
Structural Basis for Broad Substrate Selectivity of Alcohol Dehydrogenase YjgB from Escherichia coli


Abstract
1. Introduction
2. Results
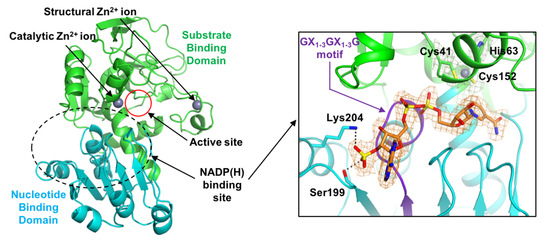
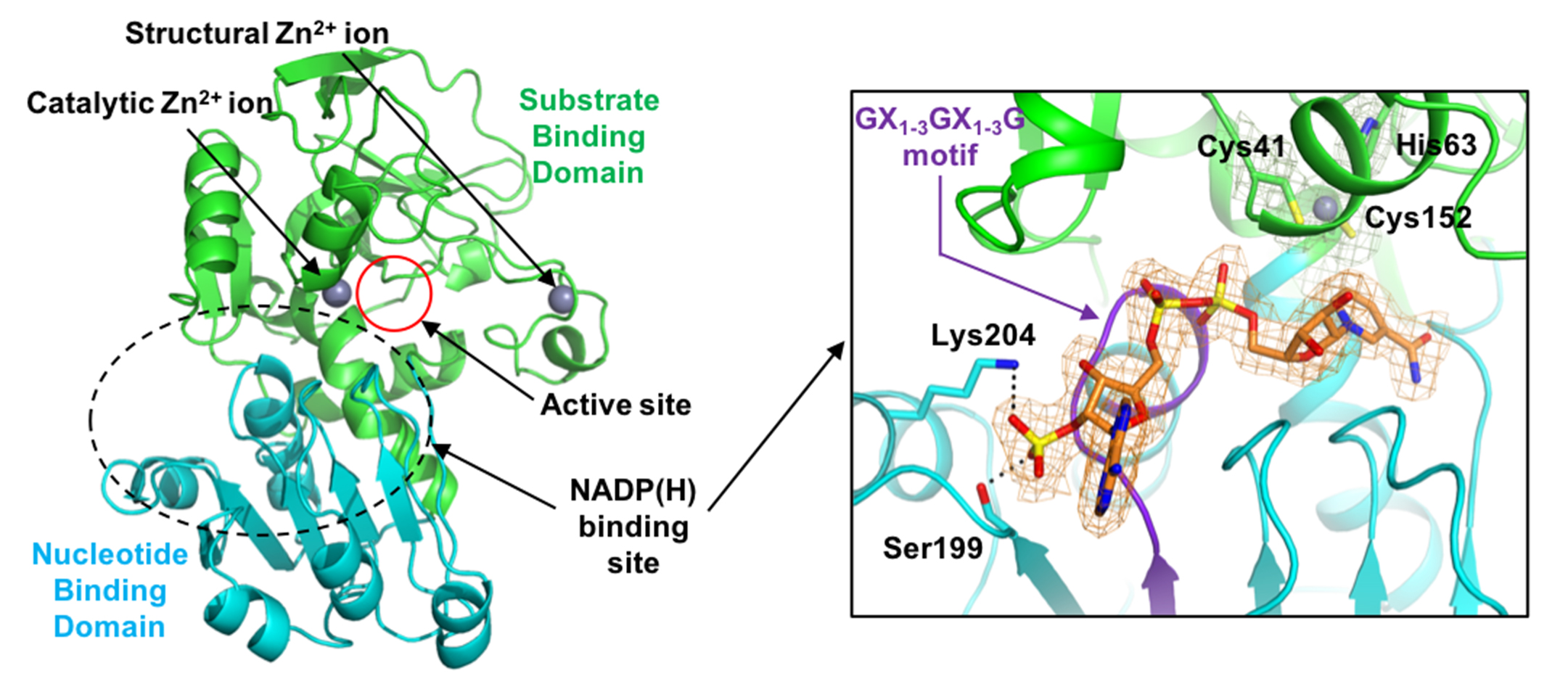
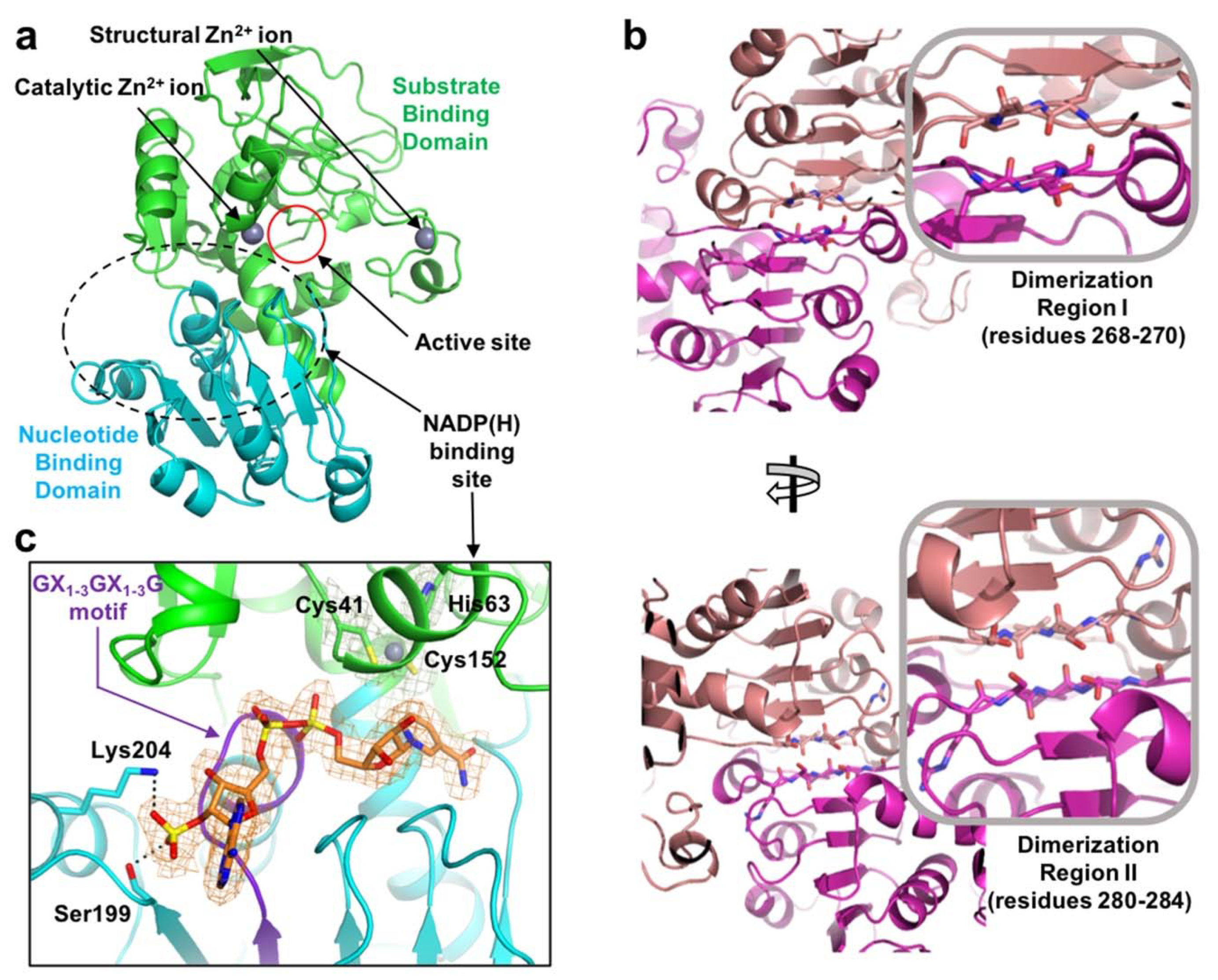
2.1. Overall Structure
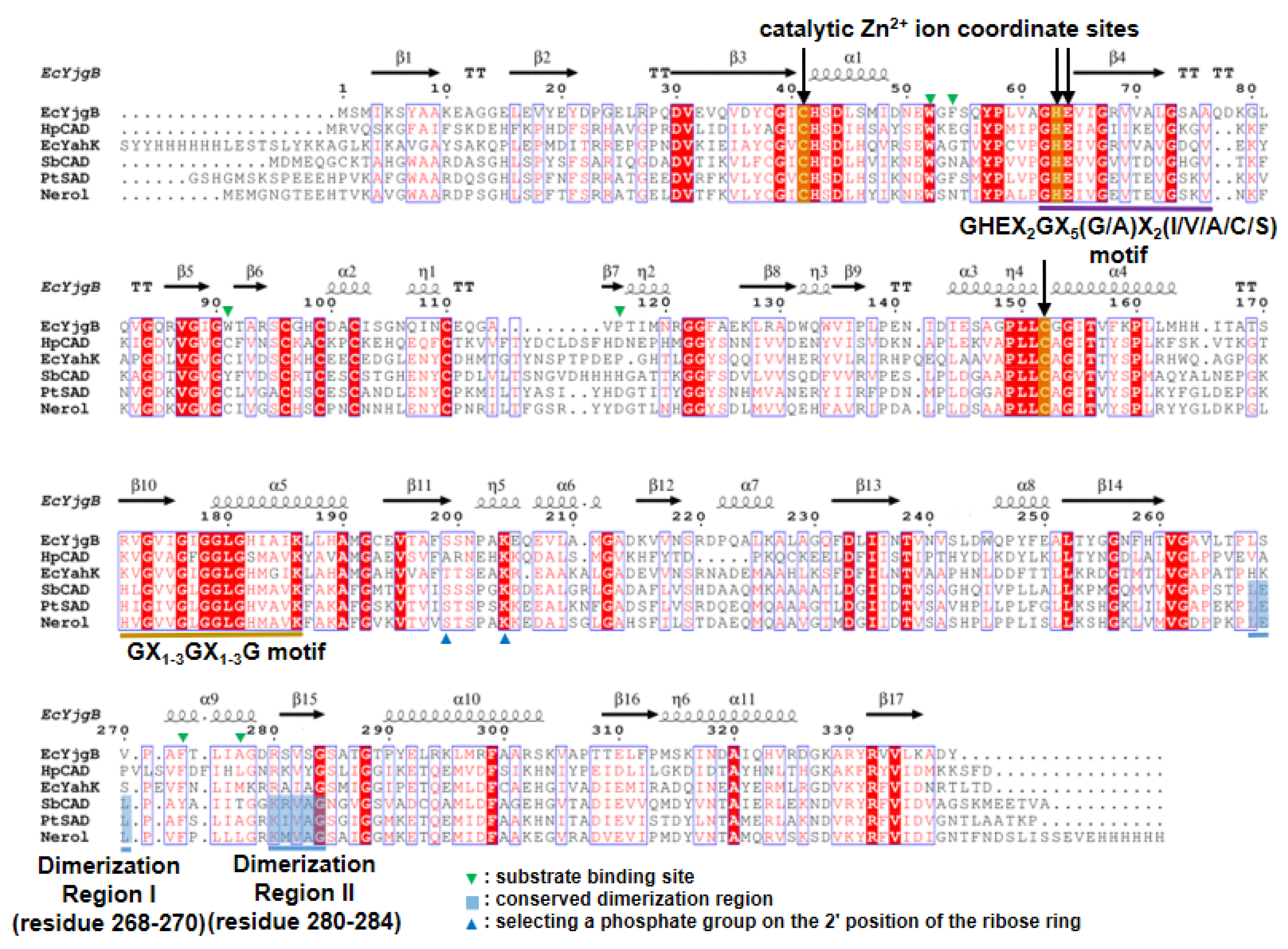
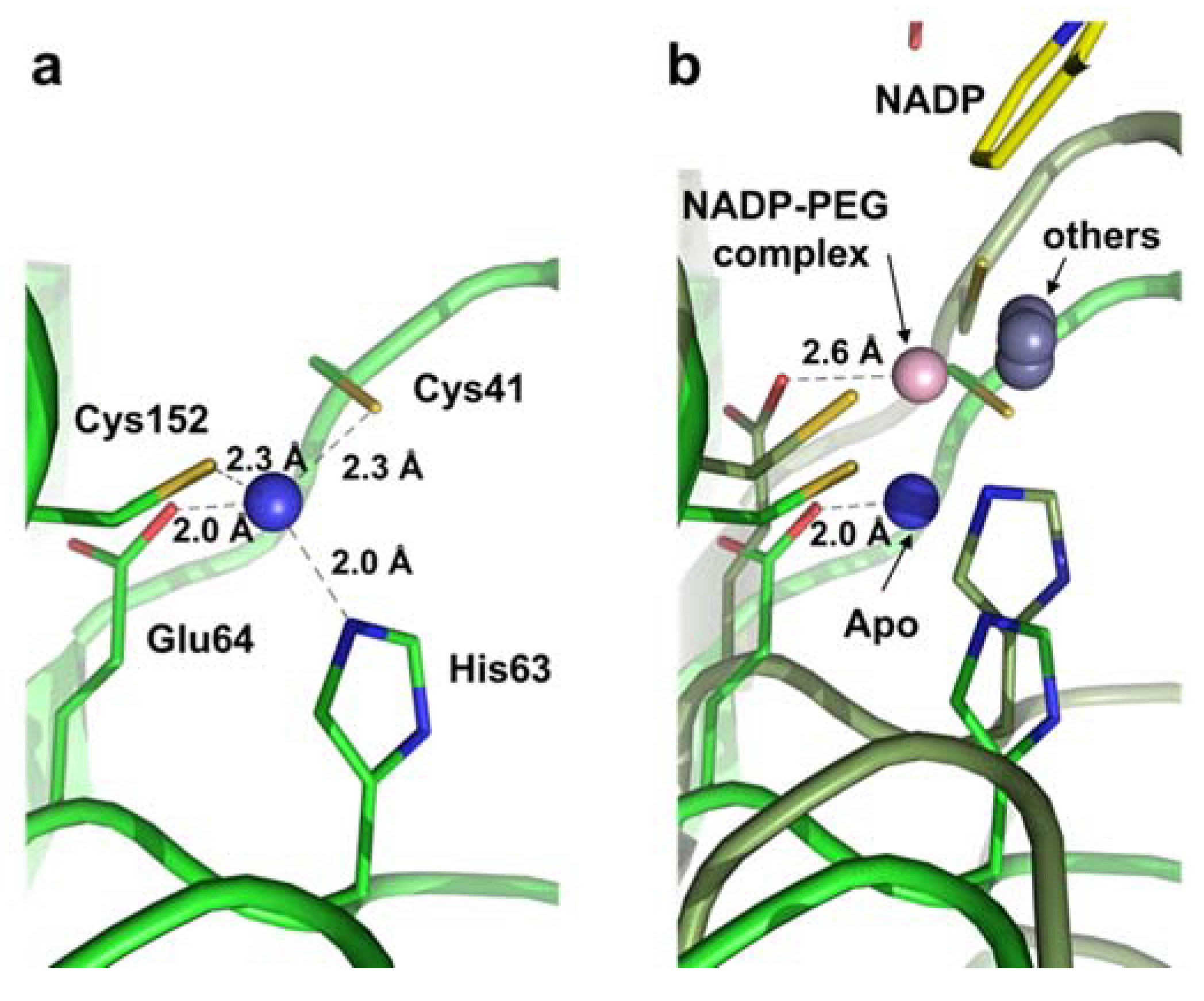
2.2. Zinc Coordination and Active Site
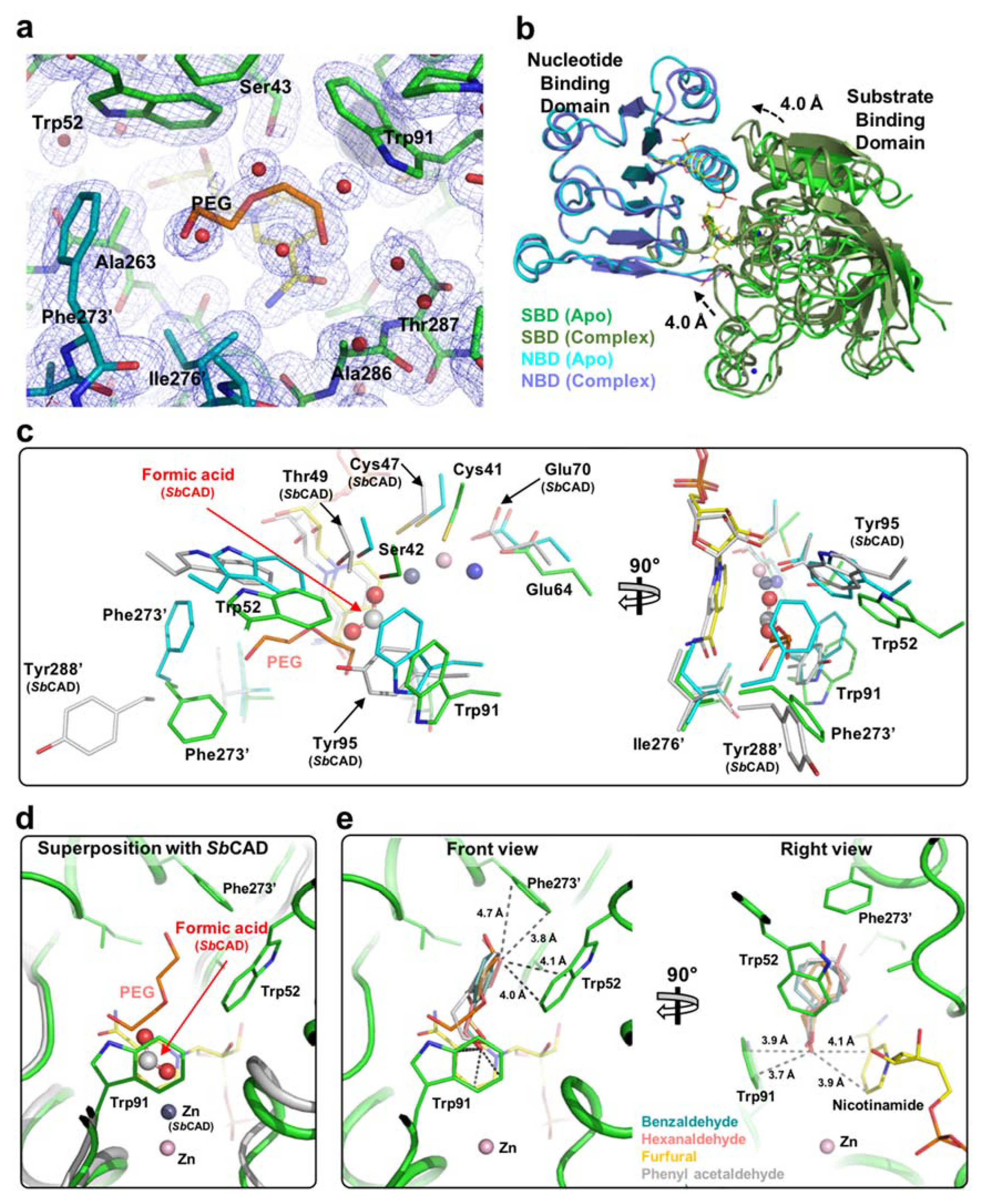
2.3. Substrate-Binding Pocket by Conformational Change
2.4. Substrate Specificity
3. Discussion
4. Materials and Methods
4.1. Cloning, Expression, and Purification
4.2. Crystallization
4.3. Data Collection and Processing
4.4. Structure Determination and Refinement
4.5. Docking Simulation
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Atsumi, S.; Wu, T.-Y.; Eckl, E.-M.; Hawkins, S.D.; Buelter, T.; Liao, J.C. Engineering the isobutanol biosynthetic pathway in Escherichia coli by comparison of three aldehyde reductase/alcohol dehydrogenase genes. Appl. Microbiol. Biotechnol. 2010, 85, 651–657. [Google Scholar] [CrossRef]
- Nakamura, C.E.; Whited, G.M. Metabolic engineering for the microbial production of 1,3-propanediol. Curr. Opin. Biotechnol. 2003, 14, 454–459. [Google Scholar] [CrossRef] [PubMed]
