Neuroprotective Effect of ent-Kaur-15-en-17-al-18-oic Acid on Amyloid Beta Peptide-Induced Oxidative Apoptosis in Alzheimer’s Disease

 , and
, and 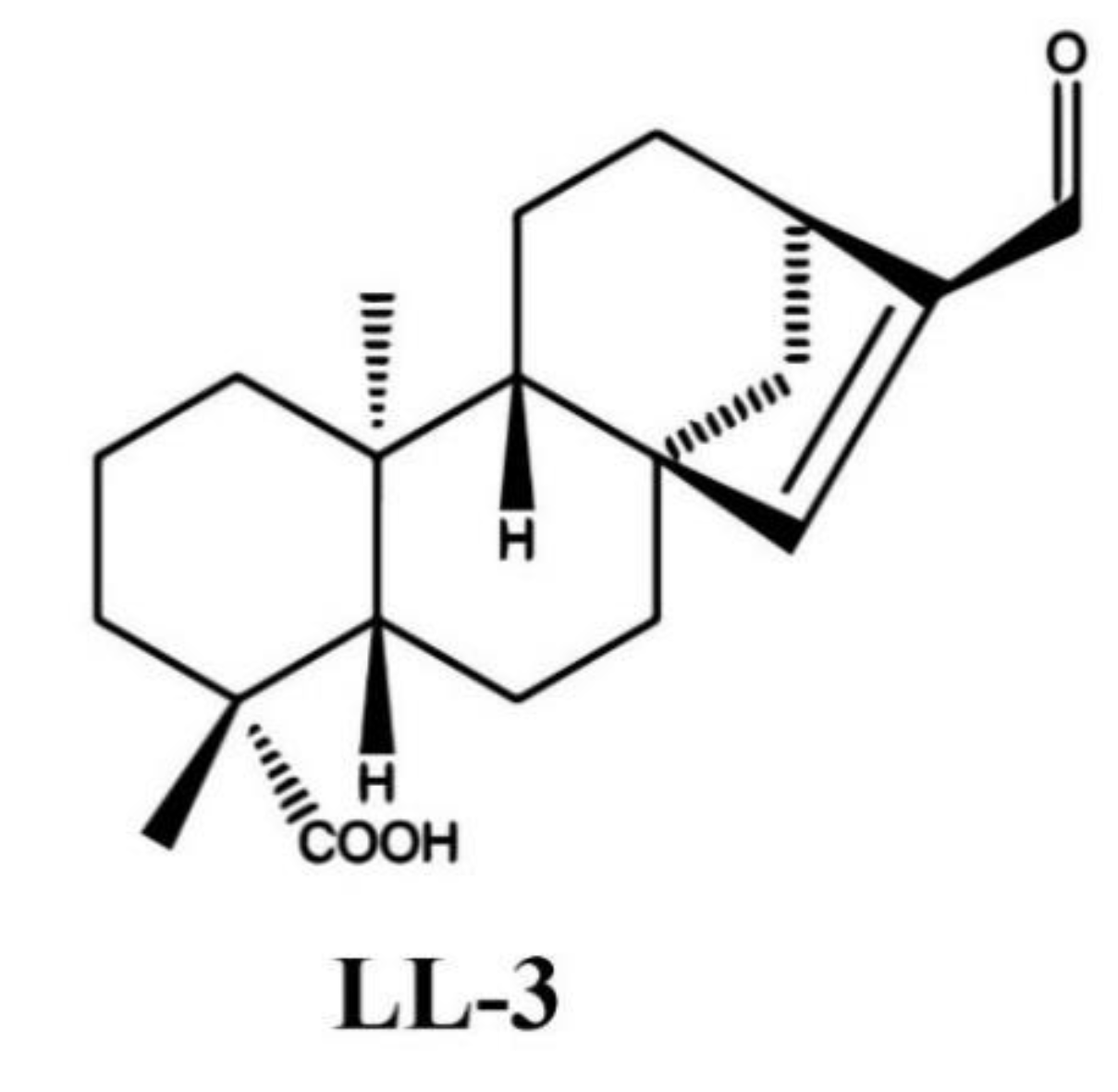{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
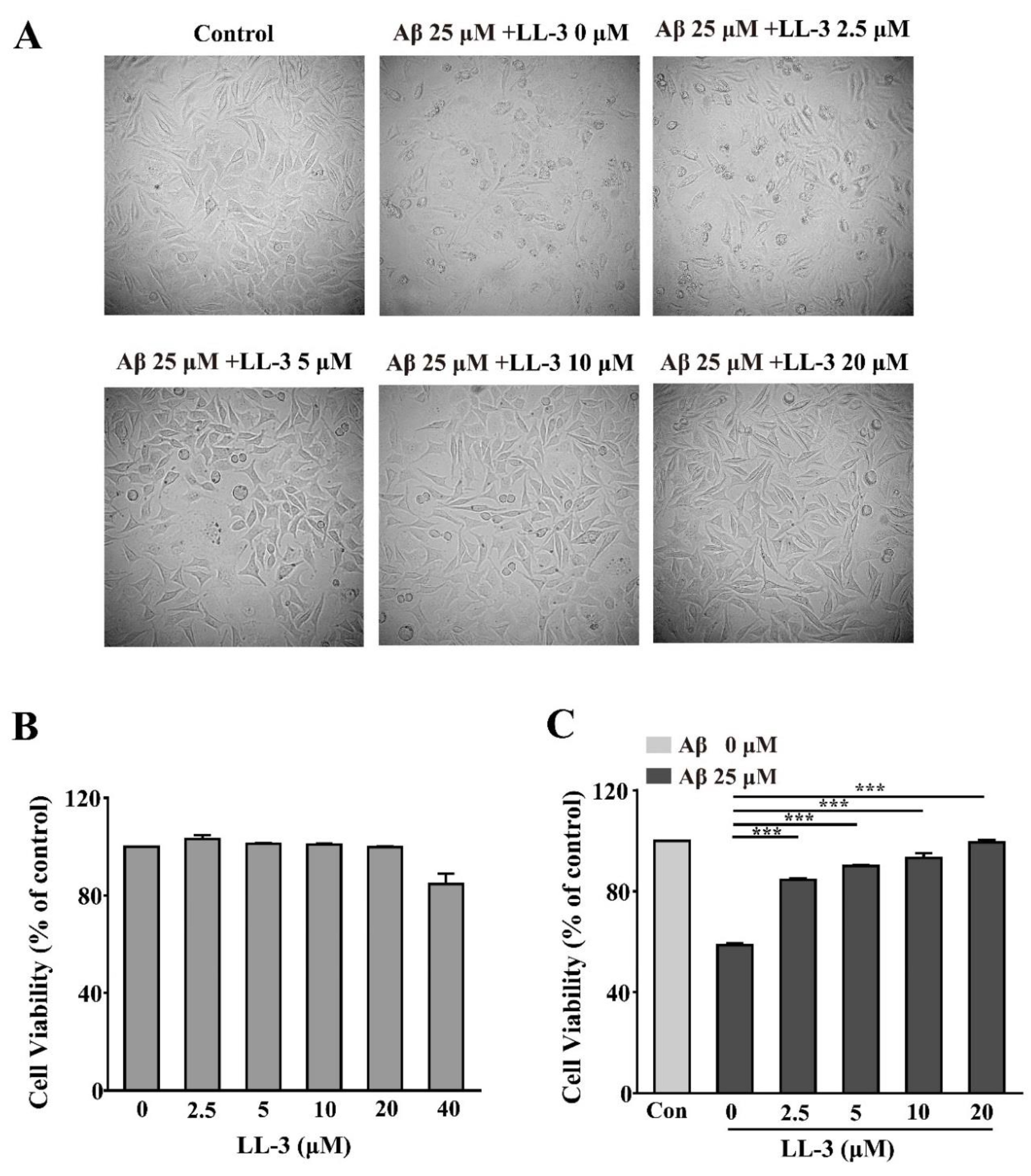
2.1. Effect of LL-3 and Amyloid Beta Peptide 25-35 on Cell Viability
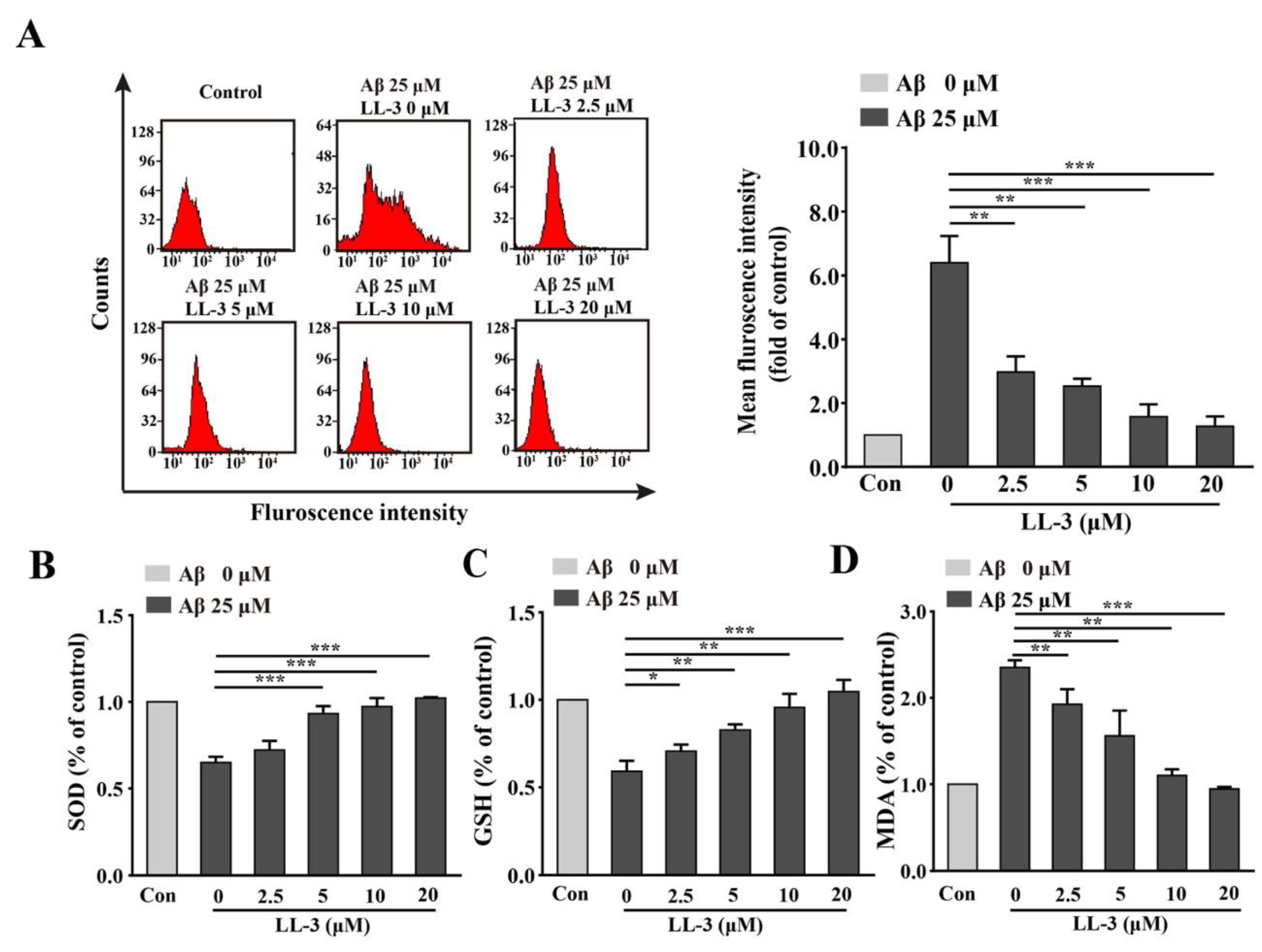
2.2. Effect of LL-3 on Aβ25-35-Induced Cellular Reactive Oxygen Species Generation
2.3. LL-3 Decreased Malondialdehyde and Increased Glutathione and Superoxide Dismutase Levels in Aβ25-35-Exposed Cells
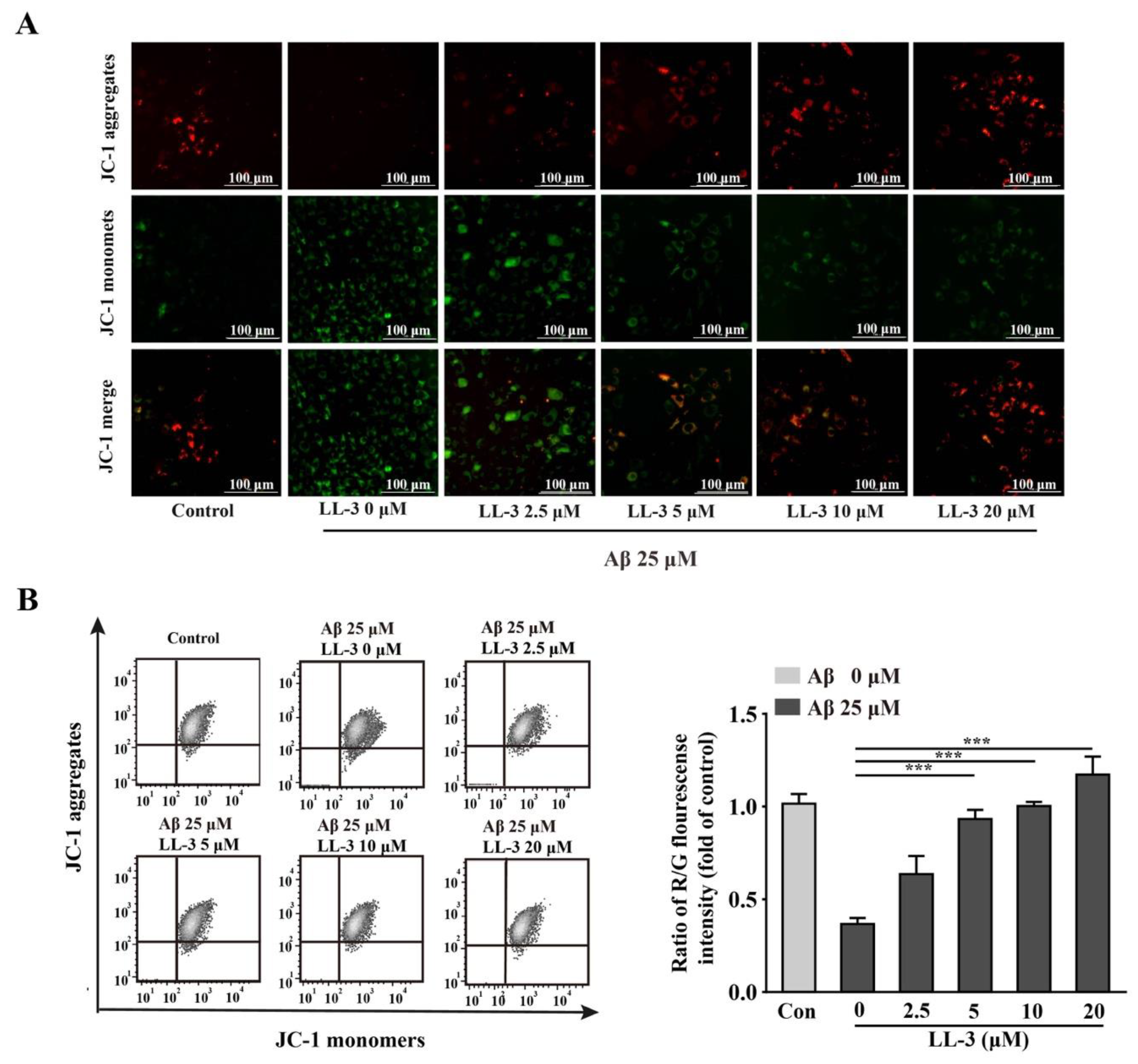
2.4. LL-3 Protected Aβ25-35-Treated Cells MMP from Damage
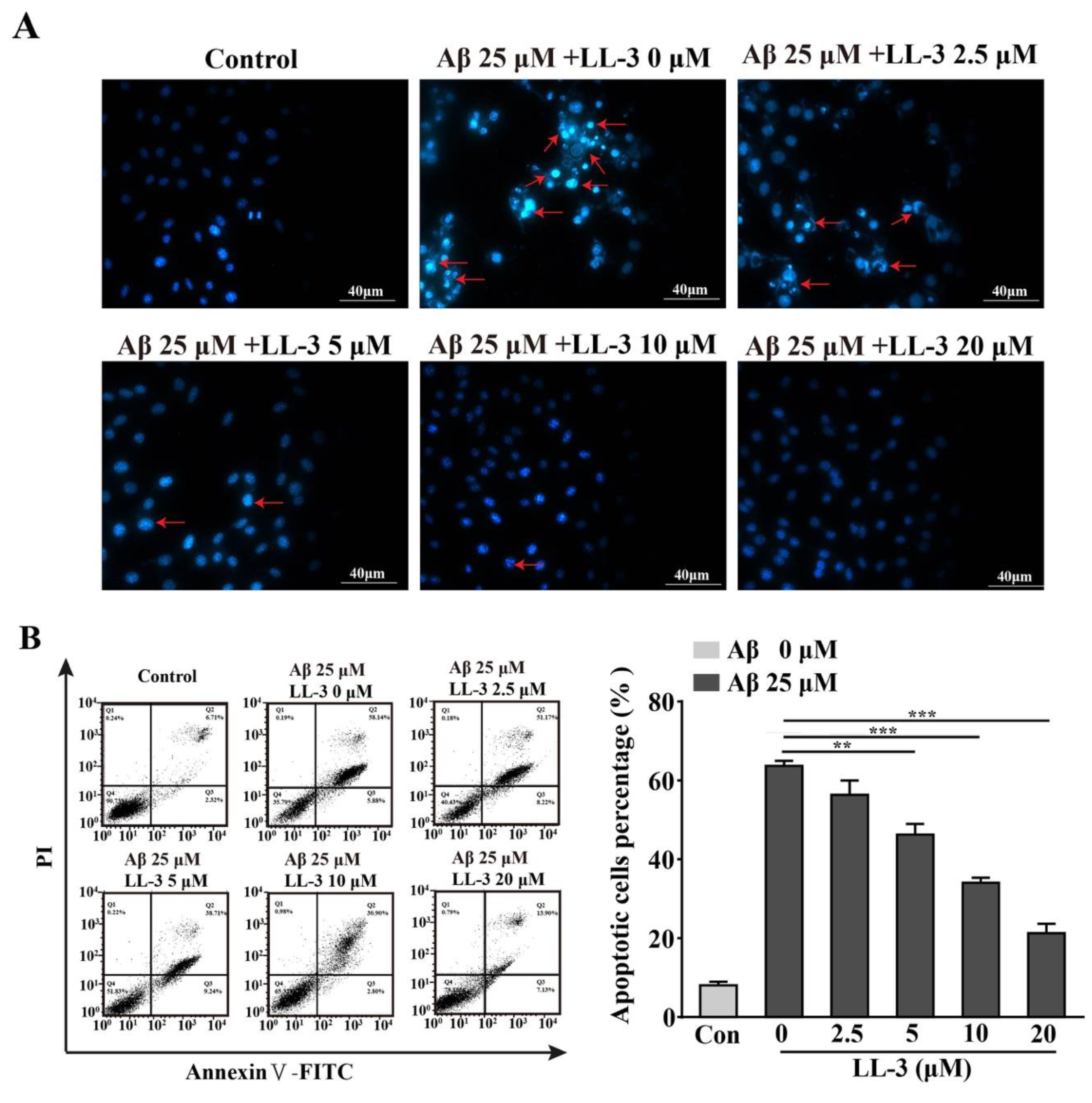
2.5. LL-3 Prevented Aβ25-35-Induced Apoptosis Adopted Diamidino-2-Phenylindole Staining
2.6. Effect of LL-3 on Aβ25-35-Induced Cellular Apoptosis
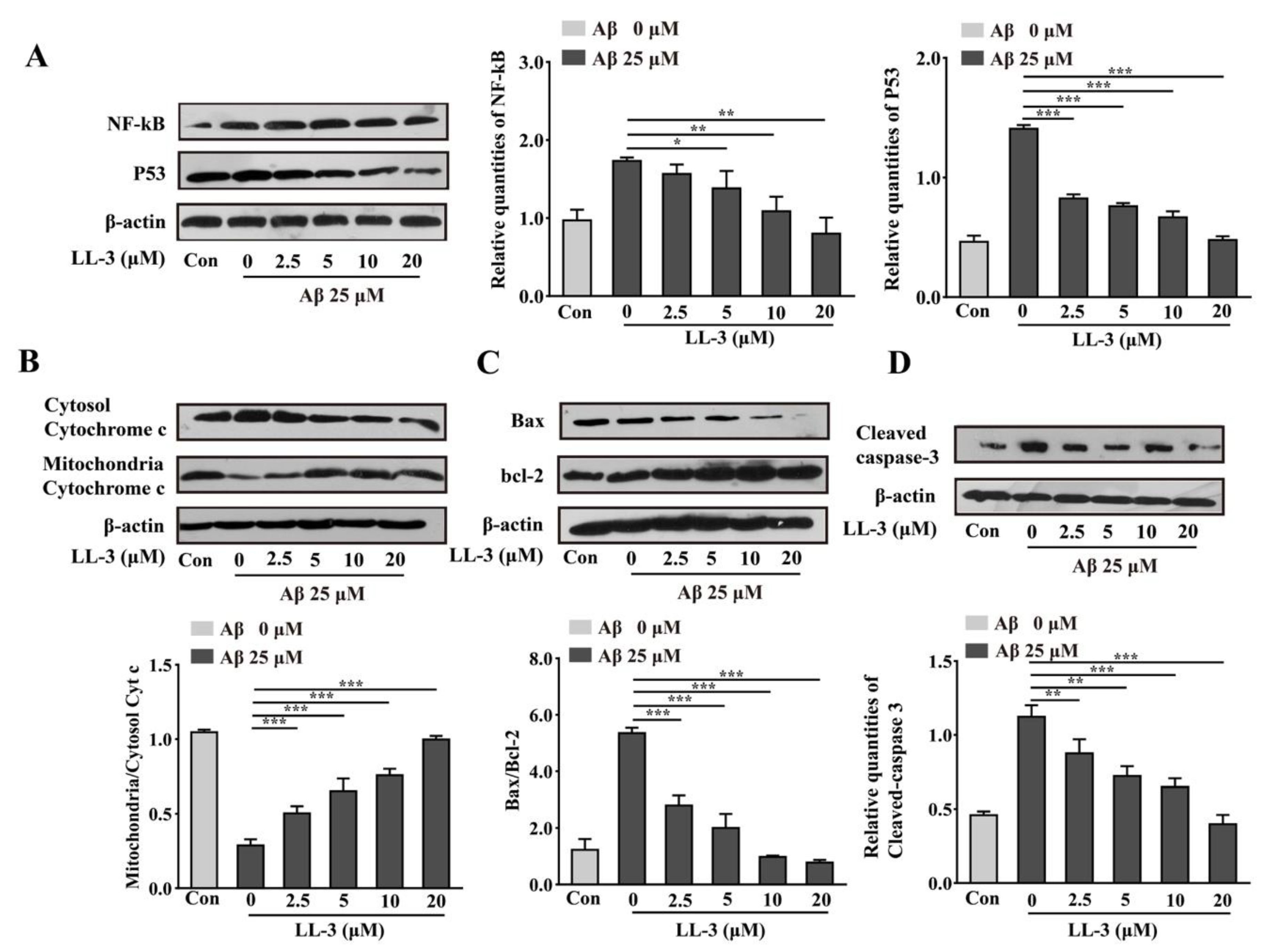
2.7. LL-3 Inhibited Aβ25-35-Treated Cells from Apoptosis Involved NF-κB and p53 Pathways
3. Discussion
4. Materials and Methods
4.1. Preparation of LL-3 Compound
4.2. Chemicals
4.3. Cell Culture
4.4. Preparation for Aβ25-35 Aggregates
4.5. Determination of Cell Cytotoxicity via MTT Assay
4.6. ROS Assay
4.7. Measurement of Intracellular SOD, GSH and MDA Levels
4.8. MMP Assay
4.9. Assessment of Morphological Alterations
4.10. Measurement of Cell Apoptosis
4.11. Western Blot Assay
