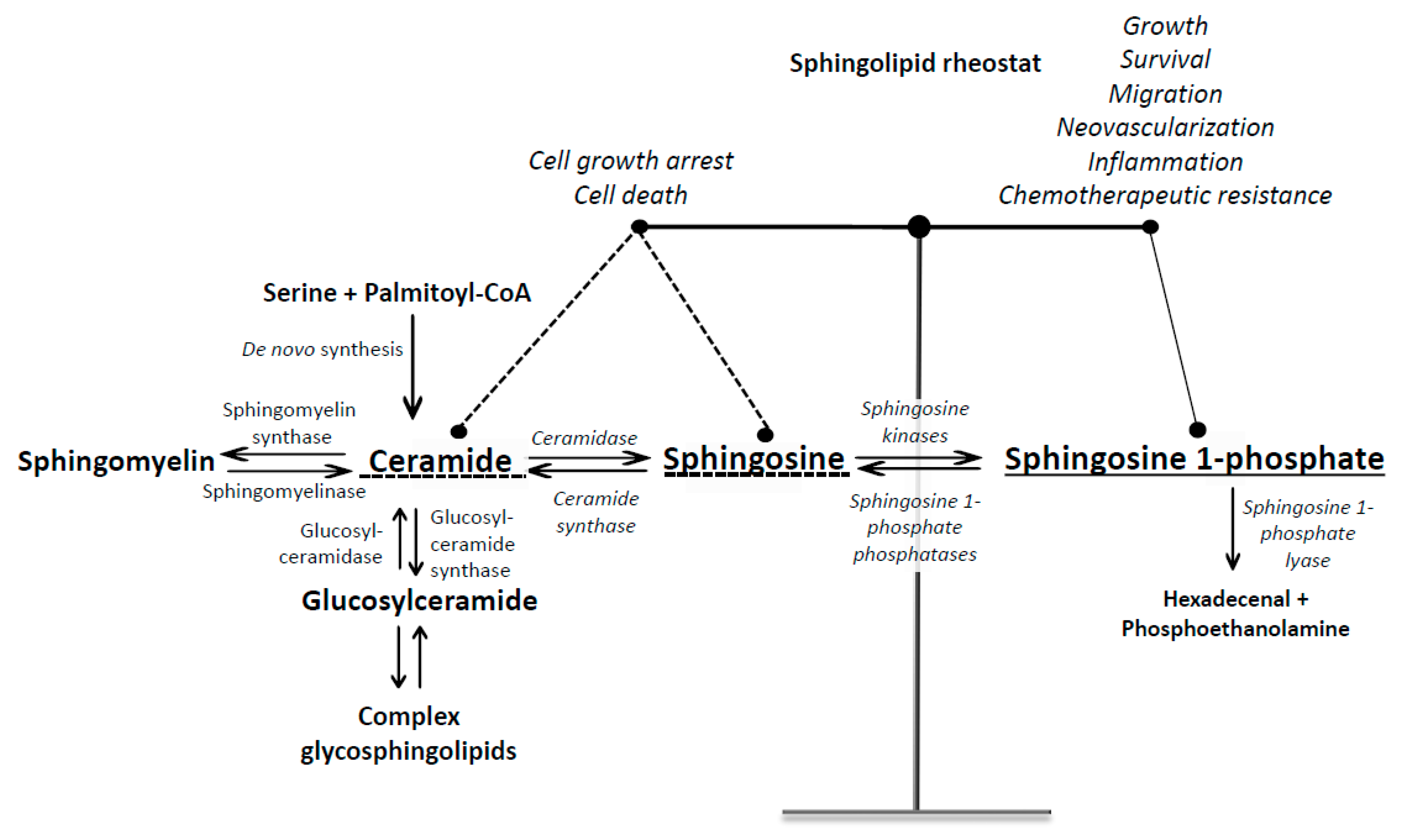
1. Introduction into Sphingosine 1-Phosphate Signaling
2. Sphingosine 1-Phosphate Transporters in Colon Cancer
3. Sphingosine 1-Phosphate Receptors in Colon Cancer
4. S1P Production and Degradation in Colon Cancer
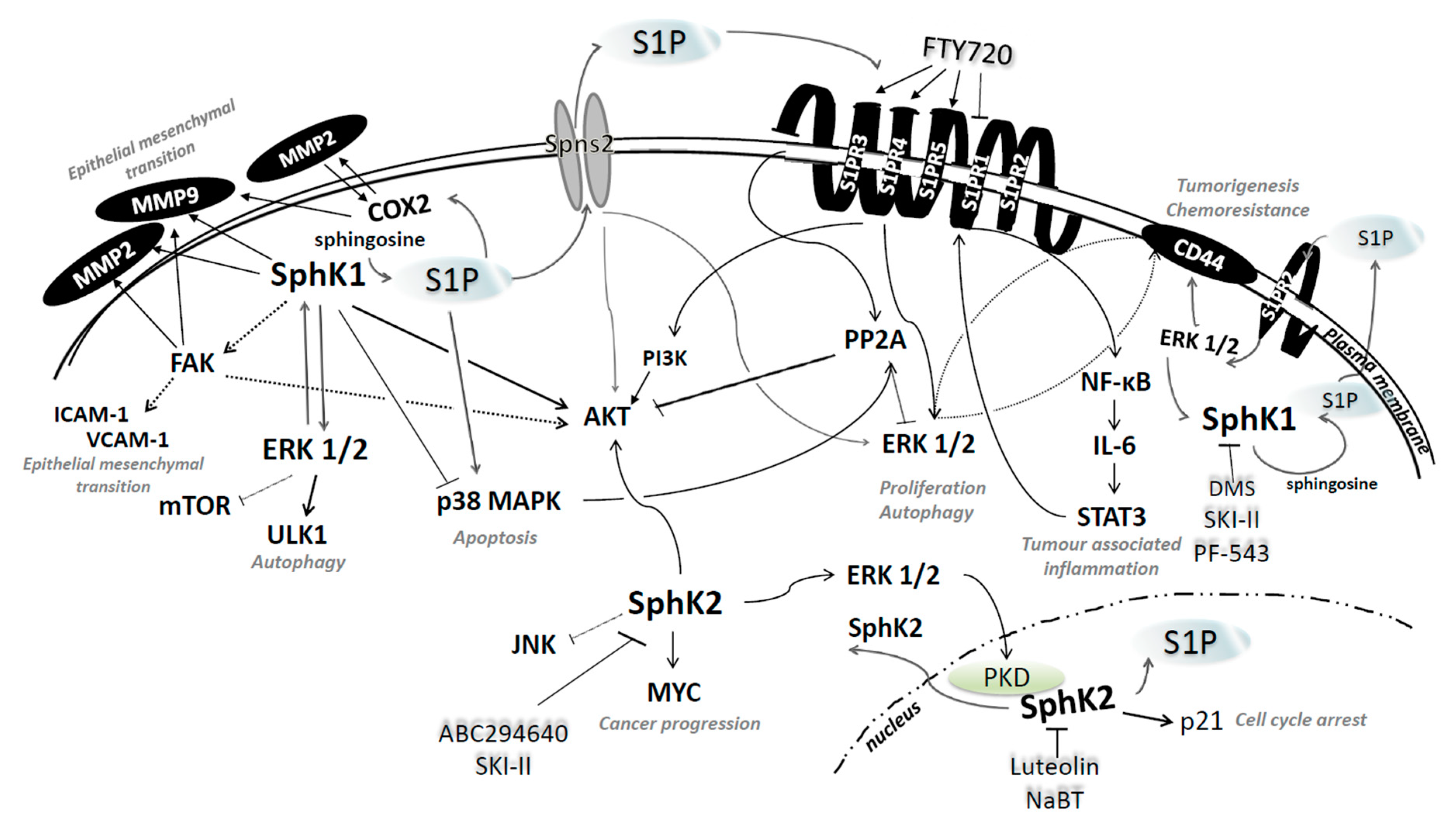
4.1. Sphingosine Kinases 1 and 2
4.2. S1P Lyase
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pyne, N.J.; El Buri, A.; Adams, D.R.; Pyne, S. Sphingosine 1-phosphate and cancer. Adv. Biol. Regul. 2018, 68, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Pyne, N.J.; Pyne, S. Sphingosine 1-phosphate and cancer. Nat. Rev. Cancer 2010, 10, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, M.; Nagahashi, M.; Rashid, O.M.; Takabe, K.; Wakai, T. The role of sphingosine-1-phosphate in the tumor microenvironment and its clinical implications. Tumour Biol. 2017, 39, 1010428317699133. [Google Scholar] [CrossRef]
- Patmanathan, S.N.; Wang, W.; Yap, L.F.; Herr, D.R.; Paterson, I.C. Mechanisms of sphingosine 1-phosphate receptor signalling in cancer. Cell. Signal. 2017, 34, 66–75. [Google Scholar] [CrossRef]
- Ancellin, N.; Colmont, C.; Su, J.; Li, Q.; Mittereder, N.; Chae, S.-S.; Stefansson, S.; Liau, G.; Hla, T. Extracellular export of sphingosine kinase-1 enzyme. Sphingosine 1-phosphate generation and the induction of angiogenic vascular maturation. J. Biol. Chem. 2002, 277, 6667–6675. [Google Scholar] [CrossRef]
- Weigert, A.; Cremer, S.; Schmidt, M.V.; von Knethen, A.; Angioni, C.; Geisslinger, G.; Brüne, B. Cleavage of sphingosine kinase 2 by caspase-1 provokes its release from apoptotic cells. Blood 2010, 115, 3531–3540. [Google Scholar] [CrossRef]
- Brindley, D.N.; Pilquil, C. Lipid phosphate phosphatases and signaling. J. Lipid Res. 2009, 50, 225–230. [Google Scholar] [CrossRef]
- Bekele, R.; David, S. Role of autotaxin and lysophosphatidate in cancer progression and resistance to chemotherapy and radiotherapy. Clin. Lipidol. 2012, 7, 313–328. [Google Scholar] [CrossRef]
- Tang, X.; Benesch, M.G.K.; Dewald, J.; Zhao, Y.Y.; Patwardhan, N.; Santos, W.L.; Curtis, J.M.; McMullen, T.P.W.; Brindley, D.N. Lipid phosphate phosphatase-1 expression in cancer cells attenuates tumor growth and metastasis in mice. J. Lipid Res. 2014, 55, 2389–2400. [Google Scholar] [CrossRef]
- Tanyi, J.L.; Morris, A.J.; Wolf, J.K.; Fang, X.; Hasegawa, Y.; Lapushin, R.; Auersperg, N.; Sigal, Y.J.; Newman, R.A.; Felix, E.A.; et al. The human lipid phosphate phosphatase-3 decreases the growth, survival, and tumorigenesis of ovarian cancer cells: Validation of the lysophosphatidic acid signaling cascade as a target for therapy in ovarian cancer. Cancer Res. 2003, 63, 1073–1082. [Google Scholar]
- Spiegel, S.; Milstien, S. Sphingosine-1-phosphate: Signaling inside and out. FEBS Lett. 2000, 476, 55–57. [Google Scholar] [CrossRef]
- van der Weyden, L.; Arends, M.J.; Campbell, A.D.; Bald, T.; Wardle-Jones, H.; Griggs, N.; Velasco-Herrera, M.D.C.; Tüting, T.; Sansom, O.J.; Karp, N.A.; et al. Genome-wide in vivo screen identifies novel host regulators of metastatic colonization. Nature 2017, 541, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Ponnusamy, S.; Selvam, S.P.; Mehrotra, S.; Kawamori, T.; Snider, A.J.; Obeid, L.M.; Shao, Y.; Sabbadini, R.; Ogretmen, B. Communication between host organism and cancer cells is transduced by systemic sphingosine kinase 1/sphingosine 1-phosphate signalling to regulate tumour metastasis. EMBO Mol. Med. 2012, 4, 761–775. [Google Scholar] [CrossRef] [PubMed]
- Takuwa, N.; Du, W.; Kaneko, E.; Okamoto, Y.; Yoshioka, K.; Takuwa, Y. Tumor-suppressive sphingosine-1-phosphate receptor-2 counteracting tumor-promoting sphingosine-1-phosphate receptor-1 and sphingosine kinase 1—Jekyll hidden behind Hyde. Am. J. Cancer Res. 2011, 1, 460–481. [Google Scholar] [PubMed]
