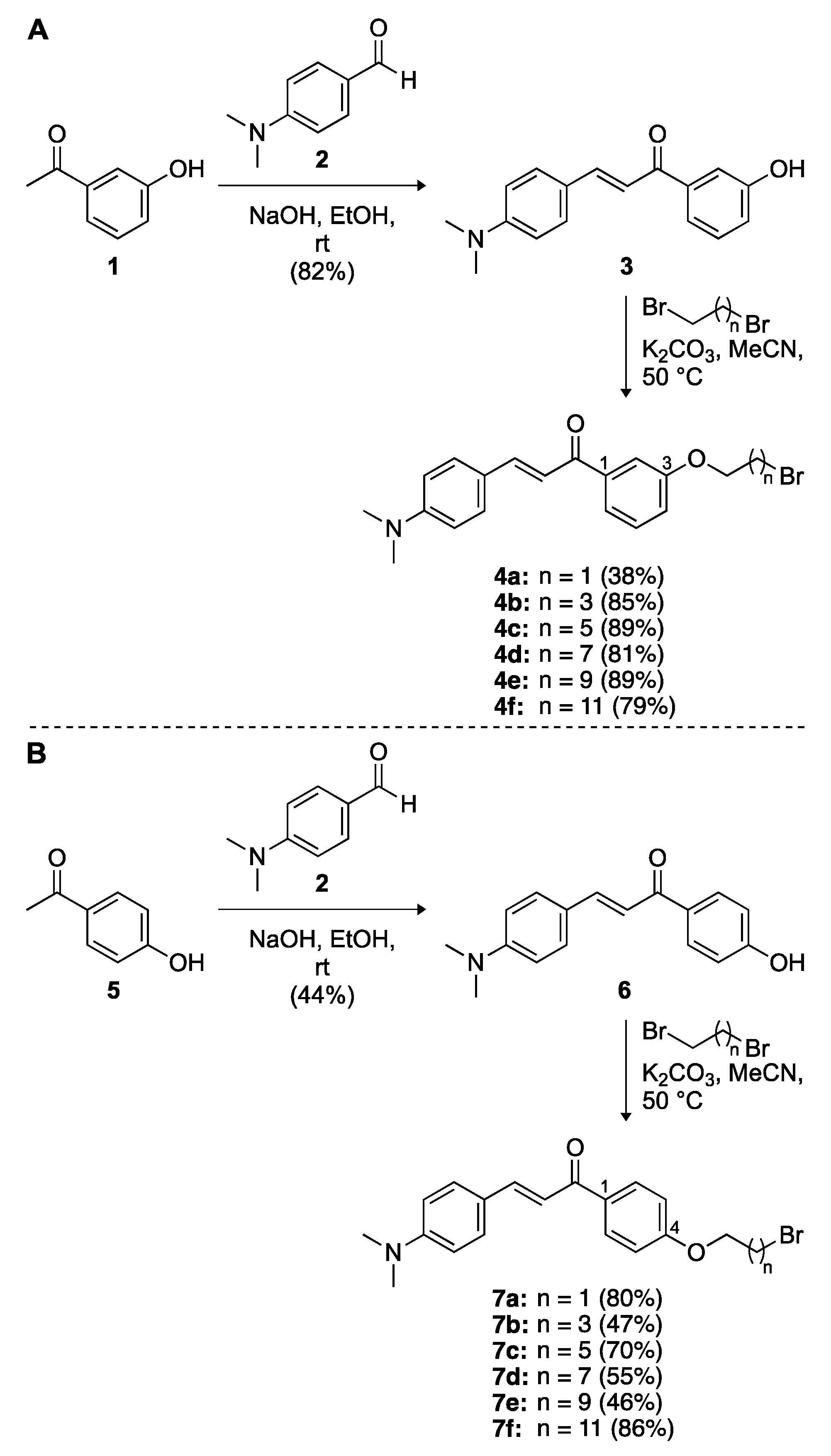
3.2. Synthesis of Compounds 3–20
(E)-3-(4-(Dimethylamino)Phenyl)-1-(3-Hydroxyphenyl)Prop-2-En-1-One (
3) (SGT9). The known compound
3 was prepared as previously described [
68]. A solution of compounds
1 (68 mg, 0.5 mmol) and
2 (75 mg, 0.5 mmol) in EtOH (1.5 mL) was treated with NaOH pellets (400 mg, 10.0 mmol). The reaction was stirred at room temperature (rt) overnight till completion. Most of the solvent was then removed and 1 N aqueous HCl was added. The precipitate was filtered to give the known compound
3 (110 mg, 82%) (R
f 0.26 in Hexanes:EtOAc/3:1) as an orange solid:
1H NMR (400 MHz, CDCl
3,
Figure S1, which matches the lit. [
68])
δ 7.78 (d,
J = 15.6 Hz, 1H, HC=C
H-Ph), 7.56–7.50 (m, 4H, aromatic), 7.35 (t,
J = 8.0 Hz, 1H, aromatic), 7.29 (d,
J = 15.6 Hz, 1H,
HC=CH-Ph), 7.03 (dd,
J1 = 8.0 Hz,
J2 = 2.0 Hz, 1H, aromatic), 6.69 (d,
J = 9.2 Hz, 2H, aromatic), 5.30 (s, 1H, O
H), 3.04 (s, 6H, N(C
H3)
2).
(E)-1-(3-(2-Bromoethoxy)Phenyl)-3-(4-(Dimethylamino)Phenyl)Prop-2-En-1-One (
4a) (SGT653). A solution of compound
3 (80 mg, 0.30 mmol) and K
2CO
3 (165 mg, 1.20 mmol) in anhydrous MeCN (5 mL) was treated with 1,2-dibromoethane (0.10 mL, 1.20 mmol) and the resulting mixture was refluxed overnight. The solvent was then removed and the obtained crude product was purified by column chromatography (SiO
2 gel, pure Hexanes to Hexanes:EtOAc/4:1; R
f 0.38 in Hexanes:EtOAc/3:1) to yield compound
4a (43 mg, 38%) as a red solid:
1H NMR (400 MHz, CDCl
3,
Figure S2)
δ 7.78 (d,
J = 15.6 Hz, 1H, HC=C
H-Ph), 7.61 (d,
J = 8.0 Hz, 1H, aromatic), 7.55–7.52 (m, 3H, aromatic), 7.39 (t,
J = 8.0 Hz, 1H, aromatic), 7.29 (d,
J = 15.6 Hz, 1H,
HC=CH-Ph), 7.10 (dd,
J1 = 8.0 Hz,
J2 = 2.8 Hz, 1H, aromatic), 6.70 (d,
J = 8.8 Hz, 2H, aromatic), 4.37 (t,
J = 6.0 Hz, 2H, BrCH
2C
H2OPh), 3.66 (t,
J = 6.0 Hz, 2H, BrC
H2CH
2OPh), 3.04 (s, 6H, N(C
H3)
2);
13C NMR (100 MHz, CDCl
3,
Figure S3)
δ 190.1, 158.3, 152.1, 146.0, 140.6, 130.5 (2 carbons), 129.5, 122.5, 121.5, 119.3, 116.7, 113.6, 111.8 (2 carbons), 68.0, 40.1 (2 carbons), 29.1;
m/
z calcd for C
19H
20BrNO
2 373.1; found 374.0 [M + H]
+.
(E)-1-(3-(4-Bromobutoxy)Phenyl)-3-(4-(Dimethylamino)Phenyl)Prop-2-En-1-One (
4b) (SGT47). A solution of compound
3 (50 mg, 0.19 mmol) and K
2CO
3 (52 mg, 0.37 mmol) in anhydrous MeCN (5 mL) was treated with 1,4-dibromobutane (0.04 mL, 0.37 mmol) and the resulting mixture was stirred at 50 °C for 4 h. The solvent was then removed and the obtained crude product was purified by column chromatography (SiO
2 gel, pure Hexanes to Hexanes:EtOAc/4:1; R
f 0.60 in Hexanes:EtOAc/3:1) to yield compound
4b (64 mg, 85%) as a red solid:
1H NMR (400 MHz, CDCl
3,
Figure S4)
δ 7.77 (d,
J = 15.6 Hz, 1H, HC=C
H-Ph), 7.57-7.50 (m, 4H, aromatic), 7.36 (t,
J = 8.4 Hz, 1H, aromatic), 7.29 (d,
J = 15.6 Hz, 1H,
HC=CH-Ph), 7.06 (d,
J = 8.4 Hz, 1H, aromatic), 6.69 (d,
J = 8.4 Hz, 2H, aromatic), 4.06 (t,
J = 6.0 Hz, 2H, BrCH
2CH
2CH
2C
H2OPh), 3.49 (t,
J = 6.4 Hz, 2H, BrC
H2CH
2CH
2CH
2OPh), 3.03 (s, 6H, N(C
H3)
2), 2.08 (p,
J = 7.2 Hz, 2H, BrCH
2CH
2C
H2CH
2OPh), 1.96 (p,
J = 7.2 Hz, 2H, BrCH
2C
H2CH
2CH
2OPh);
13C NMR (100 MHz, CDCl
3,
Figure S5)
δ 190.3, 159.0, 152.0, 145.8, 140.5, 130.4 (2 carbons), 129.4, 122.7, 120.9, 119.0, 117.0, 113.3, 111.9 (2 carbons), 67.0, 40.2 (2 carbons), 33.4, 29.4, 27.8;
m/
z calcd for C
21H
24BrNO
2 401.1; found 402.0 [M + H]
+.
