Hydrogen Tunnelling as a Probe of the Involvement of Water Vibrational Dynamics in Aqueous Chemistry?



Abstract
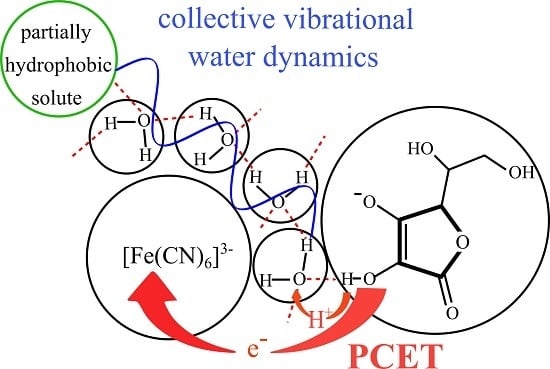
1. Introduction
2. Results and Discussion
2.1. The Temperature Dependence of Kinetic Isotope Effects. The Correlation of Isotopic Differences in the Activation Enthalpies ΔΔH‡ (D,H) and the Isotopic Differences in Entropies ΔΔS‡ (D,H)
2.2. The Water Vibrational Dynamics and Hydrogen Tunnelling in the Reaction
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ramasesha, K.; De Marco, L.; Mandal, A.; Tokmakoff, A. Water vibrations have strongly mixed intra- and intermolecular character. Nat. Chem. 2013, 5, 935–940. [Google Scholar] [CrossRef] [PubMed]
- De Marco, L.; Fournier, J.A.; Thämer, M.; Carpenter, W.; Tokmakoff, A. Anharmonic exciton dynamics and energy dissipation in liquid water from two-dimensional infrared spectroscopy. J. Chem. Phys. 2016, 145, 094501. [Google Scholar] [CrossRef] [PubMed]
- Fournier, J.A.; Carpenter, W.; De Marco, L.; Tokmakoff, A. Interplay of Ion–Water and Water–Water Interactions within the Hydration Shells of Nitrate and Carbonate Directly Probed with 2D IR Spectroscopy. J. Am. Chem. Soc. 2016, 138, 9634–9645. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, W.B.; Fournier, J.A.; Biswas, R.; Voth, G.A.; Tokmakoff, A. Delocalization and Stretch-Bend Mixing of the HOH Bend in Liquid Water. J. Chem. Phys. 2017, 147, 084503. [Google Scholar] [CrossRef] [PubMed]
- Auer, B.M.; Skinner, J.L. IR and Raman Spectra of Liquid Water: Theory and Interpretation. J. Chem. Phys. 2008, 128, 224511. [Google Scholar] [CrossRef] [PubMed]
- Paarmann, A.; Hayashi, T.; Mukamel, S.; Miller, R.J.D. Nonlinear Response of Vibrational Excitons: Simulating the Two-Dimensional Infrared Spectrum of Liquid Water. J. Chem. Phys. 2009, 130, 204110. [Google Scholar] [CrossRef]
- Falvo, C.; Palmieri, B.; Mukamel, S. Coherent Infrared Multidimensional Spectra of the OH Stretching Band in Liquid Water Simulated by Direct Nonlinear Exciton Propagation. J. Chem. Phys. 2009, 130, 184501. [Google Scholar] [CrossRef]
- Kraemer, D.; Cowan, M.L.; Paarmann, A.; Huse, N.; Nibbering, E.T.J.; Elsaesser, T.; Dwayne Miller, R.J. Temperature Dependence of the Two-Dimensional Infrared Spectrum of Liquid H2O. Proc. Natl. Acad. Sci. USA 2008, 105, 437–442. [Google Scholar] [CrossRef]
- Imoto, S.; Xantheas, S.S.; Saito, S. Ultrafast Dynamics of Liquid Water: Frequency Fluctuations of the OH Stretch and the HOH Bend. J. Chem. Phys. 2013, 139, 044503. [Google Scholar] [CrossRef]
- Nagata, Y.; Yoshimune, S.; Hsieh, C.S.; Hunger, J.; Bonn, M. Ultrafast Vibrational Dynamics of Water Disentangled by Reverse Nonequilibrium Ab Initio Molecular Dynamics Simulations. Phys. Rev. X 2015, 5, 1–11. [Google Scholar] [CrossRef]
- Formosinho, S.; Barroso, M. Proton-Coupled Electron Transfer: A Carrefour of Chemical Reactivity Traditions; Royal Society of Chemistry: Cambridge, UK, 2012. [Google Scholar]