- Shen, Y.; Zhao, S.; Wang, S.; Pan, X.; Zhang, Y.; Xu, J.; Jiang, Y.; Li, H.; Zhang, Q.; Gao, J.; et al. S1P/S1PR3 axis promotes aerobic glycolysis by YAP/c-MYC/PGAM1 axis in osteosarcoma. EBioMedicine 2019, 40, 210–223. [Google Scholar] [CrossRef]
- Watters, R.J.; Wang, H.-G.; Sung, S.-S.; Loughran, T.P.; Liu, X. Targeting sphingosine-1-phosphate receptors in cancer. Anticancer Agents Med. Chem. 2011, 11, 810–817. [Google Scholar] [CrossRef]
- Olesch, C.; Sirait-Fischer, E.; Brüne, B.; Weigert, A. Abstract A209: Targeting immune cell-specific sphingosine-1-phosphate receptor 4 to restore antitumor immunity resulting in improved therapy response. Cancer Immunol. Res. 2019, 7, 209. [Google Scholar]
- Andrieu, G.; Ledoux, A.; Branka, S.; Bocquet, M.; Gilhodes, J.; Walzer, T.; Kasahara, K.; Inagaki, M.; Sabbadini, R.A.; Cuvillier, O.; et al. Sphingosine 1-phosphate signaling through its receptor S1P5 promotes chromosome segregation and mitotic progression. Sci. Signal. 2017, 10, eaah4007. [Google Scholar] [CrossRef]
- Chang, C.-L.; Ho, M.-C.; Lee, P.-H.; Hsu, C.-Y.; Huang, W.-P.; Lee, H. S1P(5) is required for sphingosine 1-phosphate-induced autophagy in human prostate cancer PC-3 cells. Am. J. Physiol. Cell Physiol. 2009, 297, 451–458. [Google Scholar] [CrossRef]
- Huang, Y.-L.; Chang, C.-L.; Tang, C.-H.; Lin, Y.-C.; Ju, T.-K.; Huang, W.-P.; Lee, H. Extrinsic sphingosine 1-phosphate activates S1P5 and induces autophagy through generating endoplasmic reticulum stress in human prostate cancer PC-3 cells. Cell. Signal. 2014, 26, 611–618. [Google Scholar] [CrossRef]
- Maceyka, M.; Sankala, H.; Hait, N.C.; Le Stunff, H.; Liu, H.; Toman, R.; Collier, C.; Zhang, M.; Satin, L.S.; Merrill, A.H.; et al. SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J. Biol. Chem. 2005, 280, 37118–37129. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S.E.; Harikumar, K.B.; Hait, N.C.; Allegood, J.; Strub, G.M.; Kim, E.Y.; Maceyka, M.; Jiang, H.; Luo, C.; Kordula, T.; et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 2010, 465, 1084–1088. [Google Scholar] [CrossRef] [PubMed]
- Parham, K.A.; Zebol, J.R.; Tooley, K.L.; Sun, W.Y.; Moldenhauer, L.M.; Cockshell, M.P.; Gliddon, B.L.; Moretti, P.A.; Tigyi, G.; Pitson, S.M.; et al. Sphingosine 1-phosphate is a ligand for peroxisome proliferator-activated receptor-γ that regulates neoangiogenesis. FASEB J. 2015, 29, 3638–3653. [Google Scholar] [CrossRef] [PubMed]
- Hait, N.C.; Allegood, J.; Maceyka, M.; Strub, G.M.; Harikumar, K.B.; Singh, S.K.; Luo, C.; Marmorstein, R.; Kordula, T.; Milstien, S.; et al. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 2009, 325, 1254–1257. [Google Scholar] [CrossRef] [PubMed]
- Strub, G.M.; Paillard, M.; Liang, J.; Gomez, L.; Allegood, J.C.; Hait, N.C.; Maceyka, M.; Price, M.M.; Chen, Q.; Simpson, D.C.; et al. Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 2011, 25, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Arya, N. Mfsd2b, a Novel Sphingosine-1-Phosphate transporter: Implication in cancer therapeutics. J. Clin. Exp. Oncol. 2018, 7, 2. [Google Scholar] [CrossRef]
- Hisano, Y.; Kobayashi, N.; Yamaguchi, A.; Nishi, T. Mouse SPNS2 functions as a sphingosine-1-phosphate transporter in vascular endothelial cells. PLoS ONE 2012, 7, e38941. [Google Scholar] [CrossRef]
- Adamska, A.; Falasca, M. ATP-binding cassette transporters in progression and clinical outcome of pancreatic cancer: What is the way forward? World J. Gastroenterol. 2018, 24, 3222–3238. [Google Scholar] [CrossRef]
- Kruh, G.D.; Belinsky, M.G. The MRP family of drug efflux pumps. Oncogene 2003, 22, 7537–7552. [Google Scholar] [CrossRef]
- Pilorget, A.; Demeule, M.; Barakat, S.; Marvaldi, J.; Luis, J.; Béliveau, R. Modulation of P-glycoprotein function by sphingosine kinase-1 in brain endothelial cells. J. Neurochem. 2007, 100, 1203–1210. [Google Scholar] [CrossRef]
