
Combined X-ray Crystallographic, IR/Raman Spectroscopic, and Periodic DFT Investigations of New Multicomponent Crystalline Forms of Anthelmintic Drugs: A Case Study of Carbendazim Maleate

 ,
,  and
and
Abstract
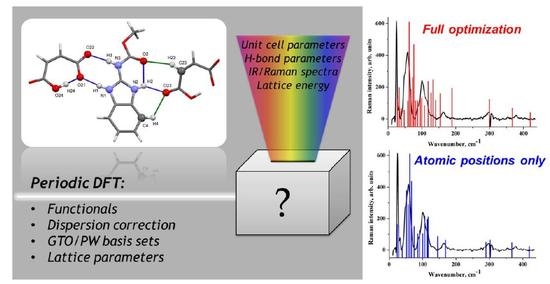
1. Introduction
2. Results and Discussion
2.1. Hydrogen Bond Patterns
2.2. Effect of Optimization on Cell Parameters
2.3. Metric Parameters of Conventional and Non-Conventional H-Bonds
2.4. IR Spectrum in the Low-Frequency Region
2.5. Raman Spectrum in the Low-Frequency Region
2.6. Lattice Energy Evaluation
3. Materials and Methods
3.1. Compounds and Solvents
3.2. Cocrystal Preparation
3.3. Thermal Analysis
3.4. Single-Crystal and Powder X-ray Diffraction (XRD) Experiments
3.5. IR and Raman Spectroscopy
3.6. Periodic (Solid-State) DFT Computations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Paulekuhn, G.S.; Dressman, J.B.; Saal, C. Trends in Active Pharmaceutical Ingredient Salt Selection based on Analysis of the Orange Book Database. J. Med. Chem. 2007, 50, 6665–6672. [Google Scholar] [CrossRef] [PubMed]
- Marder, S.R.; Perry, J.W.; Schaefer, W.P. Synthesis of Organic Salts with Large Second-Order Optical Nonlinearities. Science 1989, 245, 626–628. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Wu, M.; Kou, Y.; Min, E. Ionic liquids: Applications in catalysis. Catal. Today 2002, 74, 157–189. [Google Scholar] [CrossRef]
- Kubisa, P. Application of ionic liquids as solvents for polymerization processes. Prog. Polym. Sci. 2004, 29, 3–12. [Google Scholar] [CrossRef]
- Otero-de-la-Roza, A.; Johnson, E.R. A benchmark for non-covalent interactions in solids. J. Chem. Phys. 2012, 137, 054103. [Google Scholar] [CrossRef]
- Reilly, A.M.; Tkatchenko, A. Understanding the role of vibrations, exact exchange, and many-body van der Waals interactions in the cohesive properties of molecular crystals. J. Chem. Phys. 2013, 139, 024705. [Google Scholar] [CrossRef]
- Brandenburg, J.G.; Alessio, M.; Civalleri, B.; Peintinger, M.F.; Bredow, T.; Grimme, S. Geometrical Correction for the Inter- and Intramolecular Basis Set Superposition Error in Periodic Density Functional Theory Calculations. J. Phys. Chem. A 2013, 117, 9282–9292. [Google Scholar] [CrossRef]
- Červinka, C.; Fulem, M.; Růžička, K. CCSD(T)/CBS fragment-based calculations of lattice energy of molecular crystals. J. Chem. Phys. 2016, 144, 064505. [Google Scholar] [CrossRef]
- Cutini, M.; Civalleri, B.; Corno, M.; Orlando, R.; Brandenburg, J.G.; Maschio, L.; Ugliengo, P. Assessment of Different Quantum Mechanical Methods for the Prediction of Structure and Cohesive Energy of Molecular Crystals. J. Chem. Theory Comput. 2016, 12, 3340–3352. [Google Scholar] [CrossRef]
- Wang, K.; Mishra, M.K.; Sun, C.C. Exceptionally Elastic Single-Component Pharmaceutical Crystals. Chem. Mater. 2019, 31, 1794–1799. [Google Scholar] [CrossRef]
- López-Mejías, V.; Kampf, J.W.; Matzger, A.J. Nonamorphism in Flufenamic Acid and a New Record for a Polymorphic Compound with Solved Structures. J. Am. Chem. Soc. 2012, 134, 9872–9875. [Google Scholar] [CrossRef] [PubMed]
- Greenwell, C.; McKinley, J.L.; Zhang, P.; Zeng, Q.; Sun, G.; Li, B.; Wen, S.; Beran, G.J.O. Overcoming the difficulties of predicting conformational polymorph energetics in molecular crystals via correlated wavefunction methods. Chem. Sci. 2020, 11, 2200–2214. [Google Scholar] [CrossRef] [PubMed]