(E)-1-(3-((6-Bromohexyl)Oxy)Phenyl)-3-(4-(Dimethylamino)Phenyl)Prop-2-En-1-One (
4c) (SGT684). A solution of compound
3 (100 mg, 0.37 mmol) and K
2CO
3 (207 mg, 1.50 mmol) in anhydrous MeCN (5 mL) was treated with 1,6-dibromohexane (0.23 mL, 1.50 mmol) and the resulting mixture was stirred at 50 °C overnight. The solvent was then removed and the obtained crude product was purified by column chromatography (SiO
2 gel, pure Hexanes to Hexanes:EtOAc/4:1; R
f 0.60 in Hexanes:EtOAc/3:1) to yield compound
4c (143 mg, 89%) as an orange solid:
1H NMR (400 MHz, CDCl
3,
Figure S6)
δ 7.78 (d,
J = 15.6 Hz, 1H, HC=C
H-Ph), 7.57-7.50 (m, 4H, aromatic), 7.36 (t,
J = 8.0 Hz, 1H, aromatic), 7.30 (d,
J = 15.6 Hz, 1H,
HC=CH-Ph), 7.07 (d,
J = 8.0 Hz, 1H, aromatic), 6.68 (d,
J = 8.4 Hz, 2H, aromatic), 4.02 (t,
J = 6.4 Hz, 2H, BrCH
2CH
2(CH
2)
2CH
2C
H2OPh), 3.42 (t,
J = 6.8 Hz, 2H, BrC
H2CH
2(CH
2)
2CH
2CH
2OPh), 3.03 (s, 6H, N(C
H3)
2), 1.89 (br p,
J = 6.4 Hz, 2H, BrCH
2CH
2(CH
2)
2C
H2CH
2OPh), 1.81 (br p,
J = 5.6 Hz, 2H, BrCH
2C
H2(CH
2)
2CH
2CH
2OPh), 1.51 (m, 4H, BrCH
2CH
2(C
H2)
2CH
2CH
2OPh);
13C NMR (100 MHz, CDCl
3,
Figure S7)
δ 190.4, 159.2, 152.0, 145.8, 140.4, 130.4 (2 carbons), 129.4, 122.6, 120.7, 119.0, 116.9, 113.4, 111.8 (2 carbons), 67.9, 40.2 (2 carbons), 33.9, 32.7, 29.0, 27.9, 25.3;
m/
z calcd for C
23H
28BrNO
2 429.1; found 430.1 [M + H]
+.
(E)-1-(3-((8-Bromooctyl)Oxy)Phenyl)-3-(4-(Dimethylamino)Phenyl)Prop-2-En-1-One (
4d) (SGT685). A solution of compound
3 (100 mg, 0.37 mmol) and K
2CO
3 (207 mg, 1.50 mmol) in anhydrous MeCN (5 mL) was treated with 1,8-dibromooctane (0.28 mL, 1.50 mmol) and the resulting mixture was stirred at 50 °C overnight. The solvent was then removed and the obtained crude product was purified by column chromatography (SiO
2 gel, pure Hexanes to Hexanes:EtOAc/4:1; R
f 0.60 in Hexanes:EtOAc/3:1) to yield compound
4d (139 mg, 81%) as a yellow solid:
1H NMR (400 MHz, CDCl
3,
Figure S8)
δ 7.78 (d,
J = 15.6 Hz, 1H, HC=C
H-Ph), 7.57-7.50 (m, 4H, aromatic), 7.36 (t,
J = 8.0 Hz, 1H, aromatic), 7.30 (d,
J = 15.6 Hz, 1H,
HC=CH-Ph), 7.07 (d,
J = 8.0 Hz, 1H, aromatic), 6.68 (d,
J = 8.4 Hz, 2H, aromatic), 4.01 (t,
J = 6.4 Hz, 2H, BrCH
2CH
2(CH
2)
4CH
2C
H2OPh), 3.40 (t,
J = 6.4 Hz, 2H, BrC
H2CH
2(CH
2)
4CH
2CH
2OPh), 3.03 (s, 6H, N(C
H3)
2), 1.85 (p,
J = 7.6 Hz, 2H, BrCH
2CH
2(CH
2)
4C
H2CH
2OPh), 1.79 (p,
J = 7.6 Hz, 2H, BrCH
2C
H2(CH
2)
4CH
2CH
2OPh), 1.40–1.50 (m, 4H, BrCH
2CH
2(CH
2)
4CH
2CH
2OPh), 1.36 (m, 4H, BrCH
2CH
2(CH
2)
4CH
2CH
2OPh);
13C NMR (100 MHz, CDCl
3,
Figure S9)
δ 190.4, 159.2, 152.0, 145.8, 140.4, 130.4 (2 carbons), 129.3, 122.6, 120.6, 119.0, 116.9, 113.4, 111.8 (2 carbons), 68.1, 40.1 (2 carbons), 34.1, 32.8, 29.2 (2 carbons), 28.7, 28.1, 25.9;
m/
z calcd for C
25H
32BrNO
2 457.2; found 458.0 [M + H]
+.
(E)-1-(3-((10-Bromodecyl)Oxy)Phenyl)-3-(4-(Dimethylamino)Phenyl)Prop-2-En-1-One (
4e) (SGT686). A solution of compound
3 (100 mg, 0.37 mmol) and K
2CO
3 (207 mg, 1.50 mmol) in anhydrous MeCN (5 mL) was treated with 1,10-dibromodecane (0.34 mL, 1.50 mmol) and the resulting mixture was stirred at 50 °C overnight. The solvent was then removed and the obtained crude product was purified by column chromatography (SiO
2 gel, pure Hexanes to Hexanes:EtOAc/4:1; R
f 0.60 in Hexanes:EtOAc/3:1) to yield compound
4e (160 mg, 89%) as a yellow solid:
1H NMR (400 MHz, CDCl
3,
Figure S10)
δ 7.78 (d,
J = 15.2 Hz, 1H, HC=C
H-Ph), 7.57–7.50 (m, 4H, aromatic), 7.36 (t,
J = 8.0 Hz, 1H, aromatic), 7.30 (d,
J = 15.2 Hz, 1H,
HC=CH-Ph), 7.07 (d,
J = 8.4 Hz, 1H, aromatic), 6.68 (d,
J = 8.4 Hz, 2H, aromatic), 4.01 (t,
J = 6.8 Hz, 2H, BrCH
2CH
2(CH
2)
6CH
2C
H2OPh), 3.39 (t,
J = 6.8 Hz, 2H, BrC
H2CH
2(CH
2)
6CH
2CH
2OPh), 3.03 (s, 6H, N(C
H3)
2), 1.81 (m, 4H), 1.44 (m, 4H), 1.30 (m, 8H);
13C NMR (100 MHz, CDCl
3,
Figure S11)
δ 190.4, 159.3, 152.0, 145.8, 140.4, 130.4 (2 carbons), 129.3, 122.6, 120.6, 119.0, 116.9, 113.4, 111.8 (2 carbons), 68.1, 40.2 (2 carbons), 34.1, 32.8, 29.44, 29.35, 29.32, 29.2, 28.7, 28.1, 26.0;
m/
z calcd for C
27H
36BrNO
2 485.2; found 486.0 [M + H]
+.
(E)-1-(3-((12-Bromododecyl)Oxy)Phenyl)-3-(4-(Dimethylamino)Phenyl)Prop-2-En-1-One (
4f) (SGT633). A solution of compound
3 (80 mg, 0.30 mmol) and K
2CO
3 (165 mg, 1.20 mmol) in anhydrous MeCN (5 mL) was treated with 1,12-dibromododecane (392 mg, 1.20 mmol) and the resulting mixture was refluxed overnight. The solvent was then removed and the obtained crude product was purified by column chromatography (SiO
2 gel, pure Hexanes to Hexanes:EtOAc/4:1; R
f 0.53 in Hexanes:EtOAc/3:1) to yield compound
4f (122 mg, 79%) as an orange solid:
1H NMR (400 MHz, CDCl
3,
Figure S12)
δ 7.77 (d,
J = 15.6 Hz, 1H, HC=C
H-Ph), 7.56–7.50 (m, 4H, aromatic), 7.36 (t,
J = 8.0 Hz, 1H, aromatic), 7.30 (d,
J = 15.6 Hz, 1H,
HC=CH-Ph), 7.07 (dd,
J1 = 8.0 Hz,
J2 = 2.0 Hz, 1H, aromatic), 6.71 (d,
J = 8.4 Hz, 2H, aromatic), 4.02 (t,
J = 6.8 Hz, 2H, BrCH
2(CH
2)
10C
H2OPh), 3.39 (t,
J = 6.8 Hz, 2H, BrC
H2(CH
2)
10CH
2OPh), 3.04 (s, 6H, N(C
H3)
2), 1.87–1.75 (m, 4H), 1.47–1.27 (m, 16H);
13C NMR (100 MHz, CDCl
3,
Figure S13)
δ 190.4, 159.3, 152.0, 145.8, 140.4, 130.4 (2 carbons), 129.3, 122.7, 120.6, 119.0, 117.0, 113.4, 111.8 (2 carbons), 68.2, 40.1 (2 carbons), 34.1, 32.8, 29.5 (3 carbons), 29.4, 29.3, 29.2, 28.7, 28.2, 26.0;
m/
z calcd for C
29H
40BrNO
2 513.2; found 514.3 [M + H]
+.
(E)-3-(4-(Dimethylamino)Phenyl)-1-(4-Hydroxyphenyl)Prop-2-En-1-One (
6) (SGT649). The known compound
6 was prepared as previously described [
69]. A solution of compounds
5 (0.50 g, 3.67 mmol) and
2 (0.55 g, 3.67 mmol) in EtOH (10 mL) was treated with NaOH pellets (2.94 g, 73.4 mmol). The reaction was stirred at rt overnight till completion. Most of the solvent was then removed and 1 N aqueous HCl was added. The precipitate was filtered to give the known compound
6 (0.43 g, 44%) (R
f 0.20 in Hexanes:EtOAc/2:1) as a red solid:
1H NMR (400 MHz, CDCl
3,
Figure S14, which matches the lit. [
69])
δ 7.97 (d,
J = 8.4 Hz, 2H, aromatic), 7.77 (d,
J = 15.6 Hz, 1H, HC=C
H-Ph), 7.54 (d,
J = 8.8 Hz, 2H, aromatic), 7.33 (d,
J = 15.6 Hz, 1H,
HC=CH-Ph), 6.90 (d,
J = 8.4 Hz, 2H, aromatic), 6.72 (d,
J = 8.8 Hz, 2H, aromatic), 5.42 (s, 1H, O
H), 3.04 (s, 6H, N(C
H3)
2).
(E)-1-(4-(2-Bromoethoxy)Phenyl)-3-(4-(Dimethylamino)Phenyl)Prop-2-En-1-One (
7a) (SGT652). A solution of
6 (80 mg, 0.30 mmol) and K
2CO
3 (165 mg, 1.20 mmol) in anhydrous MeCN (5 mL) was treated with 1,2-dibromoethane (0.10 mL, 1.20 mmol) and the resulting mixture was refluxed overnight. The solvent was then removed and the obtained crude product was purified by column chromatography (SiO
2 gel, pure Hexanes to Hexanes:EtOAc/4:1; R
f 0.31 in Hexanes:EtOAc/3:1) to yield compound
7a (90 mg, 80%) as a yellow solid:
1H NMR (400 MHz, CDCl
3,
Figure S15)
δ 8.01 (d,
J = 8.8 Hz, 2H, aromatic), 7.77 (d,
J = 15.6 Hz, 1H, HC=C
H-Ph), 7.55 (d,
J = 8.8 Hz, 2H, aromatic), 7.34 (d,
J = 15.6 Hz, 1H,
HC=CH-Ph), 6.97 (d,
J = 8.8 Hz, 2H, aromatic), 6.75 (d,
J = 8.8 Hz, 2H, aromatic), 4.36 (t,
J = 6.0 Hz, 2H, BrCH
2C
H2OPh), 3.66 (t,
J = 6.0 Hz, 2H, BrC
H2CH
2OPh), 3.04 (s, 6H, N(C
H3)
2);
13C NMR (100 MHz, CDCl
3,
Figure S16)
δ 188.8, 161.3, 151.9, 145.1, 132.5, 130.6 (2 carbons), 130.3 (2 carbons), 122.8, 116.5, 114.2 (2 carbons), 111.8 (2 carbons), 67.8, 40.1 (2 carbons), 28.7;
m/
z calcd for C
19H
20BrNO
2 373.1; found 374.0 [M + H]
+.