- Weinberg, D.R.; Gagliardi, C.J.; Hull, J.F.; Murphy, C.F.; Kent, C.A.; Westlake, B.C.; Paul, A.; Ess, D.H.; McCafferty, D.G.; Meyer, T.J. Proton-Coupled Electron Transfer. Chem. Rev. 2012, 112, 4016–4093. [Google Scholar] [CrossRef] [PubMed]
- Warren, J.J.; Tronic, T.A.; Mayer, J.M. Thermochemistry of Proton-Coupled Electron Transfer Reagents and Its Implications. Chem. Rev. 2010, 110, 6961–7001. [Google Scholar] [CrossRef] [PubMed]
- Reece, S.Y.; Nocera, D.G. Proton-Coupled Electron Transfer in Biology: Result from Synergistic Studies in Natural and Model Systems. Annu. Rev. Biochem. 2009, 78, 673–699. [Google Scholar] [CrossRef] [PubMed]
- Huynh, M.H.V.; Meyer, T.J. Proton-Coupled Electron Transfer. Chem. Rev. 2007, 107, 5004–5064. [Google Scholar] [CrossRef]
- Hammes-Schiffer, S. Proton-Coupled Electron Transfer: Moving Together and Charging Forward. J. Am. Chem. Soc. 2015, 137, 8860–8871. [Google Scholar] [CrossRef]
- Hammes-Schiffer, S.; Stuchebrukhov, A.A. Theory of Coupled Electron and Proton Transfer Reactions. Chem. Rev. 2010, 110, 6939–6960. [Google Scholar] [CrossRef]
- Kagayama, N.; Sekiguchi, M.; Inada, Y.; Takagi, H.D.; Fanahashi, S. Mechanistic Study of the Reaction of L-Ascorbic Acid with Hexacyanometalate(III) Ions of Iron(III), Ruthenium(III), and Osmium(III) in Aqueous Acidic Solution at Elevated Pressures. Inorg. Chem. 1994, 33, 1881–1885. [Google Scholar] [CrossRef]
- Jakobušić Brala, C.; Pilepić, V.; Sajenko, I.; Karković, A.; Uršić, S. Ions Can Move a Proton-Coupled Electron-Transfer Reaction into Tunneling Regime. Helv. Chim. Acta 2011, 94, 1718–1731. [Google Scholar] [CrossRef]
- Karković, A.; Brala, C.J.; Pilepić, V.; Uršić, S. Solvent-Induced Hydrogen Tunnelling in Ascorbate Proton-Coupled Electron Transfers. Tetrahedron Lett. 2011, 52, 1757–1761. [Google Scholar] [CrossRef]
- Jakobušić Brala, C.; Karković, A.; Sajenko, I.; Pilepić, V.; Uršić, S. Sizeable Increase of Kinetic Isotope Effects and Tunnelling in Coupled Electron–Proton Transfers in Presence of the Quaternary Ions. PCET Processes and Hydrogen Tunnelling as a “Probe” for Structuring and Dynamical Phenomena in Water Solution. Z. Phys. Chem. 2012, 226, 29–46. [Google Scholar] [CrossRef]
- Jakobušić Brala, C.; Karković, A.; Klepac, K.; Vučinović, A.M.; Pilepić, V.; Uršić, S. Small Molecule Tunnelling Systems: Variation of Isotope Effects. Z. Phys. Chem. 2011, 225, 821–841. [Google Scholar] [CrossRef]
- Kandathil, S.M.; Driscoll, M.D.; Dunn, R.V.; Scrutton, N.S.; Hay, S. Proton Tunnelling and Promoting Vibrations during the Oxidation of Ascorbate by Ferricyanide? Phys. Chem. Chem. Phys. 2014, 16, 2256–2259. [Google Scholar] [CrossRef] [PubMed]
- Layfield, J.P.; Hammes-Schiffer, S. Hydrogen Tunneling in Enzymes and Biomimetic Models. Chem. Rev. 2014, 114, 3466–3494. [Google Scholar] [CrossRef] [PubMed]
