
Integrated Analytical Tools for Accessing Acridones and Unrelated Phenylacrylamides from Swinglea glutinosa

 ,
,  and
and 
Abstract
1. Introduction
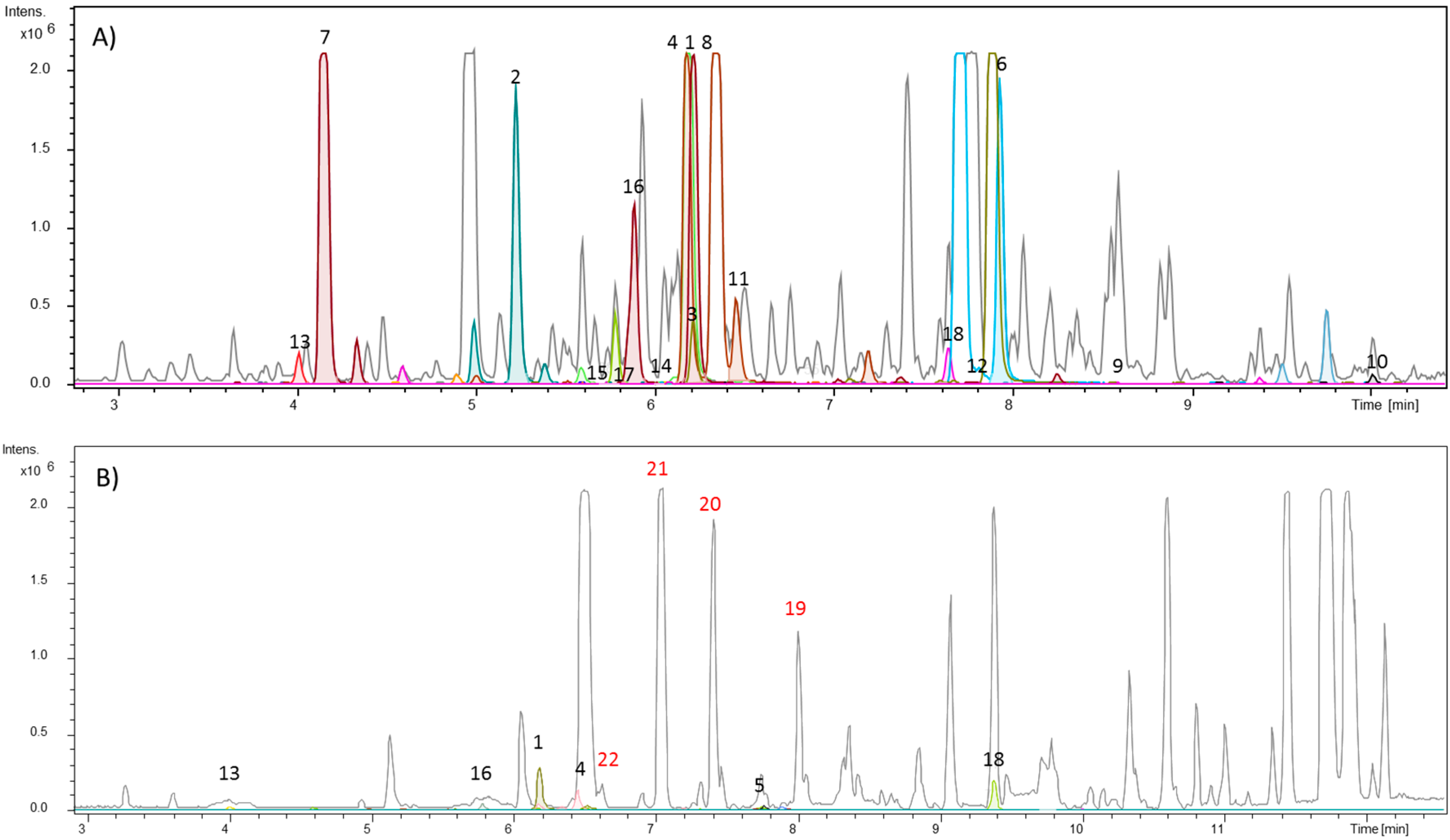
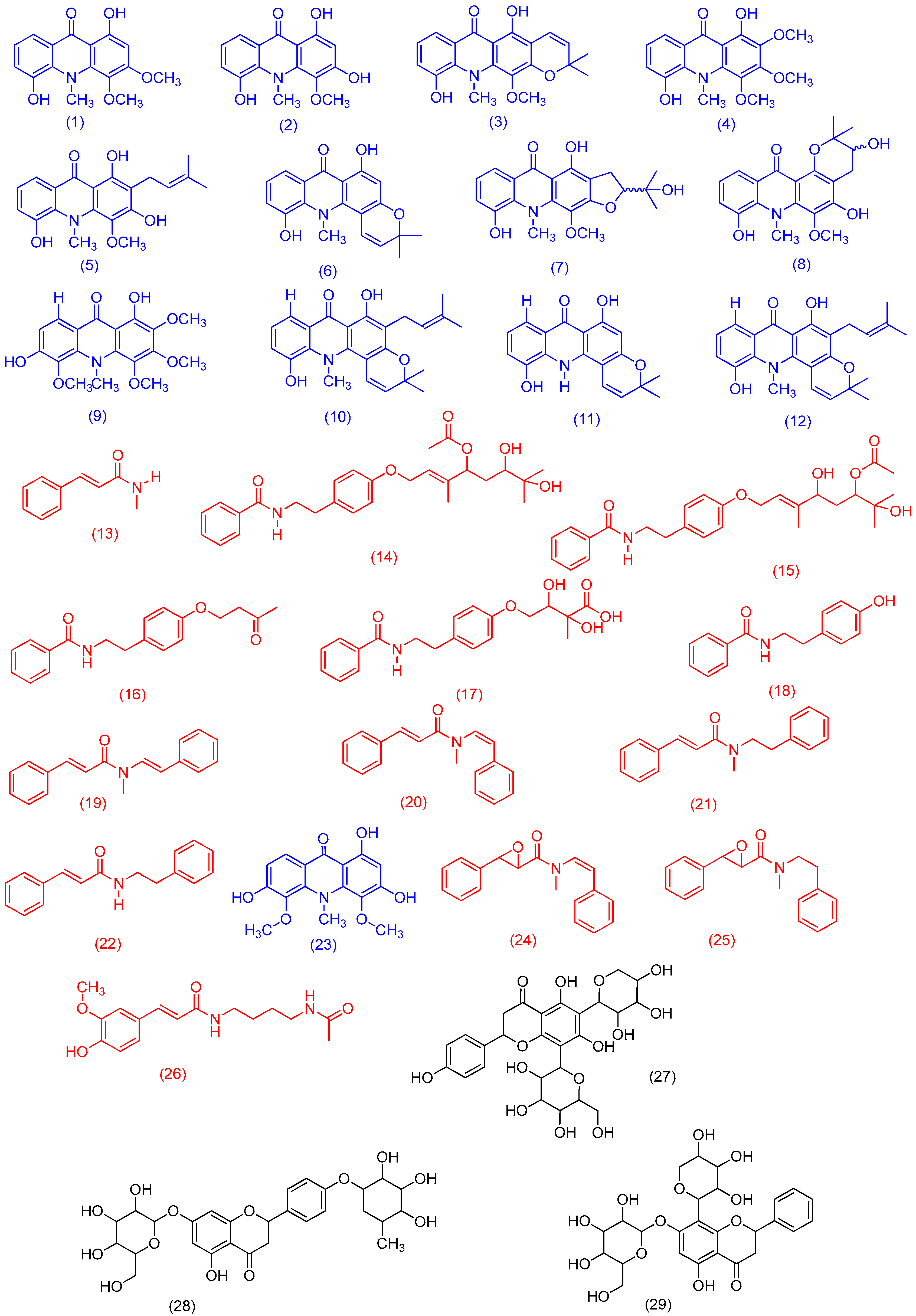
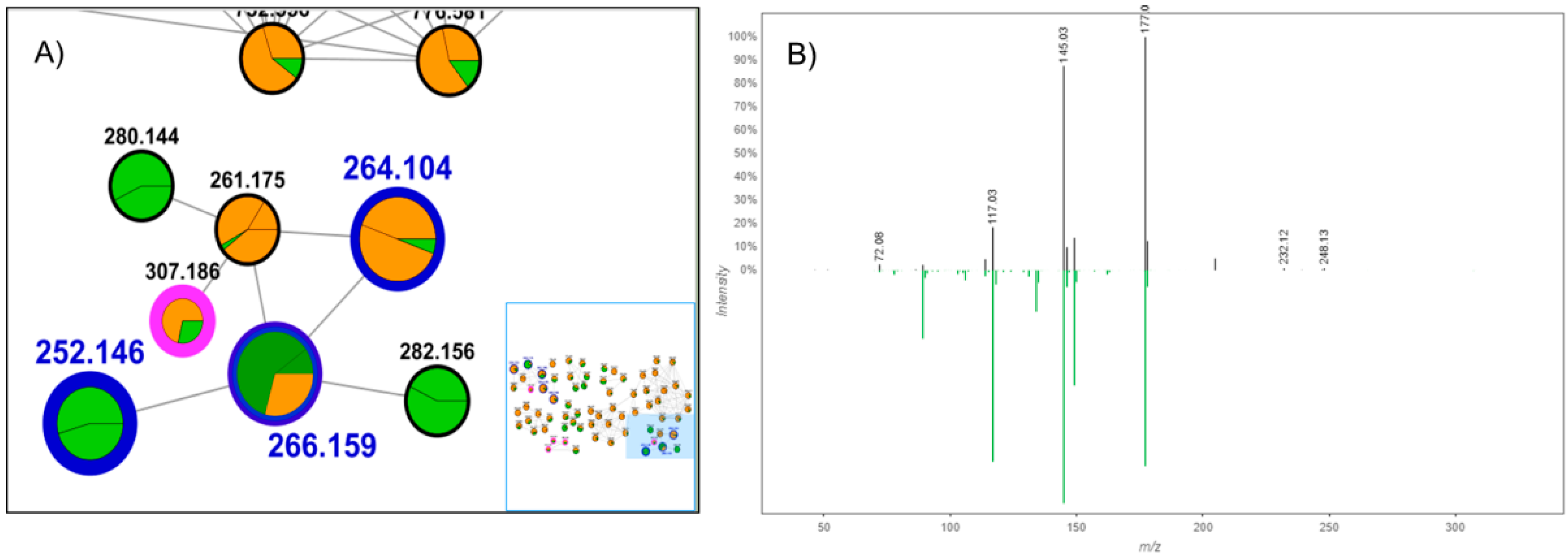
2. Results and Discussion
3. Materials and Methods
3.1. Target Analysis and Molecular MS/MS Networking-Based Dereplication
3.2. Acridone Alkaloids and Phenylacrylamides Isolation and Identification
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dinan, L. Dereplication and partial identification of compounds. In Natural Products Isolation, 2nd ed.; Sarker, S.D., Latif, Z., Gray, A.I., Eds.; Humana Press Inc.: Totowa, NJ, USA, 2005. [Google Scholar]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with GNPS. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Klitgaard, A.; Iversen, A.; Andersen, M.R.; Larsen, T.O.; Frisvad, J.C.; Nielsen, K.F. Aggressive dereplication using UHPLC-DAD-QTOF: Screning extracts for up to 3000 fungal secondary metabolites. Anal. Bioanal. Chem. 2014, 406, 1933–1943. [Google Scholar] [CrossRef] [PubMed]
- Engler, A.; Prantl, K. Die Naturlichen Pflanzenfamilien; Engler, A., Prantl, K., Eds.; Wilhelm Engelmann: Leipzig, Germany, 1931; Volume 19, pp. 187–359. [Google Scholar]
- Arato Ferreira, P.H.; Dos Santos, D.A.; Da Silva, M.F.; Vieira, P.C.; King-diaz, B.; Lotina-Hennsen, B.; Veiga, T.A. Acridone Alkaloids from Swinglea glutinosa (Rutaceae) and Their Effects on Photosynthesis. Chem. Biodivers. 2016, 13, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Do Nascimento Cerqueira, C.; Cerqueira, N.C.; Dos Santos, D.A.P.; Malaquias, K.S.; Lima, M.M.C.; Da Silva, M.F.G.; Fernandes, F.; Vieira, P.C. Novas n-benzoiltiraminas de Swinglea Glutinosa (rutaceae). Quim. Nova 2012, 35, 2181–2185. [Google Scholar] [CrossRef]