(E)-1-(4-(4-Bromobutoxy)Phenyl)-3-(4-(Dimethylamino)Phenyl)Prop-2-En-1-One (
7b) (SGT648). A solution of compound
6 (80 mg, 0.30 mmol) and K
2CO
3 (83 mg, 0.60 mmol) in anhydrous MeCN (5 mL) was treated with 1,4-dibromobutane (0.07 mL, 0.60 mmol) and the resulting mixture was refluxed overnight. The solvent was then removed and the obtained crude product was purified by column chromatography (SiO
2 gel, pure Hexanes to Hexanes:EtOAc/4:1; R
f 0.42 in Hexanes:EtOAc/3:1) to yield compound
7b (56 mg, 47%) as an orange solid:
1H NMR (400 MHz, CDCl
3,
Figure S17)
δ 8.00 (d,
J = 8.8 Hz, 2H, aromatic), 7.77 (d,
J = 15.6 Hz, 1H, HC=C
H-Ph), 7.54 (d,
J = 8.4 Hz, 2H, aromatic), 7.34 (d,
J = 15.6 Hz, 1H,
HC=CH-Ph), 6.94 (d,
J = 8.8 Hz, 2H, aromatic), 6.70 (d,
J = 8.4 Hz, 2H, aromatic), 4.07 (t,
J = 6.4 Hz, 2H, BrCH
2CH
2CH
2C
H2OPh), 3.49 (t,
J = 6.4 Hz, 2H, BrC
H2CH
2CH
2CH
2OPh), 3.03 (s, 6H, N(C
H3)
2), 2.08 (p,
J = 6.4 Hz, 2H, BrCH
2CH
2C
H2CH
2OPh), 1.97 (p,
J = 6.4 Hz, 2H, BrCH
2C
H2CH
2CH
2OPh);
13C NMR (100 MHz, CDCl
3,
Figure S18)
δ 188.8, 162.2, 151.8, 144.9, 131.8, 130.5 (2 carbons), 130.2 (2 carbons), 123.0, 116.7, 114.1 (2 carbons), 111.9 (2 carbons), 67.0, 40.2 (2 carbons), 33.3, 29.3, 27.8;
m/
z calcd for C
21H
24BrNO
2 401.1; found 402.0 [M + H]
+.
(E)-1-(4-((6-Bromohexyl)Oxy)Phenyl)-3-(4-(Dimethylamino)Phenyl)Prop-2-En-1-One (
7c) (SGT691). A solution of compound
6 (50 mg, 0.19 mmol) and K
2CO
3 (103 mg, 0.75 mmol) in anhydrous MeCN (5 mL) was treated with 1,6-dibromohexane (0.12 mL, 0.75 mmol) and the resulting mixture was stirred at 50 °C overnight. The solvent was then removed and the obtained crude product was purified by column chromatography (SiO
2 gel, pure Hexanes to Hexanes:EtOAc/4:1; R
f 0.33 in Hexanes:EtOAc/4:1) to yield compound
7c (56 mg, 70%) as an orange solid:
1H NMR (400 MHz, CDCl
3,
Figure S19)
δ 8.00 (d,
J = 8.4 Hz, 2H, aromatic), 7.77 (d,
J = 15.6 Hz, 1H, HC=C
H-Ph), 7.54 (d,
J = 8.4 Hz, 2H, aromatic), 7.35 (d,
J = 15.6 Hz, 1H,
HC=CH-Ph), 6.94 (d,
J = 8.4 Hz, 2H, aromatic), 6.71 (d,
J = 8.4 Hz, 2H, aromatic), 4.03 (t,
J = 6.4 Hz, 2H, BrCH
2CH
2CH
2C
H2OPh), 3.42 (t,
J = 6.4 Hz, 2H, BrC
H2CH
2CH
2CH
2OPh), 3.03 (s, 6H, N(C
H3)
2), 1.89 (br p,
J = 6.4 Hz, 2H, BrCH
2CH
2(CH
2)
2C
H2CH
2OPh), 1.82 (br p,
J = 6.4 Hz, 2H, BrCH
2C
H2(CH
2)
2CH
2CH
2OPh), 1.51 (m, 4H, BrCH
2CH
2(C
H2)
2CH
2CH
2OPh;
13C NMR (100 MHz, (CD
3)
2SO,
Figure S20)
δ 187.4, 162.6, 152.3, 144.7, 131.03 (2 carbons), 130.94 (2 carbons), 122.6, 116.5, 114.7 (2 carbons), 112.2 (2 carbons), 68.1, 65.0, 35.6 (2 carbons), 32.6, 28.8, 27.7, 25.0;
m/
z calcd for C
23H
28BrNO
2 429.1; found 430.0 [M + H]
+.
(E)-1-(4-((8-Bromooctyl)Oxy)Phenyl)-3-(4-(Dimethylamino)Phenyl)Prop-2-En-1-One (
7d) (SGT692). A solution of compound
6 (50 mg, 0.19 mmol) and K
2CO
3 (103 mg, 0.75 mmol) in anhydrous MeCN (5 mL) was treated with 1,8-dibromooctane (0.14 mL, 0.75 mmol) and the resulting mixture was stirred at 50 °C overnight. The solvent was then removed and the obtained crude product was purified by column chromatography (SiO
2 gel, pure Hexanes to Hexanes:EtOAc/4:1; R
f 0.33 in Hexanes:EtOAc/4:1) to yield compound
7d (47 mg, 55%) as a yellow solid:
1H NMR (400 MHz, CDCl
3,
Figure S21)
δ 8.00 (d,
J = 8.8 Hz, 2H, aromatic), 7.77 (d,
J = 15.2 Hz, 1H, HC=C
H-Ph), 7.54 (d,
J = 8.8 Hz, 2H, aromatic), 7.35 (d,
J = 15.2 Hz, 1H,
HC=CH-Ph), 6.94 (d,
J = 8.8 Hz, 2H, aromatic), 6.70 (d,
J = 8.4 Hz, 2H, aromatic), 4.02 (t,
J = 6.4 Hz, 2H, BrCH
2CH
2(CH
2)
4CH
2C
H2OPh), 3.40 (t,
J = 6.4 Hz, 2H, BrC
H2CH
2(CH
2)
4CH
2CH
2OPh), 3.03 (s, 6H, N(C
H3)
2), 1.85 (p,
J = 6.8 Hz, 2H, BrCH
2CH
2(CH
2)
4C
H2CH
2OPh), 1.80 (p,
J = 6.8 Hz, 2H, BrCH
2C
H2(CH
2)
4CH
2CH
2OPh), 1.40–1.50 (m, 4H), 1.36 (m, 4H);
13C NMR (100 MHz, CDCl
3,
Figure S22)
δ 188.8, 162.5, 144.7, 131.6, 130.5 (2 carbons), 130.2 (2 carbons), 116.9, 114.1 (2 carbons), 112.1 (2 carbons), 68.1, 40.3 (2 carbons), 34.0, 32.7, 29.14, 29.07, 28.6, 28.1, 25.9;
m/
z calcd for C
25H
32BrNO
2 457.2; found 458.4 [M + H]
+.
(E)-1-(4-((10-Bromodecyl)Oxy)Phenyl)-3-(4-(Dimethylamino)Phenyl)Prop-2-En-1-One (
7e) (SGT693). A solution of compound
6 (50 mg, 0.19 mmol) and K
2CO
3 (103 mg, 0.75 mmol) in anhydrous MeCN (5 mL) was treated with 1,10-dibromodecane (0.17 mL, 0.75 mmol) and the resulting mixture was stirred at 50 °C overnight. The solvent was then removed and the obtained crude product was purified by column chromatography (SiO
2 gel, pure Hexanes to Hexanes:EtOAc/4:1; R
f 0.41 in Hexanes:EtOAc/4:1) to yield compound
7e (41 mg, 46%) as a yellow solid:
1H NMR (400 MHz, CDCl
3,
Figure S23)
δ 8.00 (d,
J = 8.4 Hz, 2H, aromatic), 7.77 (d,
J = 15.2 Hz, 1H, HC=C
H-Ph), 7.54 (d,
J = 8.8 Hz, 2H, aromatic), 7.35 (d,
J = 15.2 Hz, 1H,
HC=CH-Ph), 6.94 (d,
J = 8.4 Hz, 2H, aromatic), 6.72 (d,
J = 8.4 Hz, 2H, aromatic), 4.02 (t,
J = 6.8 Hz, 2H, BrCH
2CH
2(CH
2)
6CH
2C
H2OPh), 3.40 (t,
J = 6.8 Hz, 2H, BrC
H2CH
2(CH
2)
6CH
2CH
2OPh), 3.03 (s, 6H, N(C
H3)
2), 1.84 (p,
J = 7.6 Hz, 2H, BrCH
2CH
2(CH
2)
6C
H2CH
2OPh), 1.79 (p,
J = 6.8 Hz, 2H, BrCH
2C
H2(CH
2)
4CH
2CH
2OPh), 1.43 (m, 4H), 1.30 (m, 8H);
13C NMR (100 MHz, CDCl
3,
Figure S24)
δ 188.8, 162.6, 144.7, 131.5, 130.5 (2 carbons), 130.2 (2 carbons), 116.9, 114.1 (2 carbons), 112.0 (2 carbons), 68.1, 40.3 (2 carbons), 34.0, 32.8, 29.4, 29.32, 29.28, 29.1, 28.7, 28.1, 25.9;
m/
z calcd for C
27H
36BrNO
2 485.2; found 486.3 [M + H]
+.