- Berrios-Rivera, S.J.; San, K.Y.; Bennett, G.N. The effect of carbon sources and lactate dehydrogenase deletion on 1,2-propanediol production in Escherichia coli. J. Ind. Microbiol. Biotechnol. 2003, 30, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Yim, H.; Haselbeck, R.; Niu, W.; Pujol-Baxley, C.; Burgard, A.; Boldt, J.; Khandurina, J.; Trawick, J.D.; Osterhout, R.E.; Stephen, R.; et al. Metabolic engineering of Escherichia coli for direct production of 1,4-butanediol. Nat. Chem. Biol. 2011, 7, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, D.R.; Yoon, S.-H.; Yuan, C.J.; Prather, K.L.J. Metabolic engineering of acetoin and meso-2,3-butanediol biosynthesis in E. Coli. Biotechnol. J. 2010, 5, 274–284. [Google Scholar] [CrossRef]
- Heer, D.; Heine, D.; Sauer, U. Resistance of Saccharomyces cerevisiae to High. Concentrations of Furfural Is Based on NADPH-Dependent Reduction by at Least Two Oxireductases. Appl. Environ. Microbiol. 2009, 75, 7631–7638. [Google Scholar] [CrossRef]
- Pick, A.; Rühmann, B.; Schmid, J.; Sieber, V. Novel CAD-like enzymes from Escherichia coli K-12 as additional tools in chemical production. Appl. Microbiol. Biotechnol. 2013, 97, 5815–5824. [Google Scholar] [CrossRef] [PubMed]