- Santos, D.A.P.; Vieira, P.C.; Silva, M.F.G.F.; Fernandes, J.B.; Rattray, L.; Crof, S.L. Antiparasitic Activities of Acridone Alkaloids from Swinglea glutinosa (Bl.) Merr. J. Braz. Chem. Soc. 2009, 20, 644–651. [Google Scholar] [CrossRef]
- Marques, E.F.; Vieira, P.C.; Severino, R.P. Acridone alkaloids as Inhibitors of Cathepsin L and V. Quím. Nova 2016, 39, 58–62. [Google Scholar] [CrossRef]
- Kuete, V.; Fouotsa, H.; Mbaveng, A.T.; Wiench, B.; Nkengfack, A.E.; Efferth, T. Cytotoxicity of a naturally occurring furoquinoline alkaloid and four acridone alkaloids towards multi-factorial drug-resistant cancer cells. Phytomedicine 2015, 22, 946–951. [Google Scholar] [CrossRef]
- Winnikoff, J.R.; Glukhov, E.; Watrous, J.; Dorrestein, P.C.; Gerwick, W.H. Quantitative molecular networking to profile marine cyanobacterial metabolomes. J. Antibiot. 2014, 67, 105–112. [Google Scholar] [CrossRef]
- Goossen, L.J.; Gooben, L.; Blanchot, M.; Arndt, M.; Salih, K.S.M. Synthesis of botryllamides and lansiumamides via ruthenium-catalyzed hydroamidation of alkynes. Synlett 2010, 1685–1687. [Google Scholar] [CrossRef]
- Lin, J.H. Cinnamamide derivatives from Clausena Lansium. Phitochemistry 1989, 28, 621–622. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Kawano, Y.; Uebayasi, M. Induction of adiponectin by natural and synthetic phenolamides in mouse and human preadipocytes and its enhancement by docosahexaenoic acid. Life Sci. 2008, 82, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Ito, C.; Furukawa, H. Lansiumamide B and SB-204900 isolated from Clausena lansium inhibit histamine and TNF-a release from RBL-2H3 cells. Inflamm. Res. 2013, 62, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Weniger, B.; Um, B.-H.; Valentin, A.; Estrada, A.; Lobstein, A.; Anton, R.; Maille, M.; Sauvain, M. Bioactive acridone alkaloids from Swinglea glutinosa. J. Nat. Prod. 2001, 64, 1221–1223. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, H.; Yogo, M.; Wu, T.-S. Acridone Alkaloids. X. 13C-Nuclear Magnetic Resonance Spectra of Acridone Alkaloids. Chem. Pharm. Bull. 1983, 31, 3084–3090. [Google Scholar] [CrossRef][Green Version]
- Wu, T.-S.; Furukawa, H. Acridone alkaloids. VII. Constituents of Citrus sinensis OSBECK var. brasiliensis TANAKA. Isolation and characterization of three new acridone alkaloids, and a new coumarin. Chem. Pharm. Bull. 1983, 31, 901–906. [Google Scholar] [CrossRef]
- Ito, C.; Kondo, Y.; Wu, T.-S.; Furukawa, H. Chemical Constituents of Glycosmis citrifolia (Willd.) Lindl. Structures of four New Acridones and Three New Quinolone Alkaloids. Chem. Pharm. Bull. 2000, 48, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.-S.; Kuoh, C.-S.; Furukawa, H. Acridone Alkaloids. VI, The constituents of Citrus depressa. Isolation and Structure Elucidation of New Acridone Alkaloids from Citrus genus. Chem. Pharm. Bull. 1983, 31, 895–900. [Google Scholar] [CrossRef]