- Johannissen, L.O.; Hay, S.; Scrutton, N.S. Nuclear Quantum Tunnelling in Enzymatic Reactions—An Enzymologist’s Perspective. Phys. Chem. Chem. Phys. 2015, 17, 30775–30782. [Google Scholar] [CrossRef]
- Hay, S.; Scrutton, N.S. Good Vibrations in Enzyme-Catalysed Reactions. Nat. Chem. 2012, 4, 161–168. [Google Scholar] [CrossRef]
- Schwartz, S.D. Protein Dynamics and the Enzymatic Reaction Coordinate. In Dynamics in Enzyme Catalysis; Topics in Current Chemistry; Klinman, J., Hammes-Schiffer, S., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 189–208. [Google Scholar] [CrossRef]
- Klinman, J.P. Dynamically Achieved Active Site Precision in Enzyme Catalysis. Acc. Chem. Res. 2015, 48, 449–456. [Google Scholar] [CrossRef]
- Kohen, A. Role of Dynamics in Enzyme Catalysis: Substantial versus Semantic Controversies. Acc. Chem. Res. 2015, 48, 466–473. [Google Scholar] [CrossRef]
- Klinman, J.P.; Kohen, A. Hydrogen Tunneling Links Protein Dynamics to Enzyme Catalysis. Annu. Rev. Biochem. 2013, 82, 471–496. [Google Scholar] [CrossRef]
- Geddes, A.; Paul, C.E.; Hay, S.; Hollmann, F.; Scrutton, N.S. Donor–Acceptor Distance Sampling Enhances the Performance of “Better than Nature” Nicotinamide Coenzyme Biomimetics. J. Am. Chem. Soc. 2016, 138, 11089–11092. [Google Scholar] [CrossRef]
- Pudney, C.R.; Guerriero, A.; Baxter, N.J.; Johannissen, L.O.; Waltho, J.P.; Hay, S.; Scrutton, N.S. Fast Protein Motions Are Coupled to Enzyme H-Transfer Reactions. J. Am. Chem. Soc. 2013, 135, 2512–2517. [Google Scholar] [CrossRef]
- Pudney, C.R.; Johannissen, L.O.; Sutcliffe, M.J.; Hay, S.; Scrutton, N.S. Direct Analysis of Donor-Acceptor Distance and Relationship to Isotope Effects and the Force Constant for Barrier Compression in Enzymatic H-Tunneling Reactions. J. Am. Chem. Soc. 2010, 132, 11329–11335. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.J.; Soudackov, A.V.; Hammes-Schiffer, S. Analysis of Kinetic Isotope Effects for Proton-Coupled Electron Transfer Reactions †. J. Phys. Chem. A 2009, 113, 2117–2126. [Google Scholar] [CrossRef] [PubMed]
- Hammes-Schiffer, S.; Soudackov, A.V. Proton-Coupled Electron Transfer in Solution, Proteins, and Electrochemistry. J. Phys. Chem. B 2008, 112, 14108–14123. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.P. The Tunnel Effect in Chemistry; Chapman & Hall: London, UK, 1980. [Google Scholar]
- Romesberg, F.E.; Schowen, R.L. Isotope Effects and Quantum Tunneling in Enzyme-Catalyzed Hydrogen Transfer. Part I. The Experimental Basis. In Advances in Physical Organic Chemistry; Richard, J.P., Ed.; Elsevier Academic Press: London, UK, 2004; pp. 27–77. [Google Scholar]
- Kohen, A. Kinetic Isotope Effects as Probes for Hydrogen Tunneling in Enzyme Catalysis. In Isotope Effects in Chemistry and Biology; Kohen, A., Limbach, H.H., Eds.; Taylor & Francis/CRC Press: New York, NY, USA, 2006; pp. 744–764. [Google Scholar]