- Chen, M.; Li, D.; Gong, N.; Wu, H.; Su, C.; Xie, C.; Xiang, H.; Lin, C.; Li, X. miR-133b down-regulates ABCC1 and enhances the sensitivity of CRC to anti-tumor drugs. Oncotarget 2017, 8, 52983–52994. [Google Scholar] [CrossRef] [PubMed]
- Candeil, L.; Gourdier, I.; Peyron, D.; Vezzio, N.; Copois, V.; Bibeau, F.; Orsetti, B.; Scheffer, G.L.; Ychou, M.; Khan, Q.A.; et al. ABCG2 overexpression in colon cancer cells resistant to SN38 and in irinotecan-treated metastases. Int. J. Cancer 2004, 109, 848–854. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, S.; Kamoi, M.; Watanabe, Y.; Suzuki, H.; Hori, S.; Terasaki, T. Correlation of induction of ATP binding cassette transporter A5 (ABCA5) and ABCB1 mRNAs with differentiation state of human colon tumor. Biol. Pharm. Bull. 2007, 30, 1144–1146. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Aguirre-Portolés, C.; Feliu, J.; Reglero, G.; Ramírez de Molina, A. ABCA1 overexpression worsens colorectal cancer prognosis by facilitating tumour growth and caveolin-1-dependent invasiveness, and these effects can be ameliorated using the BET inhibitor apabetalone. Mol. Oncol. 2018, 12, 1735–1752. [Google Scholar] [CrossRef] [PubMed]
- Pulli, I.; Blom, T.; Löf, C.; Magnusson, M.; Rimessi, A.; Pinton, P.; Törnquist, K. A novel chimeric aequorin fused with caveolin-1 reveals a sphingosine kinase 1-regulated Ca2+ microdomain in the caveolar compartment. Biochim. Et Biophys. Acta (BBA)—Mol. Cell Res. 2015, 1853, 2173–2182. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, G.T.; Maceyka, M.; Milstien, S.; Spiegel, S. Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat. Rev. Drug Discov. 2013, 12, 688–702. [Google Scholar] [CrossRef]
- Pyne, N.J.; McNaughton, M.; Boomkamp, S.; MacRitchie, N.; Evangelisti, C.; Martelli, A.M.; Jiang, H.-R.; Ubhi, S.; Pyne, S. Role of sphingosine 1-phosphate receptors, sphingosine kinases and sphingosine in cancer and inflammation. Adv. Biol. Regul. 2016, 60, 151–159. [Google Scholar] [CrossRef]
- Ogretmen, B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer 2018, 18, 33–50. [Google Scholar] [CrossRef]
- Adada, M.M.; Canals, D.; Jeong, N.; Kelkar, A.D.; Hernandez-Corbacho, M.; Pulkoski-Gross, M.J.; Donaldson, J.C.; Hannun, Y.A.; Obeid, L.M. Intracellular sphingosine kinase 2-derived sphingosine-1-phosphate mediates epidermal growth factor-induced ezrin-radixin-moesin phosphorylation and cancer cell invasion. FASEB J. 2015, 29, 4654–4669. [Google Scholar] [CrossRef]
- Liang, J.; Nagahashi, M.; Kim, E.Y.; Harikumar, K.B.; Yamada, A.; Huang, W.-C.; Hait, N.C.; Allegood, J.C.; Price, M.M.; Avni, D.; et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell 2013, 23, 107–120. [Google Scholar] [CrossRef]
- Gault, C.R.; Eblen, S.T.; Neumann, C.A.; Hannun, Y.A.; Obeid, L.M. Oncogenic K-Ras regulates bioactive sphingolipids in a sphingosine kinase 1-dependent manner. J. Biol. Chem. 2012, 287, 31794–31803. [Google Scholar] [CrossRef] [PubMed]
- Nagahashi, M.; Takabe, K.; Terracina, K.P.; Soma, D.; Hirose, Y.; Kobayashi, T.; Matsuda, Y.; Wakai, T. Sphingosine-1-Phosphate Transporters as Targets for Cancer Therapy. Available online: https://www.hindawi.com/journals/bmri/2014/651727/ (accessed on 9 April 2020).
- Gu, X.; Jiang, Y.; Xue, W.; Song, C.; Wang, Y.; Liu, Y.; Cui, B. SPNS2 promotes the malignancy of colorectal cancer cells via regulating Akt and ERK pathway. Clin. Exp. Pharmacol. Physiol. 2019, 46, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Uranbileg, B.; Nishikawa, T.; Ikeda, H.; Kurano, M.; Sato, M.; Saigusa, D.; Aoki, J.; Watanabe, T.; Yatomi, Y. Evidence suggests Sphingosine 1-Phosphate might be actively generated, degraded, and transported to extracellular spaces with increased S1P2 and S1P3 expression in colon cancer. Clin. Colorectal Cancer 2018, 17, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Bradley, E.; Dasgupta, S.; Jiang, X.; Zhao, X.; Zhu, G.; He, Q.; Dinkins, M.; Bieberich, E.; Wang, G. Critical role of Spns2, a Sphingosine-1-Phosphate transporter, in lung cancer cell survival and migration. PLoS ONE 2014, 9, e110119. [Google Scholar] [CrossRef]