(E)-1-(4-((12-Bromododecyl)Oxy)Phenyl)-3-(4-(Dimethylamino)Phenyl)Prop-2-En-1-One (
7f) (SGT654). A solution of compound
6 (80 mg, 0.30 mmol) and K
2CO
3 (165 mg, 1.20 mmol) in anhydrous MeCN (5 mL) was treated with 1,12-dibromododecane (392 mg, 1.20 mmol) and the resulting mixture was refluxed overnight. The solvent was then removed and the obtained crude product was purified by column chromatography (SiO
2 gel, pure Hexanes to Hexanes:EtOAc/4:1; R
f 0.47 in Hexanes:EtOAc/3:1) to yield compound
7f (132 mg, 86%) as an orange solid:
1H NMR (400 MHz, CDCl
3,
Figure S25)
δ 8.00 (d,
J = 8.8 Hz, 2H, aromatic), 7.77 (d,
J = 15.2 Hz, 1H, HC=C
H-Ph), 7.54 (d,
J = 8.4 Hz, 2H, aromatic), 7.35 (d,
J = 15.2 Hz, 1H,
HC=CH-Ph), 6.94 (d,
J = 8.8 Hz, 2H, aromatic), 6.73 (d,
J = 8.4 Hz, 2H, aromatic), 4.02 (t,
J = 6.8 Hz, 2H, BrCH
2(CH
2)
10C
H2OPh), 3.39 (t,
J = 6.8 Hz, 2H, BrC
H2(CH
2)
10CH
2OPh), 3.03 (s, 6H, N(C
H3)
2), 1.87-1.75 (m, 4H), 1.47–1.27 (m, 16H);
13C NMR (100 MHz, CDCl
3,
Figure S26)
δ 188.8, 162.6, 151.8, 144.8, 131.6, 130.5 (2 carbons), 130.2 (2 carbons), 123.0, 116.8, 114.1 (2 carbons), 111.9 (2 carbons), 68.2, 40.2 (2 carbons), 34.1, 32.8, 29.5 (3 carbons), 29.4, 29.3, 29.1, 28.7, 28.2, 26.0;
m/
z calcd for C
29H
40BrNO
2 513.2; found 514.1 [M + H]
+.
3-(4-Hydroxy-3-Methoxyphenyl)Propanoic Acid (
9). To a solution of ferulic acid (6.0 g, 30.9 mmol) in degassed EtOAc (100 mL) was added a catalytic amount of 10% Pd/C (0.43 g). The reaction flask was then sealed with a rubber septum and freed of air. The reaction mixture was stirred at rt overnight under H
2 atmosphere. Upon completion, the reaction mixture was filtered through a bed of celite, and concentrated to afford the known compound
9 [
70] (6.1 g, quant.) as an off-white solid:
1H NMR (400 MHz, CDCl
3,
Figure S27, which matches the lit. [
70])
δ 10.50 (very br s, 1H, CO
2H), 6.82 (d,
J = 7.6 Hz, 1H, aromatic), 6.69 (s, 1H, aromatic), 6.68 (d,
J = 7.6 Hz, 1H, aromatic), 5.60 (very br s, 1H, O
H), 3.85 (s, 3H, PhOC
H3), 2.87 (t,
J = 7.2 Hz, 2H, PhC
H2CH
2CO
2H), 2.64 (t,
J = 7.2 Hz, 2H, PhCH
2C
H2CO
2H).
6-Hydroxy-5-Methoxy-2,3-Dihydro-1H-Inden-1-One (
10). A solution of
9 (6.3 g, 32.1 mmol) in methanesulfonic acid (50 mL) was refluxed at 120 °C for 1 h. After cooling to rt, the reaction mixture was poured into ice-water, stirred for 5 min, and filtered to afford a crude dark brown solid, which was recrystallized from EtOH to afford the known compound
10 [
71] (3.8 g, 67%) as a yellow solid:
1H NMR (400 MHz, (CD
3)
2SO,
Figure S28, which matches the lit. [
71])
δ 9.38 (s, 1H, O
H), 7.03 (s, 1H, aromatic), 6.89 (s, 1H, aromatic), 3.83 (s, 3H, OC
H3), 2.92 (t,
J = 5.6 Hz, 2H, C
H2CH
2C=O), 2.49 (t,
J = 5.6 Hz, 2H, CH
2C
H2C=O).
6-((Tert-Butyldimethylsilyl)Oxy)-5-Methoxy-2,3-Dihydro-1H-Inden-1-One (
11) (SGT640). TBDMSCl (3.2 g, 21.3 mmol) was added to a solution of
10 (1.9 g, 10.7 mmol), DMAP (0.5 g, 4.3 mmol), and Et
3N (3.0 mL, 21.3 mmol) in freshly distilled CH
2Cl
2 (100 mL). The reaction mixture was stirred at rt overnight before being quenched with H
2O (100 mL). The organic layer was separated, washed with H
2O (2 × 100 mL) and brine (100 mL), dried over anhydrous MgSO
4, filtered, and concentrated under reduced pressure to afford a crude dark brown solid, which was purified by flash column chromatography (SiO
2 gel, pure Hexanes to Hexanes:EtOAc/3:1, R
f 0.44 in Hexanes:EtOAc/3:1) to afford a brown solid, which was further triturated in Hexanes to give compound
11 (2.8 g, 90%) as a white solid:
1H NMR (400 MHz, CDCl
3,
Figure S29)
δ 7.17 (s, 1H, aromatic), 6.84 (s, 1H, aromatic), 3.87 (s, 3H, PhOC
H3), 3.02 (app. t,
J = 5.6 Hz, 2H, C
H2CH
2C=O), 2.64 (app. t,
J = 5.6 Hz, 2H, CH
2C
H2C=O), 0.98 (s, 9H, SiC(C
H3)
3), 0.14 (s, 6H, Si(C
H3)
2);
13C NMR (100 MHz, CDCl
3,
Figure S30)
δ 205.7, 157.5, 150.9, 145.2, 130.0, 114.1, 107.8, 55.6, 36.6, 25.62 (3 carbons), 25.56, 18.4, −4.7 (2 carbons);
m/z calcd for C
16H
24O
3Si 292.2; found 293.2 [M + H]
+.
(Z)-2-((1-Benzylpiperidin-4-Yl)Methylene)-6-Hydroxy-5-Methoxy-2,3-Dihydro-1H-Inden-1-One (
13) (SGT641). To a solution of compounds
11 (1.00 g, 3.42 mmol) and
12 (0.68 mL, 3.42 mmol) in EtOH (10 mL) was added KOH (0.5 g), and the mixture was refluxed at 65 °C. After 1 h, the reaction was analyzed by TLC (CH
2Cl
2:MeOH/19:1, R
f 0.30 in CH
2Cl
2:MeOH/19:1). The reaction mixture was concentrated under reduced pressure to give a crude yellow solid, which was re-dissolved in H
2O (10 mL). Then, 1 N aqueous HCl was then slowly added until pH 5 to give a yellow precipitate, which was recrystallized in MeCN to afford compound
13 (0.81 g, 65%) as a yellow solid:
1H NMR (400 MHz, CDCl
3,
Figure S31)
δ 7.32–7.24 (m, 6H, aromatic), 6.87 (s, 1H, aromatic), 6.63 (d,
J = 10.0 Hz, 1H, C=C
H), 5.70 (br s, 1H, O
H), 3.98 (s, 3H, OC
H3), 3.56 (s, 2H), 3.51 (s, 2H), 2.91 (d,
J = 11.6 Hz, 2H), 2.30 (m, 1H), 2.04 (t,
J = 11.6 Hz, 2H), 1.70-1.60 (m, 4H);
13C NMR (100 MHz, CDCl
3,
Figure S32)
δ 192.5, 152.6, 145.8, 143.4, 139.8, 138.2, 135.5, 132.5, 129.2 (2 carbons), 128.2 (2 carbons), 127.0, 108.7, 106.8, 63.5, 56.2, 53.1 (2 carbons), 37.2, 31.2 (2 carbons), 29.5;
m/
z calcd for C
23H
25NO
3 363.2; found 364.2 [M + H]
+.
2-((1-Benzylpiperidin-4-Yl)Methyl)-6-Hydroxy-5-Methoxy-2,3-Dihydro-1H-Inden-1-One (
14) (SGT332). To a solution of
13 (101 mg, 0.28 mmol) in degassed THF (2.5 mL) was added 10% Pd/C (wet support, Sigma 520829-10G, 10 mg). The reaction flask was then sealed with a rubber septum and freed of air. Thioanisole (14.2 × 10
−7 mL, obtained using 5 μL of a stock solution comprising 14.2 μL of thioanisole in 50 mL of anhydrous THF) was added and the reaction mixture was stirred at rt overnight under H
2 atmosphere. Upon completion, the reaction mixture was filtered through a bed of celite, and concentrated to afford the known compound
14 (96 mg, 94%) as a yellow solid:
1H NMR (400 MHz, CDCl
3,
Figure S33)
δ 7.30-7.20 (m, 6H, aromatic), 6.82 (s, 1H, aromatic), 3.96 (s, 3H, OC
H3), 3.49 (s, 2H, NC
H2Ph), 3.20 (dd,
J1 = 18.0 Hz,
J2 = 7.6 Hz, 1H), 2.87 (m, 2H), 2.66 (dt,
J1 = 13.6 Hz,
J2 = 3.6 Hz, 2H), 1.98–1.82 (m, 3H), 1.72–1.63 (m, 2H), 1.48 (m, 1H), 1.39–1.24 (m, 3H);
13C NMR (100 MHz, CDCl
3,
Figure S34)
δ 207.8, 152.9, 147.5, 145.8, 138.3, 130.0, 129.3 (2 carbons), 128.1 (2 carbons), 126.9, 108.0, 106.9, 63.4, 56.2, 53.7 (2 carbons), 45.3, 38.7, 34.4, 33.4, 32.9, 31.7;
m/
z calcd for C
23H
27NO
3 365.2; found 366.2 [M + H]
+.