- Jarboe, L.R. YqhD: A broad-substrate range aldehyde reductase with various applications in production of biorenewable fuels and chemicals. Appl. Microbiol. Biotechnol. 2011, 89, 249–257. [Google Scholar] [CrossRef]
- Guo, D.; Zhang, L.; Pan, H.; Li, X. Metabolic engineering of Escherichia coli for production of 2-Phenylethylacetate from L-phenylalanine. Microbiologyopen 2017, 6, e00486. [Google Scholar] [CrossRef]
- Guo, D.; Zhang, L.; Kong, S.; Liu, Z.; Li, X.; Pan, H. Metabolic Engineering of Escherichia coli for Production of 2-Phenylethanol and 2-Phenylethyl Acetate from Glucose. J. Agric. Food Chem. 2018, 66, 5886–5891. [Google Scholar] [CrossRef]
- Kuo, C.H.; Chen, G.-J.; Chen, C.-I.; Liu, Y.-C.; Shieh, C.-J. Kinetics and optimization of lipase-catalyzed synthesis of rose fragrance 2-phenylethyl acetate through transesterification. Process. Biochem. 2014, 49, 437–444. [Google Scholar] [CrossRef]
- Serra, S.; Fuganti, C.; Brenna, E. Biocatalytic preparation of natural flavours and fragrances. Trends Biotechnol. 2005, 23, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Brenna, E. Flavours and fragrances by biocatalytic routes. Agro. Food Ind. Hi-Tech. 2005, 16, 18–20. [Google Scholar]