- Shida, D.; Inoue, S.; Yoshida, Y.; Kodaka, A.; Tsuji, T.; Tsuiji, M. Sphingosine kinase 1 is upregulated with lysophosphatidic acid receptor 2 in human colorectal cancer. World J. Gastroenterol. 2016, 22, 2503–2511. [Google Scholar] [CrossRef]
- Lin, Q.; Wei, Y.; Zhong, Y.; Zhu, D.; Ren, L.; Xu, P.; Zheng, P.; Feng, Q.; Ji, M.; Lv, M.; et al. Aberrant expression of sphingosine-1-phosphate receptor 1 correlates with metachronous liver metastasis and poor prognosis in colorectal cancer. Tumour Biol. 2014, 35, 9743–9750. [Google Scholar] [CrossRef]
- Chen, T.; Huang, Z.; Liu, R.; Yang, J.; Hylemon, P.B.; Zhou, H. Sphingosine-1 phosphate promotes intestinal epithelial cell proliferation via S1PR2. Front. Biosci. (Landmark Ed.) 2017, 22, 596–608. [Google Scholar]
- Petti, L.; Piontini, A.; Arena, V.; Danese, S.; Vetrano, S. Sphingosine-1-phosphate receptor 2 is a negative regulator of epithelial cell proliferation and intestinal tumorigenesis. FASEB J. 2017, 31, 1046. [Google Scholar]
- Aktas, O.; Küry, P.; Kieseier, B.; Hartung, H.-P. Fingolimod is a potential novel therapy for multiple sclerosis. Nat. Rev. Neurol. 2010, 6, 373–382. [Google Scholar] [CrossRef]
- LaMontagne, K.; Littlewood-Evans, A.; Schnell, C.; O’Reilly, T.; Wyder, L.; Sanchez, T.; Probst, B.; Butler, J.; Wood, A.; Liau, G.; et al. Antagonism of sphingosine-1-phosphate receptors by FTY720 inhibits angiogenesis and tumor vascularization. Cancer Res. 2006, 66, 221–231. [Google Scholar] [CrossRef]
- Nagaoka, Y.; Otsuki, K.; Fujita, T.; Uesato, S. Effects of phosphorylation of immunomodulatory agent FTY720 (fingolimod) on antiproliferative activity against breast and colon cancer cells. Biol. Pharm. Bull. 2008, 31, 1177–1181. [Google Scholar] [CrossRef] [PubMed]
- Perrotti, D.; Neviani, P. Targeting a tumor suppressor to suppress tumor growth: News and views on protein phosphatase 2A (PP2A) as a target for anti-cancer therapy. Lancet Oncol. 2013, 14, 229–238. [Google Scholar] [CrossRef]
- White, C.; Alshaker, H.; Cooper, C.; Winkler, M.; Pchejetski, D. The emerging role of FTY720 (Fingolimod) in cancer treatment. Oncotarget 2016, 7, 23106–23127. [Google Scholar] [CrossRef] [PubMed]
- Ushitora, Y.; Tashiro, H.; Ogawa, T.; Tanimoto, Y.; Kuroda, S.; Kobayashi, T.; Miyata, Y.; Itamoto, T.; Asahara, T.; Ohdan, H. Suppression of hepatocellular carcinoma recurrence after rat liver transplantation by FTY720, a sphingosine-1-phosphate analog. Transplantation 2009, 88, 980–986. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.V.; Wu, Y.-Y.; Lin, E.Y. STAT3 and sphingosine-1-phosphate in inflammation-associated colorectal cancer. World J. Gastroenterol. 2014, 20, 10279–10287. [Google Scholar] [CrossRef]
- Liu, S.-Q.; Xu, C.-Y.; Wu, W.-H.; Fu, Z.-H.; He, S.-W.; Qin, M.-B.; Huang, J.-A. Sphingosine kinase 1 promotes the metastasis of colorectal cancer by inducing the epithelial-mesenchymal transition mediated by the FAK/AKT/MMPs axis. Int. J. Oncol. 2018, 54, 41–52. [Google Scholar] [CrossRef]
- Liu, Y.; Deng, J.; Wang, L.; Lee, H.; Armstrong, B.; Scuto, A.; Kowolik, C.; Weiss, L.M.; Forman, S.; Yu, H. S1PR1 is an effective target to block STAT3 signaling in activated B cell-like diffuse large B-cell lymphoma. Blood 2012, 120, 1458–1465. [Google Scholar] [CrossRef]
- Cristóbal, I.; Manso, R.; Rincón, R.; Caramés, C.; Senin, C.; Borrero, A.; Martínez-Useros, J.; Rodriguez, M.; Zazo, S.; Aguilera, O.; et al. PP2A inhibition is a common event in colorectal cancer and its restoration using FTY720 shows promising therapeutic potential. Mol. Cancer Ther. 2014, 13, 938–947. [Google Scholar] [CrossRef]