(E)-2-((1-Benzylpiperidin-4-Yl)Methyl)-6-(2-(3-(3-(4-(Dimethylamino)Phenyl)Acryloyl)Phenoxy)Ethoxy)-5-Methoxy-2,3-Dihydro-1H-Inden-1-One (
15a) (SGT656). A solution of compound
14 (15 mg, 0.041 mmol), compound
4a (19 mg, 0.049 mmol), and K
2CO
3 (17 mg, 0.12 mmol) in anhydrous DMF (5 mL) was heated at 65 °C overnight. The reaction mixture was then diluted with H
2O, and extracted with EtOAc (3×). The combined organic layers were washed with H
2O (3×) and brine (3×), dried over anhydrous MgSO
4, filtered, and concentrated under reduced pressure. The crude product obtained was purified by column chromatography (SiO
2 gel, pure CH
2Cl
2 to CH
2Cl
2:MeOH/19:1; R
f 0.39 in CH
2Cl
2:MeOH/19:1) to yield compound
15a (25 mg, 93%) as an orange oil:
1H NMR (400 MHz, CDCl
3,
Figure S35)
δ 7.77 (d,
J = 15.6 Hz, 1H, HC=C
H-Ph), 7.59–7.56 (m, 2H, aromatic), 7.52 (d,
J = 8.4 Hz, 2H, aromatic), 7.37 (t,
J = 8.0 Hz, 1H, aromatic), 7.30–7.23 (m, 7H, aromatic), 7.13 (dd,
J1 = 8.0 Hz,
J2 = 2.4 Hz, 1H, aromatic), 6.83 (s, 1H, aromatic), 6.66 (d,
J = 8.4 Hz, 2H, aromatic), 4.41 (m, 4H), 3.89 (s, 3H, OC
H3), 3.52 (s, 2H, NC
H2Ph), 3.20 (dd,
J1 = 17.6 Hz,
J2 = 8.0 Hz, 1H), 3.02 (s, 6H, N(C
H3)
2), 2.91 (m, 2H), 2.66 (dt,
J1 = 14.0 Hz,
J2 = 2.4 Hz, 2H), 1.99 (m, 2H), 1.89 (m, 1H), 1.65–1.55 (m, 2H), 1.55–1.40 (m, 1H), 1.40–1.25 (m, 3H);
13C NMR (100 MHz, CDCl
3,
Figure S36)
δ 207.7, 190.1, 158.8, 155.9, 152.0, 149.2, 148.4 (2 carbons), 145.9, 140.4, 130.4 (2 carbons), 129.41, 129.37 (2 carbons), 129.1, 128.2 (2 carbons), 127.1, 122.6, 121.2, 119.2, 116.7, 113.8, 111.8 (2 carbons), 107.7, 106.3, 67.6, 66.5, 63.2, 56.1, 53.6, 53.5, 45.4, 40.1 (2 carbons), 38.6, 34.3, 33.4, 32.7, 31.5;
m/z calcd for C
42H
46N
2O
5 658.3; found 659.3 [M + H]
+.
(E)-2-((1-Benzylpiperidin-4-Yl)Methyl)-6-(4-(3-(3-(4-(Dimethylamino)Phenyl)Acryloyl)Phenoxy)Butoxy)-5-Methoxy-2,3-Dihydro-1H-Inden-1-One (
15b) (SGT681). A solution of compound
14 (30 mg, 0.082 mmol), compound
4b (40 mg, 0.099 mmol), and K
2CO
3 (34 mg, 0.25 mmol) in anhydrous DMF (5 mL) was heated at 65 °C overnight. The reaction mixture was then diluted with H
2O, and extracted with EtOAc (3×). The combined organic layers were washed with H
2O (3×) and brine (3×), dried over anhydrous MgSO
4, filtered, and concentrated under reduced pressure. The crude product obtained was purified by column chromatography (SiO
2 gel, pure CH
2Cl
2 to CH
2Cl
2:MeOH/19:1; R
f 0.39 in CH
2Cl
2:MeOH/19:1) to yield compound
15b (48 mg, 86%) as an orange oil:
1H NMR (400 MHz, CDCl
3,
Figure S37)
δ 7.77 (d,
J = 15.2 Hz, 1H, HC=C
H-Ph), 7.58–7.46 (m, 4H, aromatic), 7.38–7.22 (m, 7H, aromatic), 7.14 (s, 1H, aromatic), 7.06 (dd,
J1 = 8.4 Hz,
J2 = 2.0 Hz, 1H, aromatic), 6.80 (s, 1H, aromatic), 6.67 (d,
J = 8.8 Hz, 2H, aromatic), 4.11 (m, 4H), 3.89 (s, 3H, OC
H3), 3.57 (s, 2H, NC
H2Ph), 3.19 (dd,
J1 = 17.6 Hz,
J2 = 8.4 Hz, 1H), 3.02 (s, 6H, N(C
H3)
2), 2.96 (m, 2H), 2.65 (dt,
J1 = 14.0 Hz,
J2 = 4.4 Hz, 2H), 2.05-1.98 (m, 6H), 1.89–1.84 (m, 1H), 1.70 (m, 2H), 1.60–1.22 (m, 4H);
13C NMR (100 MHz, CDCl
3,
Figure S38)
δ 207.7, 190.3, 159.1, 155.8, 152.0, 148.7 (3 carbons), 145.8, 140.4, 130.4 (2 carbons), 129.8 (2 carbons), 129.3, 129.0, 128.4 (2 carbons), 127.7, 122.5, 120.7, 119.0, 116.8, 113.4, 111.8 (2 carbons), 107.4, 105.4, 68.6, 67.6, 62.7, 56.2, 53.3 (2 carbons), 45.1, 40.1 (2 carbons), 38.4, 33.7, 33.4, 31.8, 31.0, 26.1, 25.6;
m/z calcd for C
44H
50N
2O
5 686.4; found 687.3 [M + H]
+.
(E)-2-((1-Benzylpiperidin-4-Yl)Methyl)-6-((6-(3-(3-(4-(Dimethylamino)Phenyl)Acryloyl)Phenoxy)Hexyl)Oxy)-5-Methoxy-2,3-Dihydro-1H-Inden-1-One (
15c) (SGT687). A solution of compound
14 (30 mg, 0.082 mmol), compound
4c (42 mg, 0.099 mmol), and K
2CO
3 (34 mg, 0.24 mmol) in anhydrous DMF (5 mL) was heated at 80 °C overnight. The reaction mixture was then diluted with H
2O, and extracted with EtOAc (3×). The combined organic layers were washed with H
2O (3×) and brine (3×), dried over anhydrous MgSO
4, filtered, and concentrated under reduced pressure. The crude product obtained was purified by column chromatography (SiO
2 gel, pure CH
2Cl
2 to CH
2Cl
2:MeOH/19:1; R
f 0.39 in CH
2Cl
2:MeOH/19:1) to yield compound
15c (61 mg, quantitative yield) as an orange solid:
1H NMR (400 MHz, CDCl
3,
Figure S39)
δ 7.77 (d,
J = 15.6 Hz, 1H, HC=C
H-Ph), 7.58–7.48 (m, 4H, aromatic), 7.38–7.24 (m, 7H, aromatic), 7.13 (s, 1H, aromatic), 7.06 (d,
J = 8.4 Hz, 1H, aromatic), 6.81 (s, 1H, aromatic), 6.67 (d,
J = 8.4 Hz, 2H, aromatic), 4.02 (m, 4H), 3.91 (s, 3H, OC
H3), 3.60 (s, 2H, NC
H2Ph), 3.20 (dd,
J1 = 17.2 Hz,
J2 = 8.4 Hz, 1H), 3.03 (s, 6H, N(C
H3)
2), 2.97 (m, 2H), 2.65 (m, 2H), 2.06 (m, 2H), 1.92–1.78 (m, 5H), 1.71 (m, 2H), 1.54 (m, 4H), 1.45–1.23 (m, 4H);
13C NMR (100 MHz, CDCl
3,
Figure S40)
δ 207.8, 190.4, 159.2, 155.8, 152.0, 148.8, 148.6 (2 carbons), 145.8, 140.4, 130.4 (2 carbons), 129.6, 129.3 (2 carbons), 129.1, 128.3 (2 carbons), 127.5, 122.6, 120.6, 119.0, 116.9, 113.4, 111.8 (2 carbons), 107.4, 105.4, 68.9, 67.9, 62.9, 56.2, 53.4 (2 carbons), 45.2, 40.1 (2 carbons), 38.5, 34.0, 33.4, 32.2, 31.2, 29.1, 28.8, 25.82, 25.75;
m/z calcd for C
46H
54N
2O
5 714.4; found 715.2 [M + H]
+.
(E)-2-((1-Benzylpiperidin-4-Yl)Methyl)-6-((8-(3-(3-(4-(Dimethylamino)Phenyl)Acryloyl)Phenoxy)Octyl)Oxy)-5-Methoxy-2,3-Dihydro-1H-Inden-1-One (
15d) (SGT688). A solution of compound
14 (30 mg, 0.082 mmol), compound
4d (45 mg, 0.099 mmol), and K
2CO
3 (34 mg, 0.24 mmol) in anhydrous DMF (5 mL) was heated at 80 °C overnight. The reaction mixture was then diluted with H
2O, and extracted with EtOAc (3×). The combined organic layers were washed with H
2O (3×) and brine (3×), dried over anhydrous MgSO
4, filtered, and concentrated under reduced pressure. The crude product obtained was purified by column chromatography (SiO
2 gel, pure CH
2Cl
2 to CH
2Cl
2:MeOH/19:1; R
f 0.39 in CH
2Cl
2:MeOH/19:1) to yield compound
15d (58 mg, 95%) as an orange solid:
1H NMR (400 MHz, CDCl
3,
Figure S41)
δ 7.77 (d,
J = 15.2 Hz, 1H, HC=C
H-Ph), 7.56–7.48 (m, 4H, aromatic), 7.38–7.24 (m, 7H, aromatic), 7.13 (s, 1H, aromatic), 7.06 (d,
J = 8.4 Hz, 1H, aromatic), 6.82 (s, 1H, aromatic), 6.67 (d,
J = 8.4 Hz, 2H, aromatic), 4.01 (t,
J = 6.4 Hz, 4H), 3.92 (s, 3H, OC
H3), 3.57 (s, 2H, NC
H2Ph), 3.20 (dd,
J1 = 17.2 Hz,
J2 = 8.0 Hz, 1H), 3.03 (s, 6H, N(C
H3)
2), 2.95 (m, 2H), 2.65 (m, 2H), 2.03 (m, 2H), 1.92–1.65 (m, 7H), 1.45 (m, 5H), 1.42–1.23 (m, 7H);
13C NMR (100 MHz, CDCl
3,
Figure S42)
δ 207.8, 190.4, 159.3, 155.7, 152.0, 148.8, 148.5 (2 carbons), 145.8, 140.4, 130.4 (2 carbons), 129.5, 129.3 (2 carbons), 129.1, 128.2 (2 carbons), 127.3, 122.6, 120.6, 119.0, 116.9, 113.4, 111.8 (2 carbons), 107.4, 105.4, 69.0, 68.1, 63.1, 56.2, 53.5 (2 carbons), 45.3, 40.1 (2 carbons), 38.6, 34.2, 33.4, 32.4, 31.4, 29.3 (2 carbons), 29.2, 28.9, 25.95, 25.85;
m/z calcd for C
48H
58N
2O
5 742.4; found 743.4 [M + H]
+.