- Sulzenbacher, G.; Alvarez, K.; van den Heuvel, R.H.H.; Versluis, C.; Spinelli, S.; Campanacci, V.; Valencia, C.; Cambillau, C.; Eklund, H.; Tegoni, M. Crystal structure of E. coli alcohol dehydrogenase YqhD: Evidence of a covalently modified NADP coenzyme. J. Mol. Biol. 2004, 342, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Persson, B.; Hedlund, J.; Jörnvall, H. Medium-and short-chain dehydrogenase/reductase gene and protein families: The MDR superfamily. Cell. Mol. Life Sci. 2008, 65, 3879. [Google Scholar] [CrossRef]
- Nordling, E.; Jörnvall, H.; Persson, B. Medium-chain dehydrogenases/reductases (MDR) Family characterizations including genome comparisons and active site modelling. Eur. J. Biochem. 2002, 269, 4267–4276. [Google Scholar] [CrossRef]
- Jun, S.Y.; Walker, A.M.; Kim, H.; Ralph, J.; Vermerris, W.; Sattler, S.E.; Kang, C.H. The Enzyme Activity and Substrate Specificity of Two Major Cinnamyl Alcohol Dehydrogenases in Sorghum (Sorghum bicolor), SbCAD2 and SbCAD4. Plant. Physiol. 2017, 174, 2128–2145. [Google Scholar] [CrossRef]