- Xing, Y.; Wang, Z.H.; Ma, D.H.; Han, Y. FTY720 enhances chemosensitivity of colon cancer cells to doxorubicin and etoposide via the modulation of P-glycoprotein and multidrug resistance protein 1. J. Dig. Dis. 2014, 15, 246–259. [Google Scholar] [CrossRef]
- Rosa, R.; Marciano, R.; Malapelle, U.; Formisano, L.; Nappi, L.; D’Amato, C.; D’Amato, V.; Damiano, V.; Marfè, G.; Del Vecchio, S.; et al. Sphingosine kinase 1 overexpression contributes to cetuximab resistance in human colorectal cancer models. Clin. Cancer Res. 2013, 19, 138–147. [Google Scholar] [CrossRef]
- Alshaker, H.; Sauer, L.; Monteil, D.; Ottaviani, S.; Srivats, S.; Böhler, T.; Pchejetski, D. Chapter Six—Therapeutic potential of targeting SK1 in human cancers. In Advances in Cancer Research; Norris, J.S., Ed.; Academic Press: London, UK, 2013; Volume 117, pp. 143–200. [Google Scholar]
- Furuya, H.; Shimizu, Y.; Tamashiro, P.M.; Iino, K.; Bielawski, J.; Chan, O.T.M.; Pagano, I.; Kawamori, T. Sphingosine kinase 1 expression enhances colon tumor growth. J. Transl. Med. 2017, 15, 120. [Google Scholar] [CrossRef]
- Tan, S.S.L.; Khin, L.W.; Wong, L.; Yan, B.; Ong, C.W.; Datta, A.; Salto-Tellez, M.; Lam, Y.; Yap, C.T. Sphingosine kinase 1 promotes malignant progression in colon cancer and independently predicts survival of patients with colon cancer by competing risk approach in South asian population. Clin. Transl. Gastroenterol. 2014, 5, e51. [Google Scholar] [CrossRef] [PubMed]
- Bae, G.E.; Do, S.-I.; Kim, K.; Park, J.H.; Cho, S.; Kim, H.-S. Increased Sphingosine Kinase 1 expression predicts distant metastasis and poor outcome in patients with colorectal cancer. Anticancer Res. 2019, 39, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Lima, S.; Takabe, K.; Newton, J.; Saurabh, K.; Young, M.M.; Leopoldino, A.M.; Hait, N.C.; Roberts, J.L.; Wang, H.-G.; Dent, P.; et al. TP53 is required for BECN1- and ATG5-dependent cell death induced by sphingosine kinase 1 inhibition. Autophagy 2018, 14, 942–957. [Google Scholar] [CrossRef] [PubMed]
- Ju, T.; Gao, D.; Fang, Z.-Y. Targeting colorectal cancer cells by a novel sphingosine kinase 1 inhibitor PF-543. Biochem. Biophys. Res. Commun. 2016, 470, 728–734. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Hernández, M.; Arias, A.; Martínez-García, D.; Pérez-Tomás, R.; Quesada, R.; Soto-Cerrato, V. Targeting autophagy for cancer treatment and tumor chemosensitization. Cancers 2019, 11, 1599. [Google Scholar] [CrossRef]
- Soslow, R.A.; Dannenberg, A.J.; Rush, D.; Woerner, B.M.; Khan, K.N.; Masferrer, J.; Koki, A.T. COX-2 is expressed in human pulmonary, colonic, and mammary tumors. Cancer 2000, 89, 2637–2645. [Google Scholar] [CrossRef]
- Sinicrope, F.A.; Lemoine, M.; Xi, L.; Lynch, P.M.; Cleary, K.R.; Shen, Y.; Frazier, M.L. Reduced expression of cyclooxygenase 2 proteins in hereditary nonpolyposis colorectal cancers relative to sporadic cancers. Gastroenterology 1999, 117, 350–358. [Google Scholar] [CrossRef]
- Sano, H.; Kawahito, Y.; Wilder, R.L.; Hashiramoto, A.; Mukai, S.; Asai, K.; Kimura, S.; Kato, H.; Kondo, M.; Hla, T. Expression of Cyclooxygenase-1 and -2 in Human Colorectal Cancer. Cancer Res. 1995, 55, 3785–3789. [Google Scholar]
- Sinicrope, F.A.; Gill, S. Role of cyclooxygenase-2 in colorectal cancer. Cancer Metastasis Rev. 2004, 23, 63–75. [Google Scholar] [CrossRef]
- Kawamori, T.; Osta, W.; Johnson, K.R.; Pettus, B.J.; Bielawski, J.; Tanaka, T.; Wargovich, M.J.; Reddy, B.S.; Hannun, Y.A.; Obeid, L.M.; et al. Sphingosine kinase 1 is up-regulated in colon carcinogenesis. FASEB J. 2006, 20, 386–388. [Google Scholar] [CrossRef] [PubMed]