- Kohen, A. Current Issues in Enzymatic Hydrogen Transfer from Carbon: Tunneling and Coupled Motion from Kinetic Isotope Effect Studies. In Hydrogen-Transfer Reactions; Hynes, J.T., Klinman, J.P., Limbach, H.H., Schowen, R.L., Eds.; Wiley-VCH: Weinheim, Germany, 2007; pp. 1311–1340. [Google Scholar]
- Sen, A.; Kohen, A. Quantum Effects in Enzyme Kinetics. In Quantum Tunnelling in Enzyme-Catalysed Reactions; Scrutton, N.S., Allemann, R.K., Eds.; Royal Society of Chemistry: Cambridge, UK, 2009; pp. 164–181. [Google Scholar]
- Sajenko, I.; Pilepić, V.; Jakobušić Brala, C.; Uršić, S. Solvent Dependence of the Kinetic Isotope Effect in the Reaction of Ascorbate with the 2,2,6,6-Tetramethylpiperidine-1-Oxyl Radical: Tunnelling in a Small Molecule Reaction. J. Phys. Chem. A 2010, 114, 3423–3430. [Google Scholar] [CrossRef] [PubMed]
- Fry, A.J. The Effect of Tetramethylammonium Ion on the Voltammetric Behavior of Polycyclic Aromatic Hydrocarbons: Computations Explain a Long-Standing Anomaly. Phys. Chem. Chem. Phys. 2010, 12, 14775. [Google Scholar] [CrossRef] [PubMed]
- Luong, T.Q.; Verma, P.K.; Mitra, R.K.; Havenith, M. Onset of Hydrogen Bonded Collective Network of Water in 1,4-Dioxane. J. Phys. Chem. A 2011, 115, 14462–14469. [Google Scholar] [CrossRef]
- Mizuno, K.; Imafuji, S.; Fujiwara, T.; Ohta, T.; Tamiya, Y. Hydration of the CH Groups in 1,4-Dioxane Probed by NMR and IR: Contribution of Blue-Shifting CH···OH2 Hydrogen Bonds. J. Phys. Chem. B 2003, 107, 3972–3978. [Google Scholar] [CrossRef]
- Sirotkin, V.A.; Solomonov, B.N.; Faizullin, D.A.; Fedotov, V.D. IR Spectroscopic Study of the State of Water in Dioxane and Acetonitrile: Relationship with the Thermodynamic Activity of Water. J. Struct. Chem. 2000, 41, 997–1003. [Google Scholar] [CrossRef]
- Chang, H.-C.; Jiang, J.-C.; Chuang, C.-W.; Lin, J.-S.; Lai, W.-W.; Yang, Y.-C.; Lin, S.H. Hydrogen Bond-like Equatorial C–H⋯O Interactions in Aqueous 1,3-Dioxane: A Combined High-Pressure Infrared and Raman Spectroscopy Study. Chem. Phys. Lett. 2005, 410, 42–48. [Google Scholar] [CrossRef]
- Cringus, D.; Jansen, T.L.C.; Pshenichnikov, M.S.; Wiersma, D.A. Ultrafast Anisotropy Dynamics of Water Molecules Dissolved in Acetonitrile. J. Chem. Phys. 2007, 127, 084507. [Google Scholar] [CrossRef]
- Chang, H.; Jiang, J.; Su, C.; Lu, L.; Hsiao, C.; Chuang, C.; Lin, S.H. Pressure-Enhanced C−H···O Interactions in Aqueous Tert -Butyl Alcohol. J. Phys. Chem. A 2004, 108, 11001–11005. [Google Scholar] [CrossRef]
- Chang, H.-C.; Jiang, J.-C.; Chang, C.-Y.; Su, J.-C.; Hung, C.-H.; Liou, Y.-C.; Lin, S.H. Structural Organization in Aqueous Solutions of 1-Butyl-3-Methylimidazolium Halides: A High-Pressure Infrared Spectroscopic Study on Ionic Liquids. J. Phys. Chem. B 2008, 112, 4351–4356. [Google Scholar] [CrossRef]
- Chang, H.-C.; Chang, C.-Y.; Su, J.-C.; Chu, W.-C.; Jiang, J.-C.; Lin, S.H. Conformations of 1-Butyl-3-Methylimidazolium Chloride Probed by High Pressure Raman Spectroscopy. Int. J. Mol. Sci. 2006, 7, 417–424. [Google Scholar] [CrossRef]