- Rao, S.T.; Rossmann, M.G. Comparison of super-secondary structures in proteins. J. Mol. Biol. 1973, 76, 241–256. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Gouet, P.; Courcelle, E.; Stuart, D.I.; Metoz, F. ESPript: Analysis of multiple sequence alignments in PostScript. Bioinformatics 1999, 15, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Raj, S.B.; Ramaswamy, S.; Plapp, B.V. Yeast Alcohol Dehydrogenase Structure and Catalysis. Biochemistry 2014, 53, 5791–5803. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.M.; Harper, A.R.; Miner, W.A.; Ajufo, H.O.; Branscum, K.M.; Kao, L.; Sims, P.A. Structure of Escherichia coli AdhP (ethanol-inducible dehydrogenase) with bound NAD. Acta Cryst. Sect. F-Struct. Biol. Cryst. Commun. 2013, 69, 730–732. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- McQueen, C. Comprehensive Toxicology (Second Edition); Elsevier: Amsterdam, The Netherlands, 2017; pp. 291–330. [Google Scholar]
- Singh, S.; Brocker, C.; Koppaka, V.; Chen, Y.; Jackson, B.C.; Matsumoto, A.; Thompson, D.C.; Vasiliou, V. Aldehyde dehydrogenases in cellular responses to oxidative/electrophilic stress. Free Radic. Biol. Med. 2013, 56, 89–101. [Google Scholar] [CrossRef]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzym. 1997, 276, 307–326. [Google Scholar]