4.12. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Arriagada, P.V.; Growdon, J.H.; Hedleywhyte, E.T.; Hyman, B.T. Neurofibrillary tangles but not senile plaques parallel duration and severity of alzheimers-disease. Neurology 1992, 42, 631–639. [Google Scholar] [CrossRef]
- Ghoshal, N.; Garcia-Sierra, F.; Wuu, J.; Leurgans, S.; Bennett, D.A.; Berry, R.W.; Binder, L.I. Tau conformational changes correspond to impairments of episodic memory in mild cognitive impairment and Alzheimer’s disease. Exp. Neurol. 2002, 177, 475–493. [Google Scholar] [CrossRef] [PubMed]
- Heo, H.J.; Cho, H.Y.; Hong, B.; Kim, H.K.; Heo, T.R.; Kim, E.K.; Kim, S.K.; Kim, C.J.; Shin, D.H. Ursolic acid of Origanum majorana L. reduces A beta-induced oxidative injury. Mol. Cells 2002, 13, 5–11. [Google Scholar] [PubMed]
- Lee, H.M.; Jang, J.Y.; Seong, Y.H. Protective effect of the aerial parts of Silybum marianum against amyloid β protein (25-35)-induced neuronal death in cultured neurons. J. Biomed. Transl. Res. 2016, 17, 109–114. [Google Scholar] [CrossRef]
- Varadarajan, S.; Kanski, J.; Aksenova, M.; Lauderback, C.; Butterfield, D.A. Different mechanisms of oxidative stress and neurotoxicity for Alzheimer’s A beta(1-42) and A beta(25-35). J. Am. Chem. Soc. 2001, 123, 5625–5631. [Google Scholar] [CrossRef] [PubMed]
- Arancibia, S.; Silhol, M.; Mouliere, F.; Meffre, J.; Hollinger, I.; Maurice, T.; Tapia-Arancibia, L. Protective effect of BDNF against beta-amyloid induced neurotoxicity in vitro and in vivo in rats. Neurobiol. Dis. 2008, 31, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.; Choi, J.M.; Lee, J.; Lee, M.H.; Lee, S.; Cho, E.J. Effects