- Gu, Y.; Kar, T.; Scheiner, S. Fundamental Properties of the CH···O Interaction: Is It a True Hydrogen Bond? J. Am. Chem. Soc. 1999, 121, 9411–9422. [Google Scholar] [CrossRef]
- Stern, M.J.; Weston, R.E. Phenomenological Manifestations of Quantum-mechanical Tunneling. II. Effect on Arrhenius Pre-exponential Factors for Primary Hydrogen Kinetic Isotope Effects. J. Chem. Phys. 1974, 60, 2808–2814. [Google Scholar] [CrossRef]
- Levy, Y.; Onuchic, J.N. Water Mediation in Protein Folding and Molecular Recognition. Annu. Rev. Biophys. Biomol. Struct. 2006, 35, 389–415. [Google Scholar] [CrossRef]
- Lum, K.; Chandler, D.; Weeks, J.D. Hydrophobicity at Small and Large Length Scales. J. Phys. Chem. B 1999, 103, 4570–4577. [Google Scholar] [CrossRef]
- Chandler, D. Interfaces and the Driving Force of Hydrophobic Assembly. Nature 2005, 437, 640–647. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solute Added a | KIE b | DDH‡/kJ mol−1 | AH/AD |
|---|---|---|---|
| none (neat water c) | 4.60 (0.06) | 3.9 (0.4) | 0.97 (0.15) |
| 1,4-diox:water = 0.05:0.95 v/v | 5.42 (0.10) | 6.5 (0.2) | 0.39 (0.03) |
| 1,4-diox:water = 0.10:0.90 v/v | 6.17 (0.08) | 8.4 (0.6) | 0.21 (0.05) |
| MeCN:water = 0.05:0.95 v/v d | 5.61 (0.16) | 7.1 (0.4) | 0.32 (0.04) |
| MeCN:water = 0.10:0.90 v/v | 6.50 (0.10) | 8.6 (0.2) | 0.21 (0.02) |
| EtOH:water = 0.10:0.90 v/v | 5.72 (0.06) | 6.4 (0.3) | 0.43 (0.05) |
| c(TMACl) = 0.005 mol dm−3 | 5.28 (0.03) | 6.2 (0.4) | 0.44 (0.03) |
| c(TPACl) = 0.005 mol dm−3 | 5.38 (0.05) | 6.5 (0.4) | 0.41 (0.04) |
| c(TBACl) = 0.005 mol dm−3 | 5.28 (0.08) | 6.0 (0.4) | 0.48 (0.07) |
| c(TEACl) = 0.01 mol dm−3 | 5.15 (0.02) | 5.9 (0.3) | 0.48 (0.06) |
| c(TEACl) = 0.1 mol dm−3 e | 7.25 (0.02) | 9.5 (0.2) | 0.16 (0.01) |
| c(BTMACl) = 0.1 mol dm−3 | 6.46 (0.09) e | 8.7 (0.2) | 0.19 (0.02) |
| c(AChCl) = 0.1 mol dm−3 | 5.91 (0.16) | 7.5 (0.3) | 0.29 (0.03) |
| c(PQCl2) = 0.006 mol dm−3 | 5.57 (0.17) | 6.9 (0.2) | 0.35 (0.03) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karković Marković, A.; Jakobušić Brala, C.; Pilepić, V.; Uršić, S. Hydrogen Tunnelling as a Probe of the Involvement of Water Vibrational Dynamics in Aqueous Chemistry? Molecules 2020, 25, 172. https://doi.org/10.3390/molecules25010172
Karković Marković A, Jakobušić Brala C, Pilepić V, Uršić S. Hydrogen Tunnelling as a Probe of the Involvement of Water Vibrational Dynamics in Aqueous Chemistry? Molecules. 2020; 25(1):172. https://doi.org/10.3390/molecules25010172
Chicago/Turabian StyleKarković Marković, Ana, Cvijeta Jakobušić Brala, Viktor Pilepić, and Stanko Uršić. 2020. "Hydrogen Tunnelling as a Probe of the Involvement of Water Vibrational Dynamics in Aqueous Chemistry?" Molecules 25, no. 1: 172. https://doi.org/10.3390/molecules25010172
APA StyleKarković Marković, A., Jakobušić Brala, C., Pilepić, V., & Uršić, S. (2020). Hydrogen Tunnelling as a Probe of the Involvement of Water Vibrational Dynamics in Aqueous Chemistry? Molecules, 25(1), 172. https://doi.org/10.3390/molecules25010172