- Zwart, P.H.; Afonine, P.V.; Grosse-Kunstleve, R.W.; Hung, L.W.; Ioerger, T.R.; McCoy, A.J.; McKee, E.; Moriarty, N.W.; Read, R.J.; Sacchettini, J.C.; et al. Automated structure solution with the PHENIX suite. Methods Mol. Biol. 2008, 426, 419–435. [Google Scholar]
- Langer, G.; Cohen, S.X.; Lamzin, V.S.; Perrakis, A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 2008, 3, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Cryst. D-Biol. Cryst. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Cryst. D-Biol. Cryst. 2012, 68, 352–367. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D-Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Vaguine, A.A.; Richelle, J.; Wodak, S.J. SFCHECK: A unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. Acta Crystallogr. Sect. D-Biol. Crystallogr. 1999, 55, 191–205. [Google Scholar] [CrossRef] [PubMed]
- McNicholas, S.; Potterton, E.; Wilson, K.S.; Noble, M.E. Presenting your structures: The CCP4mg molecular-graphics software. Acta Crystallogr. D-Biol. Crystallogr. 2011, 67, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of are not available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Apo | PEG-NADP Complex | |
|---|---|---|
| Data collection | ||
| Wavelength (Å) | 0.97934 | 1.00003 |
| Space group | C2 | C2221 |
| Cell dimensions | ||
| a, b, c (Å) | 133.14, 64.47, 81.66 | 64.97, 138.99, 168.51 |
| α, β, γ (°) | 90.00, 106.14, 90.00 | 90.00, 90.00, 90.00 |
| Resolution (Å) | 50.00–1.55 (1.58–1.55) a | 30.00–2.00 (2.06–2.00) |
| Rmerge (%) b | 7.2 (57.3) | 10.9 (59.2) |
| I/σ (I) | 36.2 (3.8) | 19.2 (5.3) |
| Unique reflection | 95566 (4760) | 51912 (4358) |
| Completeness (%) | 100.0 (100.0) | 99.9 (100.0) |
| Redundancy | 7.4 (7.4) | 13.8 (14.1) |
| CC1/2 | 97.5 (89.4) | 99.9 (96.5) |
| Refinement | ||
| Resolution (Å) | 25.02–1.55 | 29.55–2.00 |
| No. reflections | 95547 | 99594 |
| Rwork (%) c/Rfree (%) d | 16.1/19.4 | 16.0/20.0 |
| No. atoms | 6093 | 5493 |
| Zn | 4 | 4 |
| Glycerol | 36 | 12 |
| NO3 | 8 | |
| NADP | 96 | |
| PEG | 14 | |
| Water | 790 | 591 |
| Averaged B-factors | 24.8 | 27.9 |
| R.m.s. deviations from ideal value | ||
| Bond lengths (Å) | 0.013 | 0.008 |
| Bond angles (°) | 1.400 | 0.903 |
| PDB ID | 7BU2 | 7BU3 |
| Apo (Å2) | NADP-PEG Complex (Å2) | Ratio (Complex/Apo) | |