of Vegetable Oils with Different Fatty Acid Compositions on Cognition and Memory Ability in Abeta25-35-Induced Alzheimer’s Disease Mouse Model. J. Med. Food 2016, 19, 912–921. [Google Scholar] [CrossRef]
- Ahn, J.Y.; Kim, S.; Jung, S.E.; Ha, T.Y. Effect of Licorice (Glycyrrhiza uralensis Fisch) on Amyloid-beta-induced Neurotoxicity in PC12 Cells. Food Sci. Biotechnol. 2010, 19, 1391–1395. [Google Scholar] [CrossRef]
- Wang, Q.; Xu, Z.; Tang, J.; Sun, J.; Gao, J.; Wu, T.; Xiao, M. Voluntary exercise counteracts A beta 25-35-induced memory impairment in mice. Behav. Brain Res. 2013, 256, 618–625. [Google Scholar] [CrossRef]
- Choi, Y.S.; Kim, S.H. Mitochondrial Dysfunction and Apoptosis Related Gene Expression in Aβ25-35-Treated Human Neuroblastoma Cell Line, SK-N-SH. J. Korean Geriatr. Soc. 2009, 13, 141–151. [Google Scholar] [CrossRef]
- Yamada, K.; Nabeshima, T. Animal models of Alzheimer’s disease and evaluation of anti-dementia drugs. Pharmacol. Ther. 2000, 88, 93–113. [Google Scholar] [CrossRef]
- Rojas-Gutierrez, E.; Munoz-Arenas, G.; Trevino, S.; Espinosa, B.; Chavez, R.; Rojas, K.; Flores, G.; Diaz, A.; Guevara, J. Alzheimer’s disease and metabolic syndrome: A link from oxidative stress and inflammation to neurodegeneration. Synapse 2017, 71, e21990. [Google Scholar] [CrossRef]
- Coppede, F.; Stoccoro, A. Mitoepigenetics and Neurodegenerative Diseases. Front. Endocrinol. 2019, 10. [Google Scholar] [CrossRef]