- Geng, W.-J.; Ma, Q.; Chen, Y.; Yang, W.; Jia, Y.-F.; Li, J.-S.; Zhang, Z.-Q.; Fan, G.-J.; Wang, S.-M. Structure–Performance Relationship in Thermally Stable Energetic Materials: Tunable Physical Properties of Benzopyridotetraazapentalene by Incorporating Amino Groups, Hydrogen Bonding, and π–π Interactions. Cryst. Growth Des. 2020, 20, 2106–2114. [Google Scholar] [CrossRef]
- Srirambhatla, V.K.; Guo, R.; Dawson, D.M.; Price, S.L.; Florence, A.J. Reversible, Two-Step Single-Crystal to Single-Crystal Phase Transitions between Desloratadine Forms I, II, and III. Cryst. Growth Des. 2020, 20, 1800–1810. [Google Scholar] [CrossRef]
- Usta, H.; Kim, D.; Ozdemir, R.; Zorlu, Y.; Kim, S.; Ruiz Delgado, M.C.; Harbuzaru, A.; Kim, S.; Demirel, G.; Hong, J.; et al. High Electron Mobility in [1]Benzothieno [3,2-b][1]benzothiophene-Based Field-Effect Transistors: Toward n-Type BTBTs. Chem. Mater. 2019, 31, 5254–5263. [Google Scholar] [CrossRef]
- Komissarova, E.A.; Dominskiy, D.I.; Zhulanov, V.E.; Abashev, G.G.; Siddiqui, A.; Singh, S.P.; Sosorev, A.Y.; Paraschuk, D.Y. Unraveling the unusual effect of fluorination on crystal packing in an organic semiconductor. Phys. Chem. Chem. Phys. 2020, 22, 1665–1673. [Google Scholar] [CrossRef]
- Zhu, L.; Yi, Y.; Li, Y.; Kim, E.-G.; Coropceanu, V.; Brédas, J.-L. Prediction of Remarkable Ambipolar Charge-Transport Characteristics in Organic Mixed-Stack Charge-Transfer Crystals. J. Am. Chem. Soc. 2012, 134, 2340–2347. [Google Scholar] [CrossRef]
- Manin, A.N.; Voronin, A.P.; Shishkina, A.V.; Vener, M.V.; Churakov, A.V.; Perlovich, G.L. Influence of Secondary Interactions on the Structure, Sublimation Thermodynamics, and Solubility of Salicylate:4-Hydroxybenzamide Cocrystals. Combined Experimental and Theoretical Study. J. Phys. Chem. B 2015, 119, 10466–10477. [Google Scholar] [CrossRef]
- Sharada, D.; Saha, A.; Saha, B.K. Charge transfer complexes as colour changing and disappearing–reappearing colour materials. New J. Chem. 2019, 43, 7562–7566. [Google Scholar] [CrossRef]
- Vishweshwar, P.; Jagadeesh Babu, N.; Nangia, A.; Mason, S.A.; Puschmann, H.; Mondal, R.; Howard, J.A.K. Variable Temperature Neutron Diffraction Analysis of a Very Short O−H···O Hydrogen Bond in 2,3,5,6-Pyrazinetetracarboxylic Acid Dihydrate: Synthon-Assisted Short Oacid−H···Owater Hydrogen Bonds in a Multicenter Array. J. Phys. Chem. A 2004, 108, 9406–9416. [Google Scholar] [CrossRef]
- Vener, M.V.; Manaev, A.V.; Egorova, A.N.; Tsirelson, V.G. QTAIM Study of Strong H-Bonds with the O−H···A Fragment (A = O, N) in Three-Dimensional Periodical Crystals. J. Phys. Chem. A 2007, 111, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Surov, A.O.; Voronin, A.P.; Vener, M.V.; Churakov, A.V.; Perlovich, G.L. Specific features of supramolecular organisation and hydrogen bonding in proline cocrystals: A case study of fenamates and diclofenac. CrystEngComm 2018, 20, 6970–6981. [Google Scholar] [CrossRef]
- Gavezzotti, A. Calculation of Intermolecular Interaction Energies by Direct Numerical Integration over Electron Densities. 2. An Improved Polarization Model and the Evaluation of Dispersion and Repulsion Energies. J. Phys. Chem. B 2003, 107, 2344–2353. [Google Scholar] [CrossRef]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer model energies and energy frameworks: Extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017, 4, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Day, G.M. Current approaches to predicting molecular organic crystal structures. Crystallogr. Rev. 2011, 17, 3–52. [Google Scholar] [CrossRef]