- Wu, T.-S.; Chen, C.-M. Acridone Alkaloids from the Root Bark of Severinia buxifolia in Hainan. Chem. Pharm. Bull. 2000, 48, 85–90. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name (Code) | Molecular Formula | Exact Mass | Accurate Mass [M + H]+ |
|---|---|---|---|
| citrusinine-I (1) | C16H15NO5 | 301.0950 | 302.1018 |
| citrusinine-II (2) | C15H13NO5 | 287.0794 | 288.0862 |
| pyranofoline (3) | C20H19NO5 | 353.1263 | 354.1334 |
| citibrasine (4) | C17H17NO6 | 331.1056 | 332.1131 |
| glycotrycine IV (5) | C20H21NO5 | 355.1420 | 356.1488 |
| 5-hydroxynoracronycine (6) | C19H17NO4 | 323.1158 | 324.1230 |
| 2,3-dihydro-4,9-dihydroxy-2-(2-hydroxypropan-2-yl)-11-methoxy-10-methylfuro[3,2-b]acridin-5(10H)-oneI (7) | C20H21NO6 | 371.1369 | 372.1439 |
| 3,4-dihydro-3,5,8-trihydroxy-6-methoxy-2,2,7-trimethyl-2H-pyrano[2,3-a]acridin-12(7H)-one (8) | C20H21NO6 | 371.1369 | 372.1452 |
| des-N-methylnoracronycine (9) | C19H17NO3 | 307.1208 | 308.1250 |
| 5-hydroxy-N-methylseverifoline (10) | C24H25NO4 | 391.1784 | 392.1842 |
| glyfoline (11) | C18H19NO7 | 361.1162 | 362.1272 |
| atalaphyllinine (12) | C23H23NO | 377.1627 | 378.1446 |
| (E)-N-methylcinnamamide [(E)-N-methylphenylacrylamide] (13) | C10H11NO | 161.0841 | 162.0912 |
| N-benzoyl-O-(4-acetoxyl-6,7-dihydroxy)geranylthiramine (14) | C27H35NO6 | 469.2464 | 470.2522 |
| N-benzoyl-O-(6-acetoxyl-4,7-dihydroxy)geranylthiramine (15) | C27H35NO6 | 469.2464 | 470.2522 |
| N-{2-[4-(butoxy-3-one) phenyl]ethylbenzamide (16) | C19H21NO3 | 311.1521 | 312.1591 |
| N-{2 [4-(2,3-dihydroxy-2-methylbutoxyethyl)phenyl] ethylbenzamide (17) | C20H23NO6 | 373.1525 | 374.1573 |
| N-benzoyltyramine (18) | C15H15NO2 | 241.1103 | 242.1175 |