- Hauptmann, S.; Scherping, I.; Droese, S.; Brandt, U.; Schulz, K.L.; Jendrach, M.; Leuner, K.; Eckert, A.; Mueller, W.E. Mitochondrial dysfunction: An early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol. Aging 2009, 30, 1574–1586. [Google Scholar] [CrossRef]
- Chao, D.T.; Korsmeyer, S.J. BCL-2 FAMILY: Regulators of cell death. Annu. Rev. Immunol. 1998, 16, 395–419. [Google Scholar] [CrossRef]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xu, Y.; Yan, J.; Zhao, X.; Sun, X.; Zhang, Y.; Guo, J.; Zhu, C. Acteoside protects human neuroblastoma SH-SY5Y cells against beta-amyloid-induced cell injury. Brain Res. 2009, 1283, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Y.; Xu, K.D.; Yan, M.; Wang, Y.P.; Zheng, X.X. Protective effects of galantamine against A beta-induced PC12 cell apoptosis by preventing mitochondrial dysfunction and endoplasmic reticulum stress. Neurochem. Int. 2010, 57, 588–599. [Google Scholar] [CrossRef] [PubMed]
- Morrison, R.S.; Kinoshita, Y.; Johnson, M.D.; Guo, W.Q.; Garden, G.A. p53-dependent cell death signaling in neurons. Neurochem. Res. 2003, 28, 15–27. [Google Scholar] [CrossRef]
- Youn, K.; Lee, S.; Jeong, W.-S.; Ho, C.-T.; Jun, M. Protective Role of Corilagin on Abeta25-35-Induced Neurotoxicity: Suppression of NF-kappaB Signaling Pathway. J. Med. Food 2016, 19, 901–911. [Google Scholar] [CrossRef]
- Picone, P.; Nuzzo, D.; Di Carlo, M. Ferulic Acid: A Natural Antioxidant Against Oxidative Stress Induced by Oligomeric A-beta on Sea Urchin Embryo. Biol. Bull. 2013, 224, 18–28. [Google Scholar] [CrossRef]