- Chickos, J.S.; Gavezzotti, A. Sublimation Enthalpies of Organic Compounds: A Very Large Database with a Match to Crystal Structure Determinations and a Comparison with Lattice Energies. Cryst. Growth Des. 2019, 19, 6566–6576. [Google Scholar] [CrossRef]
- Colombo, V.; Presti, L.L.; Gavezzotti, A. Two-component organic crystals without hydrogen bonding: Structure and intermolecular interactions in bimolecular stacking. CrystEngComm 2017, 19, 2413–2423. [Google Scholar] [CrossRef]
- Manin, A.N.; Voronin, A.P.; Manin, N.G.; Vener, M.V.; Shishkina, A.V.; Lermontov, A.S.; Perlovich, G.L. Salicylamide Cocrystals: Screening, Crystal Structure, Sublimation Thermodynamics, Dissolution, and Solid-State DFT Calculations. J. Phys. Chem. B 2014, 118, 6803–6814. [Google Scholar] [CrossRef]
- King, M.D.; Buchanan, W.D.; Korter, T.M. Identification and Quantification of Polymorphism in the Pharmaceutical Compound Diclofenac Acid by Terahertz Spectroscopy and Solid-State Density Functional Theory. Anal. Chem. 2011, 83, 3786–3792. [Google Scholar] [CrossRef]
- Bedoya-Martínez, N.; Schrode, B.; Jones, A.O.F.; Salzillo, T.; Ruzié, C.; Demitri, N.; Geerts, Y.H.; Venuti, E.; Della Valle, R.G.; Zojer, E.; et al. DFT-Assisted Polymorph Identification from Lattice Raman Fingerprinting. J. Phys. Chem. Lett. 2017, 8, 3690–3695. [Google Scholar] [CrossRef]
- Levina, E.O.; Chernyshov, I.Y.; Voronin, A.P.; Alekseiko, L.N.; Stash, A.I.; Vener, M.V. Solving the enigma of weak fluorine contacts in the solid state: A periodic DFT study of fluorinated organic crystals. Rsc. Adv. 2019, 9, 12520–12537. [Google Scholar] [CrossRef]
- Vener, M.V.; Medvedev, A.G.; Churakov, A.V.; Prikhodchenko, P.V.; Tripol’skaya, T.A.; Lev, O. H-Bond Network in Amino Acid Cocrystals with H2O or H2O2. The DFT Study of Serine–H2O and Serine–H2O2. J. Phys. Chem. A 2011, 115, 13657–13663. [Google Scholar] [CrossRef] [PubMed]
- Červinka, C.; Fulem, M. Cohesive properties of the crystalline phases of twenty proteinogenic α-aminoacids from first-principles calculations. Phys. Chem. Chem. Phys. 2019, 21, 18501–18515. [Google Scholar] [CrossRef] [PubMed]
- Tukachev, N.V.; Maslennikov, D.R.; Sosorev, A.Y.; Tretiak, S.; Zhugayevych, A. Ground-State Geometry and Vibrations of Polyphenylenevinylene Oligomers. J. Phys. Chem. Lett. 2019, 10, 3232–3239. [Google Scholar] [CrossRef]
- Tosoni, S.; Tuma, C.; Sauer, J.; Civalleri, B.; Ugliengo, P. A comparison between plane wave and Gaussian-type orbital basis sets for hydrogen bonded systems: Formic acid as a test case. J. Chem. Phys. 2007, 127, 154102. [Google Scholar] [CrossRef]
- Drużbicki, K.; Mikuli, E.; Pałka, N.; Zalewski, S.; Ossowska-Chruściel, M.D. Polymorphism of Resorcinol Explored by Complementary Vibrational Spectroscopy (FT-RS, THz-TDS, INS) and First-Principles Solid-State Computations (Plane-Wave DFT). J. Phys. Chem. B 2015, 119, 1681–1695. [Google Scholar] [CrossRef]
- Zvereva, E.E.; Shagidullin, A.R.; Katsyuba, S.A. Ab Initio and DFT Predictions of Infrared Intensities and Raman Activities. J. Phys. Chem. A 2011, 115, 63–69. [Google Scholar] [CrossRef]
- Katsyuba, S.A.; Zvereva, E.E.; Burganov, T.I. Is There a Simple Way to Reliable Simulations of Infrared Spectra of Organic Compounds? J. Phys. Chem. A 2013, 117, 6664–6670. [Google Scholar] [CrossRef]
- Churakov, A.V.; Prikhodchenko, P.V.; Lev, O.; Medvedev, A.G.; Tripol’skaya, T.A.; Vener, M.V. A model proton-transfer system in the condensed phase: NH4 + OOH−, a crystal with short intermolecular H-bonds. J. Chem. Phys. 2010, 133, 164506. [Google Scholar] [CrossRef]
