Targeting Caspase 8: Using Structural and Ligand-Based Approaches to Identify Potential Leads for the Treatment of Multi-Neurodegenerative Diseases

 ,
, 
 ,
,  ,
,
Abstract
1. Introduction
2. Results and Discussion
2.1. Pharmacophore Modeling: 3D Pharmacophore Generation
2.2. Pharmacophore-Based Virtual Screening (PBVS)
2.3. Molecular Dynamics Simulation and Molecular Docking
2.4. Thermodynamics Analysis
2.5. Physical Properties of Ligands
3. Materials and Methods
3.1. Common Feature Pharmacophore Model
3.2. Common Feature Pharmacophore Generation
3.3. Pharmacophore-Based Virtual Screening (PBVS)
3.4. Molecular Docking Studies
3.5. Molecular Dynamics Simulation
3.6. Thermodynamic Analysis
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hebert, L.E.; Beckett, L.A.; Scherr, P.A.; Evans, D.A. Annual incidence of Alzheimer disease in the United States projected to the years 2000 through 2050. Alzheimer Dis. Assoc. Disord 2001, 15, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, K.; Baig, M.H.; Mushtaq, G.; Kamal, M.A.; Greig, N.H.; Choi, I. Commonalities in Biological Pathways, Genetics, and Cellular Mechanism between Alzheimer Disease and Other Neurodegenerative Diseases: An In Silico-Updated Overview. Curr. Alzheimer Res. 2017, 14, 1190–1197. [Google Scholar] [CrossRef]
- Kanazawa, I. How do neurons die in neurodegenerative diseases? Trends Mol. Med. 2001, 7, 339–344. [Google Scholar] [CrossRef]
- Fleischer, A.; Ghadiri, A.; Dessauge, F.; Duhamel, M.; Rebollo, M.P.; Alvarez-Franco, F.; Rebollo, A. Modulating apoptosis as a target for effective therapy. Mol. Immunol. 2006, 43, 1065–1079. [Google Scholar] [CrossRef]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, L.; Passer, B.J.; Tabaton, M.; Ganjei, J.K.; D’Adamio, L. Alternative, non-secretase processing of Alzheimer’s beta-amyloid precursor protein during apoptosis by caspase-6 and -8. J. Biol. Chem. 1999, 274, 21011–21016. [Google Scholar] [CrossRef] [PubMed]
- Mohr, A.; Zwacka, R.M. In situ trapping of initiator caspases reveals intermediate surprises. Cell Biol. Int. 2007, 31, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Grutter, M.G. Caspases: Key players in programmed cell death. Curr. Opin. Struct. Biol. 2000, 10, 649–655. [Google Scholar] [CrossRef]
- Fuentes-Prior, P.; Salvesen, G.S. The protein structures that shape caspase activity, specificity, activation and inhibition. Biochem J. 2004, 384, 201–232. [Google Scholar] [CrossRef] [PubMed]
- Pop, C.; Salvesen, G.S. Human caspases: Activation, specificity, and regulation. J. Biol. Chem. 2009, 284, 21777–21781. [Google Scholar] [CrossRef] [PubMed]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect Biol. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. Caspases in Alzheimer’s disease. In Neurodegenerative Diseases; InTech: Rijeka, Croatia, 2013. [Google Scholar]
- Hanisch, U.K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef]
- Burguillos, M.A.; Deierborg, T.; Kavanagh, E.; Persson, A.; Hajji, N.; Garcia-Quintanilla, A.; Cano, J.; Brundin, P.; Englund, E.; Venero, J.L.; et al. Caspase signalling controls microglia activation and neurotoxicity. Nature 2011, 472, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Podbielska, M.; Das, A.; Smith, A.W.; Chauhan, A.; Ray, S.K.; Inoue, J.; Azuma, M.; Nozaki, K.; Hogan, E.L.; Banik, N.L. Neuron-microglia interaction induced bi-directional cytotoxicity associated with calpain activation. J. Neurochem. 2016, 139, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Rehker, J.; Rodhe, J.; Nesbitt, R.R.; Boyle, E.A.; Martin, B.K.; Lord, J.; Karaca, I.; Naj, A.; Jessen, F.; Helisalmi, S.; et al. Caspase-8, association with Alzheimer’s Disease and functional analysis of rare variants. PLoS ONE 2017, 12, e0185777. [Google Scholar] [CrossRef]
- Friedlander, R.M. Apoptosis and caspases in neurodegenerative diseases. N. Engl. J. Med. 2003, 348, 1365–1375. [Google Scholar] [CrossRef]
- Howley, B.; Fearnhead, H.O. Caspases as therapeutic targets. J. Cell Mol. Med. 2008, 12, 1502–1516. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Peairs, J.J.; Tano, R.; Zhang, N.; Tyrell, J.; Jaffe, G.J. Caspase-8-mediated apoptosis in human RPE cells. Invest. Ophthalmol. Vis. Sci. 2007, 48, 3341–3349. [Google Scholar] [CrossRef] [PubMed]
- Rohn, T.T.; Head, E.; Nesse, W.H.; Cotman, C.W.; Cribbs, D.H. Activation of caspase-8 in the Alzheimer’s disease brain. Neurobiol. Dis. 2001, 8, 1006–1016. [Google Scholar] [CrossRef] [PubMed]