- Tsujii, M.; Kawano, S.; DuBois, R.N. Cyclooxygenase-2 expression in human colon cancer cells increases metastatic potential. Proc. Natl. Acad. Sci. USA 1997, 94, 3336–3340. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yang, T.; Yan, J. Cyclooxygenase-2 increased the angiogenic and metastatic potential of tumor cells. Biochem. Biophys. Res. Commun. 2002, 299, 886–890. [Google Scholar] [CrossRef]
- Liu, S.-Q.; Huang, J.-A.; Qin, M.-B.; Su, Y.-J.; Lai, M.-Y.; Jiang, H.-X.; Tang, G.-D. Sphingosine kinase 1 enhances colon cancer cell proliferation and invasion by upregulating the production of MMP-2/9 and uPA via MAPK pathways. Int. J. Colorectal Dis. 2012, 27, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.-Y.; Liu, S.-Q.; Qin, M.-B.; Zhuge, C.-F.; Qin, L.; Qin, N.; Lai, M.-Y.; Huang, J.-A. SphK1 modulates cell migration and EMT-related marker expression by regulating the expression of p-FAK in colorectal cancer cells. Int. J. Mol. Med. 2017, 39, 1277–1284. [Google Scholar] [CrossRef]
- Jafari, N.; Drury, J.; Morris, A.J.; Onono, F.O.; Stevens, P.D.; Gao, T.; Liu, J.; Wang, C.; Lee, E.Y.; Weiss, H.L.; et al. De novo fatty acid synthesis-driven sphingolipid metabolism promotes metastatic potential of colorectal cancer. Mol. Cancer Res. 2019, 17, 140–152. [Google Scholar] [CrossRef]
- Alexiou, D.; Karayiannakis, A.J.; Syrigos, K.N.; Zbar, A.; Kremmyda, A.; Bramis, I.; Tsigris, C. Serum levels of E-selectin, ICAM-1 and VCAM-1 in colorectal cancer patients: Correlations with clinicopathological features, patient survival and tumour surgery. Eur. J. Cancer 2001, 37, 2392–2397. [Google Scholar] [CrossRef]
- Wang, J.; Wei, Q.; Wang, X.; Tang, S.; Liu, H.; Zhang, F.; Mohammed, M.K.; Huang, J.; Guo, D.; Lu, M.; et al. Transition to resistance: An unexpected role of the EMT in cancer chemoresistance. Genes Dis 2016, 3, 3–6. [Google Scholar] [CrossRef]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef]
- Kawahara, S.; Otsuji, Y.; Nakamura, M.; Murakami, M.; Murate, T.; Matsunaga, T.; Kanoh, H.; Seishima, M.; Banno, Y.; Hara, A. Sphingosine kinase 1 plays a role in the upregulation of CD44 expression through extracellular signal-regulated kinase signaling in human colon cancer cells. Anticancer Drugs 2013, 24, 473–483. [Google Scholar] [CrossRef]
- Wang, Z.; Tang, Y.; Xie, L.; Huang, A.; Xue, C.; Gu, Z.; Wang, K.; Zong, S. The prognostic and clinical value of CD44 in colorectal cancer: A meta-analysis. Front. Oncol. 2019, 9, 309. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhao, S.; Karnad, A.; Freeman, J.W. The biology and role of CD44 in cancer progression: Therapeutic implications. J. Hematol. Oncol. 2018, 11, 64. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Dong, L.; Chang, P. CD44v6 engages in colorectal cancer progression. Cell Death Dis. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, S.; Nakamura, M.; Osawa, Y.; Kono, S.; Itoh, Y.; Okano, Y.; Murate, T.; Hara, A.; Ueda, H.; Nozawa, Y.; et al. Sphingosine kinase isoforms regulate oxaliplatin sensitivity of human colon cancer cells through ceramide accumulation and Akt activation. J. Biol. Chem. 2009, 284, 10422–10432. [Google Scholar] [CrossRef]
- Xun, C.; Chen, M.-B.; Qi, L.; Tie-Ning, Z.; Peng, X.; Ning, L.; Zhi-Xiao, C.; Li-Wei, W. Targeting sphingosine kinase 2 (SphK2) by ABC294640 inhibits colorectal cancer cell growth in vitro and in vivo. J. Exp. Clin. Cancer Res. 2015, 34, 94. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, X.; Zuo, Z.; Hao, C.; Ma, Y. Sphingosine kinase 2 promotes colorectal cancer cell proliferation and invasion by enhancing MYC expression. Tumour Biol. 2016, 37, 8455–8460. [Google Scholar] [CrossRef]
- Smith, D.R.; Goh, H.S. Overexpression of the c-myc proto-oncogene in colorectal carcinoma is associated with a reduced mortality that is abrogated by point mutation of the p53 tumor suppressor gene. Clin. Cancer Res. 1996, 2, 1049–1053. [Google Scholar]