- Song, Y.S.; Park, H.J.; Kim, S.Y.; Lee, S.H.; Yoo, H.S.; Lee, H.S.; Lee, M.K.; Oh, K.W.; Kang, S.K.; Lee, S.E.; et al. Protective role of Bcl-2 on beta-amyloid-induced cell death of differentiated PC 12 cells: Reduction of NF-kB and p38 MAP kinase activation. Neurosci. Res. 2004, 49, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Longpre, F.; Garneau, P.; Christen, Y.; Ramassamy, C. Protection by EGb 761 against beta-amyloid-induced neurotoxicity: Involvement of NF-kappa B, SIRT1, and MAPKs pathways and inhibition of amyloid fibril formation. Free Radic. Biol. Med. 2006, 41, 1781–1794. [Google Scholar] [CrossRef] [PubMed]
- Markesbery, W.R. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic. Biol. Med. 1997, 23, 134–147. [Google Scholar] [CrossRef]
- Anekonda, T.S.; Reddy, P.H. Can herbs provide a new generation of drugs for treating Alzheimer’s disease? Brain Res. Rev. 2005, 50, 361–376. [Google Scholar] [CrossRef]
- Shen, T.; Qian, H.; Wang, Y.-D.; Li, H.-B.; Xie, W.-D. Terpenoids from the roots of Leontopodium longifolium and their inhibitory activity on NO production in RAW264.7 cells. Nat. Prod. Res. 2018. [Google Scholar] [CrossRef]
- An, W.L.; Cowburn, R.F.; Li, L.; Braak, H.; Alafuzoff, I.; Iqbal, K.; Iqbal, I.G.; Winblad, B.; Pei, J.J. Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer’s disease. Am. J. Pathol. 2003, 163, 591–607. [Google Scholar] [CrossRef]
- Hyun, K.J. Protective effects of Cirsium japonicum var. maackii against amyloid beta-induced neurotoxicity in C6 glial cells. Korean J. Agric. Sci. 2019, 46, 369–379. [Google Scholar]