(E)-2-((1-Benzylpiperidin-4-Yl)Methyl)-6-((10-(3-(3-(4-(Dimethylamino)Phenyl)Acryloyl)Phenoxy)Decyl)Oxy)-5-Methoxy-2,3-Dihydro-1H-Inden-1-One (
15e) (SGT689). A solution of compound
14 (30 mg, 0.082 mmol), compound
4e (48 mg, 0.099 mmol), and K
2CO
3 (34 mg, 0.24 mmol) in anhydrous DMF (5 mL) was heated at 80 °C overnight. The reaction mixture was then diluted with H
2O, and extracted with EtOAc (3×). The combined organic layers were washed with H
2O (3×) and brine (3×), dried over anhydrous MgSO
4, filtered, and concentrated under reduced pressure. The crude product obtained was purified by column chromatography (SiO
2 gel, pure CH
2Cl
2 to CH
2Cl
2:MeOH/19:1; R
f 0.39 in CH
2Cl
2:MeOH/19:1) to yield compound
15e (42 mg, 67%) as a yellow solid:
1H NMR (400 MHz, CDCl
3,
Figure S43)
δ 7.77 (d,
J = 15.6 Hz, 1H, HC=C
H-Ph), 7.56–7.48 (m, 4H, aromatic), 7.38–7.24 (m, 7H, aromatic), 7.13 (s, 1H, aromatic), 7.07 (d,
J = 8.4 Hz, 1H, aromatic), 6.81 (s, 1H, aromatic), 6.67 (d,
J = 8.4 Hz, 2H, aromatic), 4.01 (t,
J = 6.8 Hz, 4H, 2×OC
H2CH
2), 3.91 (s, 3H, OC
H3), 3.54 (s, 2H, NC
H2Ph), 3.20 (dd,
J1 = 17.2 Hz,
J2 = 8.0 Hz, 1H), 3.03 (s, 6H, N(C
H3)
2), 2.92 (m, 2H), 2.65 (m, 2H), 2.01 (m, 2H), 1.92-1.65 (m, 7H), 1.43 (m, 5H), 1.31 (m, 11H);
13C NMR (100 MHz, CDCl
3,
Figure S44)
δ 207.8, 190.4, 159.3, 155.7, 152.0, 148.8, 148.5 (2 carbons), 145.8, 140.4, 130.4 (2 carbons), 129.4, 129.3 (2 carbons), 129.2, 128.2 (2 carbons), 127.2, 122.6, 120.6, 119.0, 116.9, 113.4, 111.8 (2 carbons), 107.4, 105.4, 69.0, 68.2, 63.2, 56.2, 53.6 (2 carbons), 45.3, 40.1 (2 carbons), 38.6, 34.2, 33.3, 32.6, 31.5, 29.7 (2 carbons), 29.4, 29.3, 29.2, 28.9, 26.0, 25.9;
m/z calcd for C
50H
62N
2O
5 770.5; found 771.3 [M + H]
+.
(E)-2-((1-Benzylpiperidin-4-Yl)Methyl)-6-((12-(3-(3-(4-(Dimethylamino)Phenyl)Acryloyl)Phenoxy)Dodecyl)Oxy)-5-Methoxy-2,3-Dihydro-1H-Inden-1-One (
15f) (SGT679). A solution of compound
14 (30 mg, 0.082 mmol), compound
4f (51 mg, 0.099 mmol), and K
2CO
3 (34 mg, 0.25 mmol) in anhydrous DMF (5 mL) was heated at 80 °C overnight. The reaction mixture was then diluted with H
2O, and extracted with EtOAc (3×). The combined organic layers were washed with H
2O (3×) and brine (3×), dried over anhydrous MgSO
4, filtered, and concentrated under reduced pressure. The crude product obtained was purified by column chromatography (SiO
2 gel, pure CH
2Cl
2 to CH
2Cl
2:MeOH/19:1; R
f 0.39 in CH
2Cl
2:MeOH/19:1) to yield compound
15f (32 mg, 48%) as an orange oil:
1H NMR (400 MHz, CDCl
3,
Figure S45)
δ 7.77 (d,
J = 15.2 Hz, 1H, HC=C
H-Ph), 7.56–7.50 (m, 4H, aromatic), 7.35 (t,
J = 8.0 Hz, 1H, aromatic), 7.33–7.28 (m, 6H, aromatic), 7.12 (s, 1H, aromatic), 7.06 (dd,
J1 = 8.0 Hz,
J2 = 2.0 Hz, 1H, aromatic), 6.81 (s, 1H, aromatic), 6.67 (d,
J = 8.8 Hz, 2H, aromatic), 4.00 (m, 4H), 3.91 (s, 3H, OC
H3), 3.57 (s, 2H, NC
H2Ph), 3.19 (dd,
J1 = 17.6 Hz,
J2 = 8.0 Hz, 1H), 3.02 (s, 6H, N(C
H3)
2), 2.95 (m, 2H), 2.65 (dt,
J1 = 14.0 Hz,
J2 = 4.4 Hz, 2H), 2.03 (m, 2H), 1.90–1.75 (m, 5H), 1.73–1.60 (m, 2H), 1.50–1.40 (m, 6H), 1.40–1.25 (m, 14H);
13C NMR (100 MHz, CDCl
3,
Figure S46)
δ 207.8, 190.4, 159.3, 155.7, 152.0, 148.8, 148.5 (2 carbons), 145.8, 140.4, 130.4 (2 carbons), 129.5, 129.3 (2 carbons), 129.1, 128.3 (2 carbons), 127.3, 122.6, 120.6, 119.0, 116.9, 113.4, 111.8 (2 carbons), 107.4, 105.4, 69.1, 68.2, 62.9, 56.2, 53.4 (2 carbons), 45.3, 40.1 (2 carbons), 38.6, 34.1, 33.3, 32.3, 31.3, 29.5 (3 carbons), 29.5, 29.4, 29.3, 29.2, 28.9, 26.0, 25.9;
m/z calcd for C
52H
66N
2O
5 798.5; found 799.3 [M + H]
+.
(E)-2-((1-Benzylpiperidin-4-Yl)Methyl)-6-(2-(4-(3-(4-(Dimethylamino)Phenyl)Acryloyl)Phenoxy)Ethoxy)-5-Methoxy-2,3-Dihydro-1H-Inden-1-One (
16a) (SGT683). A solution of compound
14 (20 mg, 0.055 mmol), compound
7a (25 mg, 0.066 mmol), and K
2CO
3 (23 mg, 0.16 mmol) in anhydrous DMF (5 mL) was heated at 80 °C overnight. The reaction mixture was then diluted with H
2O, and extracted with EtOAc (3×). The combined organic layers were washed with H
2O (3×) and brine (3×), dried over anhydrous MgSO
4, filtered, and concentrated under reduced pressure. The crude product obtained was purified by column chromatography (SiO
2 gel, pure CH
2Cl
2 to CH
2Cl
2:MeOH/19:1; R
f 0.39 in CH
2Cl
2:MeOH/19:1) to yield compound
16a (48 mg, quantitative yield) as a yellow solid:
1H NMR (400 MHz, CDCl
3,
Figure S47)
δ 8.00 (d,
J = 8.8 Hz, 2H, aromatic), 7.76 (d,
J = 15.6 Hz, 1H, HC=C
H-Ph), 7.53 (d,
J = 8.8 Hz, 2H, aromatic), 7.33 (d,
J = 15.6 Hz, 1H,
HC=CH-Ph), 7.32–7.24 (m, 5H, aromatic), 7.23 (s, 1H, aromatic), 7.00 (d,
J = 8.8 Hz, 2H, aromatic), 6.84 (s, 1H, aromatic), 6.67 (d,
J = 8.8 Hz, 2H, aromatic), 4.41 (m, 4H), 3.90 (s, 3H, OC
H3), 3.55 (s, 2H, NC
H2Ph), 3.21 (dd,
J1 = 17.6 Hz,
J2 = 8.0 Hz, 1H), 3.02 (s, 6H, N(C
H3)
2), 2.92 (m, 2H), 2.67 (dt,
J1 = 14.4 Hz,
J2 = 3.2 Hz, 2H), 2.02 (m, 2H), 1.89 (m, 1H), 1.70 (m, 2H), 1.50 (m, 1H), 1.40–1.25 (m, 3H);
13C NMR (100 MHz, CDCl
3,
Figure S48)
δ 207.6, 188.9, 161.9, 155.9, 151.9, 149.3, 148.4 (2 carbons), 145.0, 132.2, 130.5 (2 carbons), 130.3 (2 carbons), 129.4 (2 carbons) 129.1, 128.2 (2 carbons), 127.2, 122.8, 116.6, 114.3 (2 carbons), 111.8 (2 carbons), 107.8, 106.3, 67.4, 66.4, 63.1, 56.2, 53.5 (2 carbons), 45.3, 40.1 (2 carbons), 38.6, 34.2, 33.4, 32.5, 31.5;
m/z calcd for C
42H
46N
2O
5 658.3; found 659.3 [M + H]
+.
(E)-2-((1-Benzylpiperidin-4-Yl)Methyl)-6-(4-(4-(3-(4-(Dimethylamino)Phenyl)Acryloyl)Phenoxy)Butoxy)-5-Methoxy-2,3-Dihydro-1H-Inden-1-One (
16b) (SGT682). A solution of compound
14 (20 mg, 0.055 mmol), compound
7b (27 mg, 0.066 mmol), and K
2CO
3 (23 mg, 0.16 mmol) in anhydrous DMF (5 mL) was heated at 65 °C overnight. The reaction mixture was then diluted with H
2O, and extracted with EtOAc (3×). The combined organic layers were washed with H
2O (3×) and brine (3×), dried over anhydrous MgSO
4, filtered, and concentrated under reduced pressure. The crude product obtained was purified by column chromatography (SiO
2 gel, pure CH
2Cl
2 to CH
2Cl
2:MeOH/19:1; R
f 0.39 in CH
2Cl
2:MeOH/19:1) to yield compound
16b (14 mg, 37%) as an orange oil:
1H NMR (400 MHz, CDCl
3,
Figure S49)
δ 7.98 (d,
J = 8.8 Hz, 2H, aromatic), 7.76 (d,
J = 15.2 Hz, 1H, HC=C
H-Ph), 7.53 (d,
J = 8.8 Hz, 2H, aromatic), 7.33 (d,
J = 15.2 Hz, 1H,
HC=CH-Ph), 7.33–7.26 (m, 5H, aromatic), 7.13 (s, 1H, aromatic), 6.93 (d,
J = 8.8 Hz, 2H, aromatic), 6.81 (s, 1H, aromatic), 6.67 (d,
J = 8.8 Hz, 2H, aromatic), 4.41 (q,
J = 5.6 Hz, 4H), 3.89 (s, 3H, OC
H3), 3.62 (s, 2H, NC
H2Ph), 3.21 (dd,
J1 = 17.6 Hz,
J2 = 8.4 Hz, 1H), 3.02 (s, 6H, N(C
H3)
2), 2.92 (m, 2H), 2.67 (dt,
J1 = 14.0 Hz,
J2 = 4.0 Hz, 2H), 2.10 (m, 2H), 2.02 (m, 4H), 1.88 (m, 1H), 1.72 (m, 2H), 1.60–1.20 (m, 4H);
13C NMR (100 MHz, CDCl
3,
Figure S50)
δ 207.7, 188.9, 162.4, 155.8, 151.9, 148.7 (3 carbons), 144.9, 132.1, 131.7, 131.0, 130.5 (2 carbons), 130.2 (2 carbons), 129.7, 129.1, 128.3 (2 carbons), 122.8, 116.6, 114.1 (2 carbons), 111.8 (2 carbons), 107.5, 105.5, 68.6, 67.6, 62.7, 56.1, 53.3, 45.2, 40.1 (2 carbons), 38.5, 33.9, 33.4, 31.9, 31.1, 29.7, 26.0, 25.5;
m/z calcd for C
44H
50N
2O
5 686.4; found 687.4 [M + H]
+.