- Neubauer, H.A.; Pham, D.H.; Zebol, J.R.; Moretti, P.A.B.; Peterson, A.L.; Leclercq, T.M.; Chan, H.; Powell, J.A.; Pitman, M.R.; Samuel, M.S.; et al. An oncogenic role for sphingosine kinase 2. Oncotarget 2016, 7, 64886–64899. [Google Scholar] [CrossRef]
- Mizutani, N.; Omori, Y.; Tanaka, K.; Ito, H.; Takagi, A.; Kojima, T.; Nakatochi, M.; Ogiso, H.; Kawamoto, Y.; Nakamura, M.; et al. Increased SPHK2 transcription of human colon cancer cells in serum-depleted culture: The involvement of CREB transcription factor. J. Cell. Biochem. 2015, 116, 2227–2238. [Google Scholar] [CrossRef]
- Antoon, J.W.; White, M.D.; Slaughter, E.M.; Driver, J.L.; Khalili, H.S.; Elliott, S.; Smith, C.D.; Burow, M.E.; Beckman, B.S. Targeting NFκB mediated breast cancer chemoresistance through selective inhibition of sphingosine kinase-2. Cancer Biol. 2011, 11, 678–689. [Google Scholar] [CrossRef]
- Lewis, C.S.; Voelkel-Johnson, C.; Smith, C.D. Suppression of c-Myc and RRM2 expression in pancreatic cancer cells by the sphingosine kinase-2 inhibitor ABC294640. Oncotarget 2016, 7, 60181–60192. [Google Scholar] [CrossRef] [PubMed]
- Sundaramoorthy, P.; Gasparetto, C.; Kang, Y. The combination of a sphingosine kinase 2 inhibitor (ABC294640) and a Bcl-2 inhibitor (ABT-199) displays synergistic anti-myeloma effects in myeloma cells without a t(11;14) translocation. Cancer Med. 2018, 7, 3257–3268. [Google Scholar] [CrossRef] [PubMed]
- Sankala, H.M.; Hait, N.C.; Paugh, S.W.; Shida, D.; Lépine, S.; Elmore, L.W.; Dent, P.; Milstien, S.; Spiegel, S. Involvement of Sphingosine Kinase 2 in p53-independent induction of p21 by the chemotherapeutic drug doxorubicin. Cancer Res. 2007, 67, 10466–10474. [Google Scholar] [CrossRef] [PubMed]
- Mahyar-Roemer, M.; Roemer, K. p21 Waf1/Cip1 can protect human colon carcinoma cells against p53-dependent and p53-independent apoptosis induced by natural chemopreventive and therapeutic agents. Oncogene 2001, 20, 3387–3398. [Google Scholar] [CrossRef]
- Rejhová, A.; Opattová, A.; Čumová, A.; Slíva, D.; Vodička, P. Natural compounds and combination therapy in colorectal cancer treatment. Eur. J. Med. Chem. 2018, 144, 582–594. [Google Scholar] [CrossRef]
- Kang, K.A.; Piao, M.J.; Ryu, Y.S.; Hyun, Y.J.; Park, J.E.; Shilnikova, K.; Zhen, A.X.; Kang, H.K.; Koh, Y.S.; Jeong, Y.J.; et al. Luteolin induces apoptotic cell death via antioxidant activity in human colon cancer cells. Int. J. Oncol. 2017, 51, 1169–1178. [Google Scholar] [CrossRef]
- Yao, Y.; Rao, C.; Zheng, G.; Wang, S. Luteolin suppresses colorectal cancer cell metastasis via regulation of the miR-384/pleiotrophin axis. Oncol. Rep. 2019, 42, 131–141. [Google Scholar] [CrossRef]
- Pandurangan, A.K.; Ananda Sadagopan, S.K.; Dharmalingam, P.; Ganapasam, S. Luteolin, a bioflavonoid inhibits Azoxymethane-induced colorectal cancer through activation of Nrf2 signaling. Toxicol. Mech. Methods 2014, 24, 13–20. [Google Scholar] [CrossRef]
- Pandurangan, A.K.; Esa, N.M. Luteolin, a bioflavonoid inhibits colorectal cancer through modulation of multiple signaling pathways: A review. Asian Pac. J. Cancer Prev. 2014, 15, 5501–5508. [Google Scholar] [CrossRef]
- Abdel Hadi, L.; Di Vito, C.; Marfia, G.; Ferraretto, A.; Tringali, C.; Viani, P.; Riboni, L. Sphingosine Kinase 2 and ceramide transport as key targets of the natural flavonoid luteolin to induce apoptosis in colon cancer cells. PLoS ONE 2015, 10, e0143384. [Google Scholar]