| lansamide I (19) | C18H17NO | 263.131 | 264.1379 |
| lansiumamide B (20) | C18H17NO | 263.131 | 264.1381 |
| lansiumamide C (21) | C18H19NO | 265.147 | 266.1554 |
| N-(2-phenylethyl)cinnamamide (22) | C17H17NO | 251.131 | 252.1397 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calheiros de Carvalho, A.; De Camillis Rodrigues, L.; Ribeiro, A.I.; Fernandes da Silva, M.F.d.G.; Soman de Medeiros, L.; Moura Veiga, T.A. Integrated Analytical Tools for Accessing Acridones and Unrelated Phenylacrylamides from Swinglea glutinosa. Molecules 2020, 25, 153. https://doi.org/10.3390/molecules25010153
Calheiros de Carvalho A, De Camillis Rodrigues L, Ribeiro AI, Fernandes da Silva MFdG, Soman de Medeiros L, Moura Veiga TA. Integrated Analytical Tools for Accessing Acridones and Unrelated Phenylacrylamides from Swinglea glutinosa. Molecules. 2020; 25(1):153. https://doi.org/10.3390/molecules25010153
Chicago/Turabian StyleCalheiros de Carvalho, Ana, Luiza De Camillis Rodrigues, Alany Ingrid Ribeiro, Maria Fátima das Graças Fernandes da Silva, Lívia Soman de Medeiros, and Thiago André Moura Veiga. 2020. "Integrated Analytical Tools for Accessing Acridones and Unrelated Phenylacrylamides from Swinglea glutinosa" Molecules 25, no. 1: 153. https://doi.org/10.3390/molecules25010153
APA StyleCalheiros de Carvalho, A., De Camillis Rodrigues, L., Ribeiro, A. I., Fernandes da Silva, M. F. d. G., Soman de Medeiros, L., & Moura Veiga, T. A. (2020). Integrated Analytical Tools for Accessing Acridones and Unrelated Phenylacrylamides from Swinglea glutinosa. Molecules, 25(1), 153. https://doi.org/10.3390/molecules25010153