- Tuma, C.; Daniel Boese, A.; Handy, N.C. Predicting the binding energies of H-bonded complexes: A comparative DFT study. Phys. Chem. Chem. Phys. 1999, 1, 3939–3947. [Google Scholar] [CrossRef]
- Vener, M.V.; Levina, E.O.; Koloskov, O.A.; Rykounov, A.A.; Voronin, A.P.; Tsirelson, V.G. Evaluation of the Lattice Energy of the Two-Component Molecular Crystals Using Solid-State Density Functional Theory. Cryst. Growth Des. 2014, 14, 4997–5003. [Google Scholar] [CrossRef]
- Shishkina, A.V.; Zhurov, V.V.; Stash, A.I.; Vener, M.V.; Pinkerton, A.A.; Tsirelson, V.G. Noncovalent Interactions in Crystalline Picolinic Acid N-Oxide: Insights from Experimental and Theoretical Charge Density Analysis. Cryst. Growth Des. 2013, 13, 816–828. [Google Scholar] [CrossRef]
- Akin-Ojo, O.; Wang, F. Effects of the dispersion interaction in liquid water. Chem. Phys. Lett. 2011, 513, 59–62. [Google Scholar] [CrossRef]
- Katsyuba, S.A.; Vener, M.V.; Zvereva, E.E.; Brandenburg, J.G. The role of London dispersion interactions in strong and moderate intermolecular hydrogen bonds in the crystal and in the gas phase. Chem. Phys. Lett. 2017, 672, 124–127. [Google Scholar] [CrossRef]
- Voronin, A.P.; Perlovich, G.L.; Vener, M.V. Effects of the crystal structure and thermodynamic stability on solubility of bioactive compounds: DFT study of isoniazid cocrystals. Comput. Theor. Chem. 2016, 1092, 1–11. [Google Scholar] [CrossRef]
- Mohaček-Grošev, V.; Grdadolnik, J.; Stare, J.; Hadži, D. Identification of hydrogen bond modes in polarized Raman spectra of single crystals of α-oxalic acid dihydrate. J. Raman Spectrosc. 2009, 40, 1605–1614. [Google Scholar] [CrossRef]
- Yushina, I.D.; Kolesov, B.A.; Bartashevich, E.V. Raman spectroscopy study of new thia- and oxazinoquinolinium triodides. New J. Chem. 2015, 39, 6163–6170. [Google Scholar] [CrossRef]
- Katsyuba, S.A.; Vener, M.V.; Zvereva, E.E.; Fei, Z.; Scopelliti, R.; Laurenczy, G.; Yan, N.; Paunescu, E.; Dyson, P.J. How Strong Is Hydrogen Bonding in Ionic Liquids? Combined X-ray Crystallographic, Infrared/Raman Spectroscopic, and Density Functional Theory Study. J. Phys. Chem. B 2013, 117, 9094–9105. [Google Scholar] [CrossRef]
- Bolla, G.; Nangia, A. Novel pharmaceutical salts of albendazole. CrystEngComm 2018, 20, 6394–6405. [Google Scholar] [CrossRef]
- Chen, J.-M.; Wang, Z.-Z.; Wu, C.-B.; Li, S.; Lu, T.-B. Crystal engineering approach to improve the solubility of mebendazole. CrystEngComm 2012, 14, 6221–6229. [Google Scholar] [CrossRef]
- Chen, J.; Lu, T. New Crystalline Forms of Mebendazole with n-Alkyl Carboxylic Acids: Neutral and Ionic Status. Chin. J. Chem. 2013, 31, 635–640. [Google Scholar] [CrossRef]
- Musin, R.N.; Mariam, Y.H. An integrated approach to the study of intramolecular hydrogen bonds in malonaldehyde enol derivatives and naphthazarin: Trend in energetic versus geometrical consequences. J. Phys. Org. Chem. 2006, 19, 425–444. [Google Scholar] [CrossRef]
- Filarowski, A.; Koll, A.; Sobczyk, L. Intramolecular Hydrogen Bonding in o-hydroxy Aryl Schiff Bases. Curr. Org. Chem. 2009, 13, 172–193. [Google Scholar] [CrossRef]
- Rozenberg, M.; Loewenschuss, A.; Marcus, Y. An empirical correlation between stretching vibration redshift and hydrogen bond length. Phys. Chem. Chem. Phys. 2000, 2, 2699–2702. [Google Scholar] [CrossRef]
- Etter, M.C. Encoding and decoding hydrogen-bond patterns of organic compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Nyman, J.; Day, G.M. Static and lattice vibrational energy differences between polymorphs. CrystEngComm 2015, 17, 5154–5165. [Google Scholar] [CrossRef]