(E)-2-((1-Benzylpiperidin-4-Yl)Methyl)-6-((6-(4-(3-(4-(Dimethylamino)Phenyl)Acryloyl)Phenoxy)Hexyl)Oxy)-5-Methoxy-2,3-Dihydro-1H-Inden-1-One (
16c) (SGT694). A solution of compound
14 (25 mg, 0.068 mmol), compound
7c (35 mg, 0.082 mmol), and K
2CO
3 (28 mg, 0.21 mmol) in anhydrous DMF (5 mL) was heated at 80 °C overnight. The reaction mixture was then diluted with H
2O, and extracted with EtOAc (3×). The combined organic layers were washed with H
2O (3×) and brine (3×), dried over anhydrous MgSO
4, filtered, and concentrated under reduced pressure. The crude product obtained was purified by column chromatography (SiO
2 gel, pure CH
2Cl
2 to CH
2Cl
2:MeOH/19:1; R
f 0.39 in CH
2Cl
2:MeOH/19:1) to yield compound
16c (48 mg, 98%) as a yellow solid:
1H NMR (400 MHz, CDCl
3,
Figure S51)
δ 7.99 (d,
J = 8.8 Hz, 2H, aromatic), 7.76 (d,
J = 15.2 Hz, 1H, HC=C
H-Ph), 7.53 (d,
J = 8.4 Hz, 2H, aromatic), 7.36–7.24 (m, 6H, aromatic), 7.14 (s, 1H, aromatic), 6.93 (d,
J = 8.4 Hz, 2H, aromatic), 6.82 (s, 1H, aromatic), 6.68 (d,
J = 8.8 Hz, 2H, aromatic), 4.032 (t,
J = 6.0 Hz, 2H), 4.025 (t,
J = 6.0 Hz, 2H), 3.91 (s, 3H, OC
H3), 3.54 (s, 2H, NC
H2Ph), 3.20 (dd,
J1 = 17.6 Hz,
J2 = 8.4 Hz, 1H), 3.02 (s, 6H, N(C
H3)
2), 2.92 (m, 2H), 2.65 (m, 2H), 2.00 (m, 2H), 1.90–1.80 (m, 9H), 1.60–1.20 (m, 6H);
13C NMR (100 MHz, CDCl
3,
Figure S52)
δ 207.7, 188.8, 162.5, 155.7, 151.8, 148.8, 148.5 (3 carbons), 144.8, 131.6, 130.5, 130.2, 129.5, 129.1 (2 carbons), 128.2 (2 carbons), 127.3, 122.8 (2 carbons), 116.6, 114.1, 111.8 (2 carbons), 107.4, 105.4, 68.8, 67.9, 63.0, 56.2, 56.1, 53.4, 45.3, 40.1, 38.5 (2 carbons), 33.9, 33.3, 29.0, 28.8, 25.71, 25.66, 25.58, 24.9;
m/z calcd for C
46H
54N
2O
5 714.4; found 715.3 [M + H]
+.
(E)-2-((1-Benzylpiperidin-4-Yl)Methyl)-6-((8-(4-(3-(4-(Dimethylamino)Phenyl)Acryloyl)Phenoxy)Octyl)Oxy)-5-Methoxy-2,3-Dihydro-1H-Inden-1-One (
16d) (SGT695). A solution of compound
14 (25 mg, 0.068 mmol), compound
7d (38 mg, 0.082 mmol), and K
2CO
3 (28 mg, 0.21 mmol) in anhydrous DMF (5 mL) was heated at 80 °C overnight. The reaction mixture was then diluted with H
2O, and extracted with EtOAc (3×). The combined organic layers were washed with H
2O (3×) and brine (3×), dried over anhydrous MgSO
4, filtered, and concentrated under reduced pressure. The crude product obtained was purified by column chromatography (SiO
2 gel, pure CH
2Cl
2 to CH
2Cl
2:MeOH/19:1; R
f 0.40 in CH
2Cl
2:MeOH/19:1) to yield compound
16d (51 mg, quantitative yield) as a yellow solid:
1H NMR (400 MHz, CDCl
3,
Figure S53)
δ 7.99 (d,
J = 9.2 Hz, 2H, aromatic), 7.76 (d,
J = 15.2 Hz, 1H, HC=C
H-Ph), 7.53 (d,
J = 8.8 Hz, 2H, aromatic), 7.36–7.24 (m, 6H, aromatic), 7.13 (s, 1H, aromatic), 6.93 (d,
J = 8.8 Hz, 2H, aromatic), 6.82 (s, 1H, aromatic), 6.68 (d,
J = 8.4 Hz, 2H, aromatic), 4.01 (t,
J = 6.4 Hz, 4H), 3.92 (s, 3H, OC
H3), 3.54 (s, 2H, NC
H2Ph), 3.20 (dd,
J1 = 17.6 Hz,
J2 = 8.4 Hz, 1H), 3.02 (s, 6H, N(C
H3)
2), 2.92 (m, 2H), 2.65 (m, 2H), 2.00 (m, 2H), 1.90–1.76 (m, 5H), 1.76–1.65 (m, 2H), 1.50–1.20 (m, 12H);
13C NMR (100 MHz, CDCl
3,
Figure S54)
δ 207.8, 188.9, 162.5, 155.7, 151.8, 148.8, 148.5 (3 carbons), 144.8, 131.6, 130.5 (2 carbons), 130.2 (2 carbons), 129.4, 129.2, 128.2 (2 carbons), 127.2, 122.9, 116.7, 114.1 (2 carbons), 111.8 (2 carbons), 107.4, 105.4, 69.0, 68.1, 63.2, 56.2, 53.6 (2 carbons), 45.4, 40.1 (2 carbons), 38.6, 34.2, 33.3, 31.5, 29.7, 29.2 (2 carbons), 29.1, 28.8, 25.9, 25.8;
m/z calcd for C
48H
58N
2O
5 742.4; found 743.3 [M + H]
+.
(E)-2-((1-Benzylpiperidin-4-Yl)Methyl)-6-((10-(4-(3-(4-(Dimethylamino)Phenyl)Acryloyl)Phenoxy)Decyl)Oxy)-5-Methoxy-2,3-Dihydro-1H-Inden-1-One (
16e) (SGT696). A solution of compound
16 (20 mg, 0.055 mmol), compound
7e (32 mg, 0.066 mmol), and K
2CO
3 (23 mg, 0.16 mmol) in anhydrous DMF (5 mL) was heated at 80 °C overnight. The reaction mixture was then diluted with H
2O, and extracted with EtOAc (3×). The combined organic layers were washed with H
2O (3×) and brine (3×), dried over anhydrous MgSO
4, filtered, and concentrated under reduced pressure. The crude product obtained was purified by column chromatography (SiO
2 gel, pure CH
2Cl
2 to CH
2Cl
2:MeOH/19:1; R
f 0.40 in CH
2Cl
2:MeOH/19:1) to yield compound
16e (40 mg, 95%) as a yellow solid:
1H NMR (400 MHz, CDCl
3,
Figure S55)
δ 7.99 (d,
J = 8.8 Hz, 2H, aromatic), 7.76 (d,
J = 15.2 Hz, 1H, HC=C
H-Ph), 7.53 (d,
J = 8.8 Hz, 2H, aromatic), 7.36–7.24 (m, 6H, aromatic), 7.13 (s, 1H, aromatic), 6.93 (d,
J = 8.8 Hz, 2H, aromatic), 6.82 (s, 1H, aromatic), 6.68 (d,
J = 8.0 Hz, 2H, aromatic), 4.01 (t,
J = 6.8 Hz, 4H), 3.92 (s, 3H, OC
H3), 3.55 (s, 2H, NC
H2Ph), 3.20 (dd,
J1 = 17.6 Hz,
J2 = 8.4 Hz, 1H), 3.02 (s, 6H, N(C
H3)
2), 2.94 (m, 2H), 2.65 (m, 2H), 2.02 (m, 2H), 1.92–1.60 (m, 7H), 1.50–1.20 (m, 16H);
13C NMR (100 MHz, CDCl
3,
Figure S56)
δ 207.8, 188.9, 162.6, 155.8, 151.8, 148.8, 148.5 (3 carbons), 144.8, 131.6, 130.5 (2 carbons), 130.2 (2 carbons), 129.4, 129.2, 128.2 (2 carbons), 127.2, 122.9, 116.7, 114.1 (2 carbons), 111.8 (2 carbons), 107.4, 105.4, 69.0, 68.2, 63.1, 56.2, 53.6 (2 carbons), 45.3, 40.1 (2 carbons), 38.6, 34.2, 33.4, 31.5, 29.7, 29.42, 29.40, 29.30, 29.28, 29.1, 28.9, 26.0, 25.9;
m/z calcd for C
50H
62N
2O
5 770.5; found 771.4 [M + H]
+.