- Chung, Y.C.; Kruyer, A.; Yao, Y.; Feierman, E.; Richards, A.; Strickland, S.; Norris, E.H. Hyperhomocysteinemia exacerbates Alzheimer’s disease pathology by way of the -amyloid fibrinogen interaction. J. Thromb. Haemost. 2016, 14, 1442–1452. [Google Scholar] [CrossRef]
- Watanabe, K.; Uemura, K.; Asada, M.; Maesako, M.; Akiyama, H.; Shimohama, S.; Takahashi, R.; Kinoshita, A. The participation of insulin-like growth factor-binding protein 3 released by astrocytes in the pathology of Alzheimer’s disease. Mol. Brain 2015, 8. [Google Scholar] [CrossRef]
- Tamagnini, F.; Novelia, J.; Kerrigan, T.L.; Brown, J.T.; Tsaneva-Atanasova, K.; Randall, A.D. Altered intrinsic excitability of hippocampal CA1 pyramidal neurons in aged PDAPP mice. Front. Cell. Neurosci. 2015, 9. [Google Scholar] [CrossRef] [PubMed]
- Janota, C.S.; Brites, D.; Lemere, C.A.; Brito, M.A. Glio-vascular changes during ageing in wild-type and Alzheimer’s disease-like APP/PS1 mice. Brain Res. 2015, 1620, 153–168. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liang, W.; Zhao, X.; Feng, J.; Song, F.; Pan, Y. Ursolic acid attenuates beta-amyloid-induced memory impairment in mice. Arq. Neuro-Psiquiatr. 2016, 74, 482–488. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Surgucheva, I.; Shestopalov, V.I.; Surguchov, A. Effect of gamma-Synuclein Silencing on Apoptotic Pathways in Retinal Ganglion Cells. J. Biol. Chem. 2008, 283, 36377–36385. [Google Scholar] [CrossRef]