- Karamertzanis, P.G.; Day, G.M.; Welch, G.W.A.; Kendrick, J.; Leusen, F.J.J.; Neumann, M.A.; Price, S.L. Modeling the interplay of inter- and intramolecular hydrogen bonding in conformational polymorphs. J. Chem. Phys. 2008, 128, 244708. [Google Scholar] [CrossRef]
- Juliano, T.R.; Korter, T.M. Terahertz Vibrations of Crystalline Acyclic and Cyclic Diglycine: Benchmarks for London Force Correction Models. J. Phys. Chem. A 2013, 117, 10504–10512. [Google Scholar] [CrossRef]
- Stein, M.; Heimsaat, M. Intermolecular Interactions in Molecular Organic Crystals upon Relaxation of Lattice Parameters. Crystals 2019, 9, 665. [Google Scholar] [CrossRef]
- Barone, V.; Biczysko, M.; Bloino, J. Fully anharmonic IR and Raman spectra of medium-size molecular systems: Accuracy and interpretation. PCCP 2014, 16, 1759–1787. [Google Scholar] [CrossRef]
- Vener, M.V.; Sauer, J. Quantum anharmonic frequencies of the O⋯ H⋯ O fragment of the H5O2+ ion: A model three-dimensional study. Chem. Phys. Lett. 1999, 312, 591–597. [Google Scholar] [CrossRef]
- Brela, M.; Stare, J.; Pirc, G.; Sollner-Dolenc, M.; Boczar, M.; Wójcik, M.J.; Mavri, J. Car–Parrinello Simulation of the Vibrational Spectrum of a Medium Strong Hydrogen Bond by Two-Dimensional Quantization of the Nuclear Motion: Application to 2-Hydroxy-5-nitrobenzamide. J. Phys. Chem. B 2012, 116, 4510–4518. [Google Scholar] [CrossRef] [PubMed]
- Brela, M.Z.; Wójcik, M.J.; Witek, Ł.J.; Boczar, M.; Wrona, E.; Hashim, R.; Ozaki, Y. Born–Oppenheimer Molecular Dynamics Study on Proton Dynamics of Strong Hydrogen Bonds in Aspirin Crystals, with Emphasis on Differences between Two Crystal Forms. J. Phys. Chem. B 2016, 120, 3854–3862. [Google Scholar] [CrossRef] [PubMed]
- Sushchinskiĭ, M.M. Raman Spectra of Molecules and Crystals; Wiley: New York, NY, USA, 1972. [Google Scholar]
- Takahashi, M. Terahertz Vibrations and Hydrogen-Bonded Networks in Crystals. Crystals 2014, 4, 74–103. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, H.-W.; Tominaga, K.; Hayashi, M. Mixing of intermolecular and intramolecular vibrations in optical phonon modes: Terahertz spectroscopy and solid-state density functional theory. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2016, 6, 386–409. [Google Scholar] [CrossRef]
- Sakun, V.P.; Vener, M.V.; Sokolov, N.D. Proton tunneling assisted by the intermolecular vibration excitation. Temperature dependence of the proton spin-lattice relaxation time in benzoic acid powder. J. Chem. Phys. 1996, 105, 379–387. [Google Scholar] [CrossRef]
- Yang, J.; Li, S.; Zhao, H.; Song, B.; Zhang, G.; Zhang, J.; Zhu, Y.; Han, J. Molecular Recognition and Interaction between Uracil and Urea in Solid-State Studied by Terahertz Time-Domain Spectroscopy. J. Phys. Chem. A 2014, 118, 10927–10933. [Google Scholar] [CrossRef]
- Takahashi, M.; Okamura, N.; Ding, X.; Shirakawa, H.; Minamide, H. Intermolecular hydrogen bond stretching vibrations observed in terahertz spectra of crystalline vitamins. CrystEngComm 2018, 20, 1960–1969. [Google Scholar] [CrossRef]
- Jepsen, P.U.; Clark, S.J. Precise ab-initio prediction of terahertz vibrational modes in crystalline systems. Chem. Phys. Lett. 2007, 442, 275–280. [Google Scholar] [CrossRef]
- Ruggiero, M.T. Invited Review: Modern Methods for Accurately Simulating the Terahertz Spectra of Solids. J. InfraredMillim. Terahertz Waves 2020. [Google Scholar] [CrossRef]