(E)-2-((1-Benzylpiperidin-4-Yl)Methyl)-6-((12-(4-(3-(4-(Dimethylamino)Phenyl)Acryloyl)Phenoxy)Dodecyl)Oxy)-5-Methoxy-2,3-Dihydro-1H-Inden-1-One (
16f) (SGT680). A solution of compound
14 (25 mg, 0.068 mmol), compound
7f (42 mg, 0.082 mmol), and K
2CO
3 (28 mg, 0.21 mmol) in anhydrous DMF (5 mL) was heated at 80 °C overnight. The reaction mixture was then diluted with H
2O, and extracted with EtOAc (3×). The combined organic layers were washed with H
2O (3×) and brine (3×), dried over anhydrous MgSO
4, filtered, and concentrated under reduced pressure. The crude product obtained was purified by column chromatography (SiO
2 gel, pure CH
2Cl
2 to CH
2Cl
2:MeOH/19:1; R
f 0.39 in CH
2Cl
2:MeOH/19:1) to yield compound
16f (56 mg, quantitative yield) as an orange solid:
1H NMR (400 MHz, CDCl
3,
Figure S57)
δ 7.99 (d,
J = 8.8 Hz, 2H, aromatic), 7.76 (d,
J = 15.6 Hz, 1H, HC=C
H-Ph), 7.53 (d,
J = 8.8 Hz, 2H, aromatic), 7.38–7.26 (m, 6H, aromatic), 7.12 (s, 1H, aromatic), 6.93 (d,
J = 8.8 Hz, 2H, aromatic), 6.81 (s, 1H, aromatic), 6.67 (d,
J = 8.4 Hz, 2H, aromatic), 4.01 (t,
J = 6.4 Hz, 2H), 4.00 (t,
J = 6.4 Hz, 2H), 3.91 (s, 3H, OC
H3), 3.67 (s, 2H, NC
H2Ph), 3.21 (dd,
J1 = 17.6 Hz,
J2 = 8.4 Hz, 1H), 3.03 (m, 2H), 3.02 (s, 6H, N(C
H3)
2), 2.64 (m, 2H), 2.16 (m, 2H), 2.00 (m, 1H), 1.92–1.70 (m, 7H), 1.50–1.20 (m, 19H);
13C NMR (100 MHz, CDCl
3,
Figure S58)
δ 207.6, 188.9, 162.6, 155.8, 151.8, 148.9, 148.5 (3 carbons), 144.8, 131.6, 130.5 (2 carbons), 130.2 (2 carbons), 129.8, 129.1, 128.4 (2 carbons), 127.8, 122.9, 116.7, 114.1 (2 carbons), 111.8 (2 carbons), 107.4, 105.4, 69.1, 68.2, 62.5, 56.2, 53.2 (2 carbons), 45.1, 40.1 (2 carbons), 38.4, 33.6, 33.4, 31.0, 29.7, 29.50 (3 carbons), 29.47, 29.33, 29.30, 29.1, 28.9, 26.0, 25.9;
m/z calcd for C
52H
66N
2O
5 798.5; found 799.3 [M + H]
+.
(E)-3-(4-(Dimethylamino)Phenyl)-1-(3-(2-Hydroxyethoxy)Phenyl)Prop-2-En-1-One (
17) (SGT863). A solution of compound (50 mg, 0.19 mmol) and K
2CO
3 (52 mg, 0.37 mmol) in anhydrous MeCN (5 mL) was treated with 2-bromoethanol (30 μL, 0.37 mmol) and the resulting mixture was refluxed overnight. The solvent was then removed and the obtained crude product was purified by column chromatography (SiO
2 gel, pure Hexanes to Hexanes:EtOAc/1:1; R
f 0.32 in Hexanes:EtOAc/1:1) to yield compound
17 (14 mg, 24%) as a red solid:
1H NMR (400 MHz, CDCl
3,
Figure S59)
δ 7.78 (d,
J = 15.2 Hz, 1H, HC=C
H-Ph), 7.60–7.58 (m, 1H, aromatic), 7.55–7.51 (m, 3H, aromatic), 7.38 (t,
J = 8.4 Hz, 1H, aromatic), 7.29 (d,
J = 16.0 Hz, 1H,
HC=CH-Ph), 7.10 (ddd,
J1 = 8.4 Hz,
J2 = 2.8 Hz,
J3 = 0.8 Hz, 1H, aromatic), 6.68 (d,
J = 8.8 Hz, 2H, aromatic), 4.15 (t,
J = 4.4 Hz, 2H, HOCH
2C
H2OPh), 3.98 (q,
J = 4.0 Hz, 2H, HOC
H2CH
2OPh), 3.03 (s, 6H, N(C
H3)
2);
13C NMR (125 MHz, CDCl
3,
Figure S60)
δ 189.3, 157.9, 145.1, 139.6, 129.6 (3 carbons), 128.6, 120.3, 118.2 (2 carbons), 116.0, 112.7 (2 carbons), 111.1, 68.5, 60.5, 39.3 (2 carbons);
m/
z calcd for C
19H
21NO
3 311.2; found 312.1 [M + H]
+.
2-(2-Bromoethoxy)Tetrahydro-2H-Pyran (
18) (SGT864). A mixture of 2-bromoethanol (353 mg, 2.8 mmol), 3,4-dihydro-2
H-pyran (0.31 mL, 3.4 mmol), and
p-TsOH monohydrate (11 mg, 0.06 mmol) in anhydrous CH
2Cl
2 (10 mL) was stirred at rt overnight. The reaction mixture was washed with NaHCO
3, H
2O, and brine, dried over anhydrous MgSO
4, filtered, and concentrated under reduced pressure. The crude product obtained was purified by column chromatography (SiO
2 gel, Hexanes:EtOAc/19:1; R
f 0.52 in Hexanes:EtOAc/9:1) to yield the known compound
18 [
72] (362 mg, 62%) as a colorless oil:
1H NMR (400 MHz, CDCl
3,
Figure S61, which matches the lit. [
72])
δ 4.66 (t,
J = 3.6 Hz, 1H), 3.99 (dt,
J1 = 11.2 Hz,
J2 = 6.4 Hz, 1H), 3.87 (ddd,
J1 = 11.6 Hz,
J2 = 8.0 Hz,
J3 = 2.8 Hz, 1H), 3.75 (dt,
J1 = 11.2 Hz,
J2 = 6.8 Hz, 1H), 3.54–3.44 (m, 3H), 1.88–1.77 (m, 1H), 1.76–1.66 (m, 1H), 1.65–1.48 (m, 4H).
2-((1-Benzylpiperidin-4-Yl)Methyl)-5-Methoxy-6-(2-((Tetrahydro-2H-Pyran-2-Yl)Oxy)Ethoxy)-2,3-Dihydro-1H-Inden-1-One (
19) (SGT1729). A solution of compound
14 (50 mg, 0.14 mmol) and K
2CO
3 (39 mg, 0.27 mmol) in anhydrous DMF (5 mL) was treated with a solution of compound
18 (57 mg, 0.27 mmol) in DMF (1 mL), and the resulting mixture was stirred at rt overnight. The reaction mixture was quenched with H
2O and extracted with EtOAc (3×). The combined organic layers were washed with H
2O (3×), and brine (3×), dried over anhydrous MgSO
4, filtered, and concentrated under reduced pressure. The crude product obtained was purified by column chromatography (SiO
2 gel, pure CH
2Cl
2 to CH
2Cl
2:MeOH/19:1; R
f 0.21 in CH
2Cl
2:MeOH/19:1) to yield compound
19 (63 mg, 93%) as a yellow oil:
1H NMR (400 MHz, CDCl
3,
Figure S62)
δ 7.35–7.22 (m, 5H, aromatic), 7.21 (s, 1H, aromatic), 6.82 (s, 1H, aromatic), 4.70 (t,
J = 3.6 Hz, 2H), 4.21 (t,
J = 4.8 Hz, 2H), 4.05 (m, 1H), 3.91 (s, 3H, OC
H3), 3.87–3.82 (m, 2H), 3.49 (m, 2H, NC
H2Ph), 3.20 (dd,
J1 = 17.6 Hz,
J2 = 8.4 Hz, 1H), 2.88 (m, 2H), 2.66 (dt,
J1 = 14.4 Hz,
J2 = 3.6 Hz, 2H), 2.00–1.76 (m, 4H), 1.75–1.48 (m, 8H), 1.38–1.23 (m, 3H);
13C NMR (100 MHz, CDCl
3,
Figure S63) 207.6, 155.9, 148.8, 148.7, 129.5 (2 carbons), 129.1 (2 carbons), 128.2 (2 carbons), 127.3, 107.6, 106.3, 98.9, 68.5, 65.42, 65.41, 63.1, 62.1, 56.12, 56.10, 53.5, 45.3, 38.6, 34.1, 33.4, 31.4, 30.4, 25.4, 19.2;
m/
z calcd for C
30H
39NO
5 493.3; found 494.2 [M + H]
+.
2-((1-Benzylpiperidin-4-Yl)Methyl)-6-(2-Hydroxyethoxy)-5-Methoxy-2,3-Dihydro-1H-Inden-1-One (
20) (SGT1732). A solution of compound
19 (50 mg, 0.10 mmol) and
p-TsOH monohydrate (19 mg, 0.10 mmol) in MeOH (5 mL) was stirred at rt overnight. The solvents were removed, and the crude material obtained was dissolved in EtOAc, washed with NaHCO
3, H
2O, and brine, dried over anhydrous MgSO
4, filtered, and concentrated under reduced pressure. The crude product obtained was purified by column chromatography (SiO
2 gel, pure CH
2Cl
2 to CH
2Cl
2:MeOH/19:1; R
f 0.16 in CH
2Cl
2:MeOH/19:1) to yield compound
20 (18 mg, 44%) as a colorless oil:
1H NMR (400 MHz, CDCl
3,
Figure S64)
δ 7.35–7.22 (m, 5H, aromatic), 7.18 (s, 1H, aromatic), 6.84 (s, 1H, aromatic), 4.12 (t,
J = 4.4 Hz, 2H), 3.96 (t,
J = 4.4 Hz, 2H), 3.92 (s, 3H, OC
H3), 3.55 (br s, 2H, NC
H2Ph), 3.22 (dd,
J1 = 17.6 Hz,
J2 = 8.0 Hz, 1H), 2.93 (m, 2H), 2.67 (dt,
J1 = 14.0 Hz,
J2 = 3.2 Hz, 2H), 2.10–1.94 (m, 2H), 1.93–1.84 (m, 1H), 1.80–1.30 (m, 6H);
13C NMR (100 MHz, CDCl
3,
Figure S65) 207.5, 155.9, 149.2, 148.4, 129.6 (2 carbons), 129.2 (2 carbons), 128.3 (2 carbons), 127.5, 107.6, 106.7, 70.7, 62.9, 61.0, 56.1, 53.41, 53.35, 45.2, 38.5, 33.9, 33.4 (2 carbons), 31.2;
m/
z calcd for C
25H
31NO
4 409.23; found 410.2 [M + H]
+.
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}