- Xu, Z.; Tao, J.; Chen, P.; Chen, L.; Sharma, S.; Wang, G.; Dong, Q. Sodium butyrate inhibits colorectal cancer cell migration by downregulating Bmi-1 through enhanced miR-200c expression. Mol. Nutr. Food Res. 2018, 62, e1700844. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yi, M.; Zha, L.; Chen, S.; Li, Z.; Li, C.; Gong, M.; Deng, H.; Chu, X.; Chen, J.; et al. Sodium butyrate induces endoplasmic reticulum stress and autophagy in colorectal cells: Implications for apoptosis. PLoS ONE 2016, 11, e0147218. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Liu, Y.; Zou, F. Sensitization of human colon cancer cells to sodium butyrate-induced apoptosis by modulation of sphingosine kinase 2 and protein kinase D. Exp. Cell Res. 2012, 318, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Liu, Y.G.; Zou, M.C.; Zou, F. Sodium butyrate induces apoptosis of human colon cancer cells by modulating ERK and Sphingosine Kinase 2. Biomed. Environ. Sci. 2014, 27, 197–203. [Google Scholar]
- Siddikuzzaman, N.; Guruvayoorappan, C.; Berlin Grace, V.M. All trans retinoic acid and cancer. Immunopharmacol. Immunotoxicol. 2011, 33, 241–249. [Google Scholar] [CrossRef]
- Lotan, R. Effects of vitamin A and its analogs (retinoids) on normal and neoplastic cells. Biochim. Et Biophys. Acta (BBA)—Rev. Cancer 1980, 605, 33–91. [Google Scholar] [CrossRef]
- Chu, J.-H.; Gao, Z.-H.; Qu, X.-J. Down-regulation of sphingosine kinase 2 (SphK2) increases the effects of all-trans-retinoic acid (ATRA) on colon cancer cells. Biomed. Pharmacother. 2014, 68, 1089–1097. [Google Scholar] [CrossRef]
- Sun, D.-F.; Gao, Z.-H.; Liu, H.-P.; Yuan, Y.; Qu, X.-J. Sphingosine 1-phosphate antagonizes the effect of all-trans retinoic acid (ATRA) in a human colon cancer cell line by modulation of RARβ expression. Cancer Lett. 2012, 319, 182–189. [Google Scholar] [CrossRef]
- Colié, S.; Veldhoven, P.P.V.; Kedjouar, B.; Bedia, C.; Albinet, V.; Sorli, S.-C.; Garcia, V.; Djavaheri-Mergny, M.; Bauvy, C.; Codogno, P.; et al. Disruption of Sphingosine 1-Phosphate lyase confers resistance to chemotherapy and promotes oncogenesis through Bcl-2/Bcl-xL upregulation. Cancer Res. 2009, 69, 9346–9353. [Google Scholar] [CrossRef]
- Min, J.; Van Veldhoven, P.P.; Zhang, L.; Hanigan, M.H.; Alexander, H.; Alexander, S. Sphingosine-1-phosphate lyase regulates sensitivity of human cells to select chemotherapy drugs in a p38-dependent manner. Mol. Cancer Res. 2005, 3, 287–296. [Google Scholar] [CrossRef]
- Oskouian, B.; Sooriyakumaran, P.; Borowsky, A.D.; Crans, A.; Dillard-Telm, L.; Tam, Y.Y.; Bandhuvula, P.; Saba, J.D. Sphingosine-1-phosphate lyase potentiates apoptosis via p53- and p38-dependent pathways and is down-regulated in colon cancer. Proc. Natl. Acad. Sci. USA 2006, 103, 17384–17389. [Google Scholar] [CrossRef] [PubMed]
- Degagné, E.; Pandurangan, A.; Bandhuvula, P.; Kumar, A.; Eltanawy, A.; Zhang, M.; Yoshinaga, Y.; Nefedov, M.; de Jong, P.J.; Fong, L.G.; et al. Sphingosine-1-phosphate lyase downregulation promotes colon carcinogenesis through STAT3-activated microRNAs. J. Clin. Investig. 2014, 124, 5368–5384. [Google Scholar] [CrossRef] [PubMed]
- Schwiebs, A.; Herrero San Juan, M.; Schmidt, K.G.; Wiercinska, E.; Anlauf, M.; Ottenlinger, F.; Thomas, D.; Elwakeel, E.; Weigert, A.; Farin, H.F.; et al. Cancer-induced inflammation and inflammation-induced cancer in colon: A role for S1P lyase. Oncogene 2019, 38, 4788–4803. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grbčić, P.; Sedić, M. Sphingosine 1-Phosphate Signaling and Metabolism in Chemoprevention and Chemoresistance in Colon Cancer. Molecules 2020, 25, 2436. https://doi.org/10.3390/molecules25102436
Grbčić P, Sedić M. Sphingosine 1-Phosphate Signaling and Metabolism in Chemoprevention and Chemoresistance in Colon Cancer. Molecules. 2020; 25(10):2436. https://doi.org/10.3390/molecules25102436
Chicago/Turabian StyleGrbčić, Petra, and Mirela Sedić. 2020. "Sphingosine 1-Phosphate Signaling and Metabolism in Chemoprevention and Chemoresistance in Colon Cancer" Molecules 25, no. 10: 2436. https://doi.org/10.3390/molecules25102436
APA StyleGrbčić, P., & Sedić, M. (2020). Sphingosine 1-Phosphate Signaling and Metabolism in Chemoprevention and Chemoresistance in Colon Cancer. Molecules, 25(10), 2436. https://doi.org/10.3390/molecules25102436