- Vener, M.V.; Sauer, J. Environmental effects on vibrational proton dynamics in H5O2+: DFT study on crystalline H5O2 + ClO4−. Phys. Chem. Chem. Phys. 2005, 7, 258–263. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shishkina, A.; Stash, A.; Civalleri, B.; Ellern, A.; Tsirelson, V. Electron-density and electrostatic-potential features of orthorhombic chlorine trifluoride. Mendeleev Commun. 2010, 20, 161–164. [Google Scholar] [CrossRef]
- Sen, A.; Mitev, P.D.; Eriksson, A.; Hermansson, K. H-bond and electric field correlations for water in highly hydrated crystals. Int. J. Quantum Chem. 2016, 116, 67–80. [Google Scholar] [CrossRef]
- King, M.D.; Buchanan, W.D.; Korter, T.M. Application of London-type dispersion corrections to the solid-state density functional theory simulation of the terahertz spectra of crystalline pharmaceuticals. Phys. Chem. Chem. Phys. 2011, 13, 4250–4259. [Google Scholar] [CrossRef]
- Kim, J.; Kwon, O.P.; Jazbinsek, M.; Park, Y.C.; Lee, Y.S. First-Principles Calculation of Terahertz Absorption with Dispersion Correction of 2,2′-Bithiophene as Model Compound. J. Phys. Chem. C 2015, 119, 12598–12607. [Google Scholar] [CrossRef]
- Fonari, A.; Corbin, N.S.; Vermeulen, D.; Goetz, K.P.; Jurchescu, O.D.; McNeil, L.E.; Bredas, J.L.; Coropceanu, V. Vibrational properties of organic donor-acceptor molecular crystals: Anthracene-pyromellitic-dianhydride (PMDA) as a case study. J. Chem. Phys. 2015, 143, 224503. [Google Scholar] [CrossRef]
- Deringer, V.L.; George, J.; Dronskowski, R.; Englert, U. Plane-Wave Density Functional Theory Meets Molecular Crystals: Thermal Ellipsoids and Intermolecular Interactions. Acc. Chem. Res. 2017, 50, 1231–1239. [Google Scholar] [CrossRef]
- Beran, G.J.O.; Nanda, K. Predicting Organic Crystal Lattice Energies with Chemical Accuracy. J. Phys. Chem. Lett. 2010, 1, 3480–3487. [Google Scholar] [CrossRef]
- Moellmann, J.; Grimme, S. DFT-D3 Study of Some Molecular Crystals. J. Phys. Chem. C 2014, 118, 7615–7621. [Google Scholar] [CrossRef]
- Yang, J.; Hu, W.; Usvyat, D.; Matthews, D.; Schütz, M.; Chan, G.K.-L. Ab initio determination of the crystalline benzene lattice energy to sub-kilojoule/mole accuracy. Science 2014, 345, 640–643. [Google Scholar] [CrossRef]
- Červinka, C.; Fulem, M. State-of-the-Art Calculations of Sublimation Enthalpies for Selected Molecular Crystals and Their Computational Uncertainty. J. Chem. Theory Comput. 2017, 13, 2840–2850. [Google Scholar] [CrossRef] [PubMed]
- Gharagheizi, F.; Ilani-Kashkouli, P.; Acree, W.E.; Mohammadi, A.H.; Ramjugernath, D. A group contribution model for determining the sublimation enthalpy of organic compounds at the standard reference temperature of 298K. Fluid Phase Equilibria 2013, 354, 265–285. [Google Scholar] [CrossRef]
- Bygrave, P.J.; Allan, N.L.; Manby, F.R. The embedded many-body expansion for energetics of molecular crystals. J. Chem. Phys. 2012, 137, 164102. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, J.L.; Palmer, D.S.; Mourik, T.V.; Mitchell, J.B.O. Are the Sublimation Thermodynamics of Organic Molecules Predictable? J. Chem. Inf. Modeling 2016, 56, 2162–2179. [Google Scholar] [CrossRef]
- Perlovich, G.L.; Raevsky, O.A. Sublimation of Molecular Crystals: Prediction of Sublimation Functions on the Basis of HYBOT Physicochemical Descriptors and Structural Clusterization. Cryst. Growth Des. 2010, 10, 2707–2712. [Google Scholar] [CrossRef]
- Grabowski, S.J. What Is the Covalency of Hydrogen Bonding? Chem. Rev. 2011, 111, 2597–2625. [Google Scholar] [CrossRef]