- Gervais, F.G.; Xu, D.G.; Robertson, G.S.; Vaillancourt, J.P.; Zhu, Y.X.; Huang, J.Q.; LeBlanc, A.; Smith, D.; Rigby, M.; Shearman, M.S.; et al. Involvement of caspases in proteolytic cleavage of Alzheimer’s amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell 1999, 97, 395–406. [Google Scholar] [CrossRef]
- Li, J.-X.; Lin, C.-J.; Yang, X.-P.; Jia, Z.-J. New bisabolane sesquiterpenes and coumarin from Leontopodium longifolium. Chem. Biodivers. 2006, 3, 783–790. [Google Scholar] [CrossRef]
- Song, K.-S.; Jeong, W.-S.; Jun, M. Inhibition of beta-amyloid peptide-induced neurotoxicity by kaempferol 3-O-(6″-acetyl)-beta-glucopyranoside from butterbur (Petasites japonicus) leaves in B103 cells. Food Sci. Biotechnol. 2012, 21, 845–851. [Google Scholar] [CrossRef]
- Zhao, X.; Zeng, Z.; Gaur, U.; Fang, J.; Peng, T.; Li, S.; Zheng, W. Metformin protects PC12 cells and hippocampal neurons from H2O (2)-induced oxidative damage through activation of AMPK pathway. J. Cell. Physiol. 2019, 234, 16619–16629. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Li, J.; Liu, C.; Song, C.; Li, P.; Yin, F.; Xiao, Y.; Li, J.; Jiang, W.; Zong, A.; et al. Protective effects of low molecular weight chondroitin sulfate on amyloid beta (a beta)-induced damage in vitro and in vivo. Neuroscience 2015, 305, 169–182. [Google Scholar] [CrossRef]
- Liu, J.-F.; Yan, X.-D.; Qi, L.-S.; Li, L.; Hu, G.-Y.; Li, P.; Zhao, G. Ginsenoside Rd attenuates A beta(25-35)-induced oxidative stress and apoptosis in primary cultured hippocampal neurons. Chem. Biol. Interact. 2015, 239, 12–18. [Google Scholar] [CrossRef]