|---|---|---|---|
| Zn | 17.8 | 38.8 | 2.2 |
| Cys41 | 17.0 | 31.9 | 1.9 |
| His63 | 15.5 | 31.9 | 2.1 |
| Glu64 | 14.6 | 33.8 | 2.3 |
| Cys152 | 15.6 | 28.1 | 1.8 |
| Whole protein | 24.8 | 27.9 | 1.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, G.T.; Kim, Y.-G.; Ahn, J.-W.; Chang, J.H. Structural Basis for Broad Substrate Selectivity of Alcohol Dehydrogenase YjgB from Escherichia coli. Molecules 2020, 25, 2404. https://doi.org/10.3390/molecules25102404
Nguyen GT, Kim Y-G, Ahn J-W, Chang JH. Structural Basis for Broad Substrate Selectivity of Alcohol Dehydrogenase YjgB from Escherichia coli. Molecules. 2020; 25(10):2404. https://doi.org/10.3390/molecules25102404
Chicago/Turabian StyleNguyen, Giang Thu, Yeon-Gil Kim, Jae-Woo Ahn, and Jeong Ho Chang. 2020. "Structural Basis for Broad Substrate Selectivity of Alcohol Dehydrogenase YjgB from Escherichia coli" Molecules 25, no. 10: 2404. https://doi.org/10.3390/molecules25102404
APA StyleNguyen, G. T., Kim, Y.-G., Ahn, J.-W., & Chang, J. H. (2020). Structural Basis for Broad Substrate Selectivity of Alcohol Dehydrogenase YjgB from Escherichia coli. Molecules, 25(10), 2404. https://doi.org/10.3390/molecules25102404