- Landeros-Rivera, B.; Moreno-Esparza, R.; Hernández-Trujillo, J. Theoretical study of intermolecular interactions in crystalline arene–perhaloarene adducts in terms of the electron density. Rsc. Adv. 2016, 6, 77301–77309. [Google Scholar] [CrossRef]
- Surov, A.O.; Churakov, A.V.; Perlovich, G.L. Three Polymorphic Forms of Ciprofloxacin Maleate: Formation Pathways, Crystal Structures, Calculations, and Thermodynamic Stability Aspects. Cryst. Growth Des. 2016, 16, 6556–6567. [Google Scholar] [CrossRef]
- Jarzembska, K.N.; Hoser, A.A.; Varughese, S.; Kamiński, R.; Malinska, M.; Stachowicz, M.; Pedireddi, V.R.; Woźniak, K. Structural and Energetic Analysis of Molecular Assemblies in a Series of Nicotinamide and Pyrazinamide Cocrystals with Dihydroxybenzoic Acids. Cryst. Growth Des. 2017, 17, 4918–4931. [Google Scholar] [CrossRef]
- Tao, Q.; Hao, Q.-Q.; Voronin, A.P.; Dai, X.-L.; Huang, Y.; Perlovich, G.L.; Lu, T.-B.; Chen, J.-M. Polymorphic Forms of a Molecular Salt of Phenazopyridine with 3,5-Dihydroxybenzoic Acid: Crystal Structures, Theoretical Calculations, Thermodynamic Stability, and Solubility Aspects. Cryst. Growth Des. 2019, 19, 5636–5647. [Google Scholar] [CrossRef]
- Kong, S.; Shenderovich, I.G.; Vener, M.V. Density Functional Study of the Proton Transfer Effect on Vibrations of Strong (Short) Intermolecular O−H···N/O−···H−N+ Hydrogen Bonds in Aprotic Solvents. J. Phys. Chem. A 2010, 114, 2393–2399. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.-L.; Voronin, A.P.; Huang, Y.-L.; Perlovich, G.L.; Zhao, X.-H.; Lu, T.-B.; Chen, J.-M. 5-Fluorouracil Cocrystals with Lipophilic Hydroxy-2-Naphthoic Acids: Crystal Structures, Theoretical Computations, and Permeation Studies. Cryst. Growth Des. 2020, 20, 923–933. [Google Scholar] [CrossRef]
- Manin, A.N.; Voronin, A.P.; Drozd, K.V.; Churakov, A.V.; Perlovich, G.L. Pharmaceutical salts of emoxypine with dicarboxylic acids. Acta Crystallogr. Sect. C 2018, 74, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Preiss, U.P.; Zaitsau, D.H.; Beichel, W.; Himmel, D.; Higelin, A.; Merz, K.; Caesar, N.; Verevkin, S.P. Estimation of Lattice Enthalpies of Ionic Liquids Supported by Hirshfeld Analysis. Chem. Phys. Chem. 2015, 16, 2890–2898. [Google Scholar] [CrossRef]
- Mohamed, S.; Tocher, D.A.; Price, S.L. Computational prediction of salt and cocrystal structures—Does a proton position matter? Int. J. Pharm. 2011, 418, 187–198. [Google Scholar] [CrossRef]
- Chen, L.; Bryantsev, V.S. A density functional theory based approach for predicting melting points of ionic liquids. Phys. Chem. Chem. Phys. 2017, 19, 4114–4124. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Seddon, K.R.; Leslie, M. Hydrogen-bonding contributions to the lattice energy of salts for second harmonic generation. Struct. Chem. 1992, 3, 63–65. [Google Scholar] [CrossRef]
- Blazejowski, J.; Lubkowski, J. Lattice energy of organic ionic crystals and its importance in analysis of features, behaviour and reactivity of solid-state systems. J. Therm. Anal. 1992, 38, 2195–2210. [Google Scholar] [CrossRef]
- Sheldrick, G. SADABS, Program for Scaling and Correction of Area Detector Data; University of Göttingen: Gottingen, Germany, 1997. [Google Scholar]
- Sheldrick, G. A short history of SHELX. Acta Crystallogr. Sect. A: Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-mechanical condensed matter simulations with CRYSTAL. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Buongiorno Nardelli, M.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Seaton, C.C.; Parkin, A. Making Benzamide Cocrystals with Benzoic Acids: The Influence of Chemical Structure. Cryst. Growth Des. 2011, 11, 1502–1511. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