- Jomova, K.; Valko, M. Advances in metal-induced oxidative stress and human disease. Toxicology 2011, 283, 65–87. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Zhao, X.; Lin, S.; Liu, F.; Ma, J.; Han, Z.; Jia, F.; Xie, W.; Zhang, Q.; Li, X. Neuroprotective Effect of ent-Kaur-15-en-17-al-18-oic Acid on Amyloid Beta Peptide-Induced Oxidative Apoptosis in Alzheimer’s Disease. Molecules 2020, 25, 142. https://doi.org/10.3390/molecules25010142
Zhang C, Zhao X, Lin S, Liu F, Ma J, Han Z, Jia F, Xie W, Zhang Q, Li X. Neuroprotective Effect of ent-Kaur-15-en-17-al-18-oic Acid on Amyloid Beta Peptide-Induced Oxidative Apoptosis in Alzheimer’s Disease. Molecules. 2020; 25(1):142. https://doi.org/10.3390/molecules25010142
Chicago/Turabian StyleZhang, Caiyun, Xingming Zhao, Shiqi Lin, Fangyuan Liu, Jiahui Ma, Zhuo Han, Fujuan Jia, Weidong Xie, Qian Zhang, and Xia Li. 2020. "Neuroprotective Effect of ent-Kaur-15-en-17-al-18-oic Acid on Amyloid Beta Peptide-Induced Oxidative Apoptosis in Alzheimer’s Disease" Molecules 25, no. 1: 142. https://doi.org/10.3390/molecules25010142
APA StyleZhang, C., Zhao, X., Lin, S., Liu, F., Ma, J., Han, Z., Jia, F., Xie, W., Zhang, Q., & Li, X. (2020). Neuroprotective Effect of ent-Kaur-15-en-17-al-18-oic Acid on Amyloid Beta Peptide-Induced Oxidative Apoptosis in Alzheimer’s Disease. Molecules, 25(1), 142. https://doi.org/10.3390/molecules25010142