Sample Availability: Samples of crystalline [CRB + MLE] (1:1), input and output files of the computations are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fragment (a) | Exp. | Computations | ||||
|---|---|---|---|---|---|---|
| PBE-D3/6-31G(d,p) | B3LYP/6-31G(d,p) | B3LYP-D2/6-31G(d,p) | B3LYP-D3/6-31G(d,p) | PBE-D3/PW (b) | ||
| O24–H24···O21 (intra-) | 2.442 | 2.462 (2.481) | 2.446 (2.399) | 2.468 (2.433) | 2.457 (2.443) | 2.453 (2.456) |
| N1–H1···O21 | 2.685 | 2.662 (2.700) | 2.665 (2.622) | 2.700 (2.640) | 2.680 (2.641) | 2.675 (2.670) |
| N3–H3···O22 | 2.761 | 2.735 (2.798) | 2.768 (2.718) | 2.762 (2.746) | 2.757 (2.745) | 2.741 (2.744) |
| N2–H2···O23 | 2.756 | 2.701 (2.665) | 2.714 (2.684) | 2.750 (2.681) | 2.701 (2.677) | |
| ∆V = (Vexp − Vtheor)/Vexp (%) | 3.1 | 6.8 | −9.2 | 13.3 | 9.4 | <−0.1 |
| Exp. (a) | Normal Mode Symmetry, Assignment (b) | Computations (c) | |||
|---|---|---|---|---|---|
| PBE-D3/6-31G(d,p) | B3LYP/6-31G(d,p) | B3LYP-D2/6-31G(d,p) | PBE-D3/PW (d) | ||
| 180–167 vs, broad | Bu, ν(N3···O22) + ν(N1···O21) | 179 (453) 186 (336) (e) | 180 (353) 171 (390) | 150 (66) (e) | 177 |
| 215 s | Bu, CH3 twist | 219 (62) 228 (80) | 222 (63) 219 (51) | 202 (313) | |
| 266 s | Bu, ν(O24···O21) | 257 (172) 260 (194) | 264 (165) 263 (153) | 278 (152) | 258 |
| 302 s | Bu, CNC(=O) bending | 299 (58) 302 (43) | 303 (68) 302 (65) | 312 (95) | |
| 330–346 vs, broad | Bu, ν(N1···O21) | 362 (132) 365 (46) | 358 (223) 334 (122) | 368 (156) | 349 |
| 380 s | Bu, ν(N3···O22) | 368 (160) 376 (206) | 374 (76) 351 (183) | 383 (88) (e) | 364 |
| B3LYP-D3/6-31G(d,p) (AtomOnly) | PBE-D3/6-31G(d,p) (AtomOnly) | PBE-D3/6-31G(d,p) (FullOpt) | PBE-D3/PW (AtomOnly) | |
|---|---|---|---|---|
| Neutral molecules in the gas phase | 277.8 | 266.8 | 258.4 | 278.3 |
| Charged ions in the gas phase a | 817.2 | 655.6 | 647.2 | 625.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voronin, A.P.; Surov, A.O.; Churakov, A.V.; Parashchuk, O.D.; Rykounov, A.A.; Vener, M.V. Combined X-ray Crystallographic, IR/Raman Spectroscopic, and Periodic DFT Investigations of New Multicomponent Crystalline Forms of Anthelmintic Drugs: A Case Study of Carbendazim Maleate. Molecules 2020, 25, 2386. https://doi.org/10.3390/molecules25102386
Voronin AP, Surov AO, Churakov AV, Parashchuk OD, Rykounov AA, Vener MV. Combined X-ray Crystallographic, IR/Raman Spectroscopic, and Periodic DFT Investigations of New Multicomponent Crystalline Forms of Anthelmintic Drugs: A Case Study of Carbendazim Maleate. Molecules. 2020; 25(10):2386. https://doi.org/10.3390/molecules25102386
Chicago/Turabian StyleVoronin, Alexander P., Artem O. Surov, Andrei V. Churakov, Olga D. Parashchuk, Alexey A. Rykounov, and Mikhail V. Vener. 2020. "Combined X-ray Crystallographic, IR/Raman Spectroscopic, and Periodic DFT Investigations of New Multicomponent Crystalline Forms of Anthelmintic Drugs: A Case Study of Carbendazim Maleate" Molecules 25, no. 10: 2386. https://doi.org/10.3390/molecules25102386
APA StyleVoronin, A. P., Surov, A. O., Churakov, A. V., Parashchuk, O. D., Rykounov, A. A., & Vener, M. V. (2020). Combined X-ray Crystallographic, IR/Raman Spectroscopic, and Periodic DFT Investigations of New Multicomponent Crystalline Forms of Anthelmintic Drugs: A Case Study of Carbendazim Maleate. Molecules, 25(10), 2386. https://doi.org/10.3390/molecules25102386