- Fricker, M.; Vilalta, A.; Tolkovsky, A.M.; Brown, G.C. Caspase inhibitors protect neurons by enabling selective necroptosis of inflamed microglia. J. Biol. Chem. 2013, 288, 9145–9152. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, K.; Khan, S.; Adil, M.; Saeed, M.; Srivastava, A.K. Structure based molecular inhibition of Caspase-8 for treatment of multi-neurodegenerative diseases using known natural compounds. Bioinformation 2014, 10, 191–195. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sanchez, I.; Xu, C.J.; Juo, P.; Kakizaka, A.; Blenis, J.; Yuan, J. Caspase-8 is required for cell death induced by expanded polyglutamine repeats. Neuron 1999, 22, 623–633. [Google Scholar] [CrossRef]
- Morrice, J.R.; Gregory-Evans, C.Y.; Shaw, C.A. Necroptosis in amyotrophic lateral sclerosis and other neurological disorders. Biochim. Biophys. Acta. Mol. Basis. Dis. 2017, 1863, 347–353. [Google Scholar] [CrossRef]
- Berger, A.B.; Sexton, K.B.; Bogyo, M. Commonly used caspase inhibitors designed based on substrate specificity profiles lack selectivity. Cell Res. 2006, 16, 961–963. [Google Scholar] [CrossRef] [PubMed]
- Baig, M.H.; Ahmad, K.; Roy, S.; Ashraf, J.M.; Adil, M.; Siddiqui, M.H.; Khan, S.; Kamal, M.A.; Provaznik, I.; Choi, I. Computer Aided Drug Design: Success and Limitations. Curr. Pharm. Des. 2016, 22, 572–581. [Google Scholar] [CrossRef]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein–ligand docking using GOLD. Proteins: Struct. Funct. Bioinform. 2003, 52, 609–623. [Google Scholar] [CrossRef]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. Ccp4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Reszka, P.; Schulz, R.; Methling, K.; Lalk, M.; Bednarski, P.J. Synthesis, enzymatic evaluation, and docking studies of fluorogenic caspase 8 tetrapeptide substrates. ChemMedChem 2010, 5, 103–117. [Google Scholar] [CrossRef]
- Tian, C.; Asghar, S.; Wu, Y.; Chen, Z.; Jin, X.; Yin, L.; Huang, L.; Ping, Q.; Xiao, Y. Improving intestinal absorption and oral bioavailability of curcumin via taurocholic acid-modified nanostructured lipid carriers. Int. J. Nanomed. 2017, 12, 7897–7911. [Google Scholar] [CrossRef]
- Balaramnavar, V.M.; Srivastava, R.; Rahuja, N.; Gupta, S.; Rawat, A.K.; Varshney, S.; Chandasana, H.; Chhonker, Y.S.; Doharey, P.K.; Kumar, S.; et al. Identification of novel PTP1B inhibitors by pharmacophore based virtual screening, scaffold hopping and docking. Eur. J. Med. Chem. 2014, 87, 578–594. [Google Scholar] [CrossRef]
- Ahmad, K.; Balaramnavar, V.M.; Baig, M.H.; Srivastava, A.K.; Khan, S.; Kamal, M.A. Identification of potent caspase-3 inhibitors for treatment of multi- neurodegenerative diseases using pharmacophore modeling and docking approaches. Cns Neurol. Disord. Drug Targets 2014, 13, 1346–1353. [Google Scholar] [CrossRef]
- Sprague, P.W. Automated chemical hypothesis generation and database searching with Catalyst®. Perspect. Drug Discov. Des. 1995, 3, 1–20. [Google Scholar] [CrossRef]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.A.; Karplus, M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Thomsen, R.; Christensen, M.H. MolDock: A new technique for high-accuracy molecular docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef] [PubMed]
- Gehlhaar, D.K.; Bouzida, D.; Rejto, P.A. Fully automated and rapid flexible docking of inhibitors covalently bound to serine proteases. In Proceedings of the International Conference on Evolutionary Programming, San Diego, CA, USA, 25–27 March 1998; pp. 449–461. [Google Scholar]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; van Gunsteren, W.F. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef] [PubMed]
- Toukan, K.; Rahman, A. Molecular-dynamics study of atomic motions in water. Phys. Rev. B Condens Matter 1985, 31, 2643–2648. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Baker, M.A.; Whiley, P.A.; Arjomand, A.; Ludeman, J.; Wong, C.; Jans, D.A.; Loveland, K.L. Towards delineation of a developmental α-importome in the mammalian male germline. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Schüttelkopf, A.W.; Van Aalten, D.M. PRODRG: A tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr. Sect. D: Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar]
- Lee, S.; Chang, G.; Lee, I.; Chung, J.; Sung, K.; No, K. The PreADME: Pc-based program for batch prediction of adme properties. EuroQSAR 2004, 9, 5–10. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
Sample Availability: Not available. |





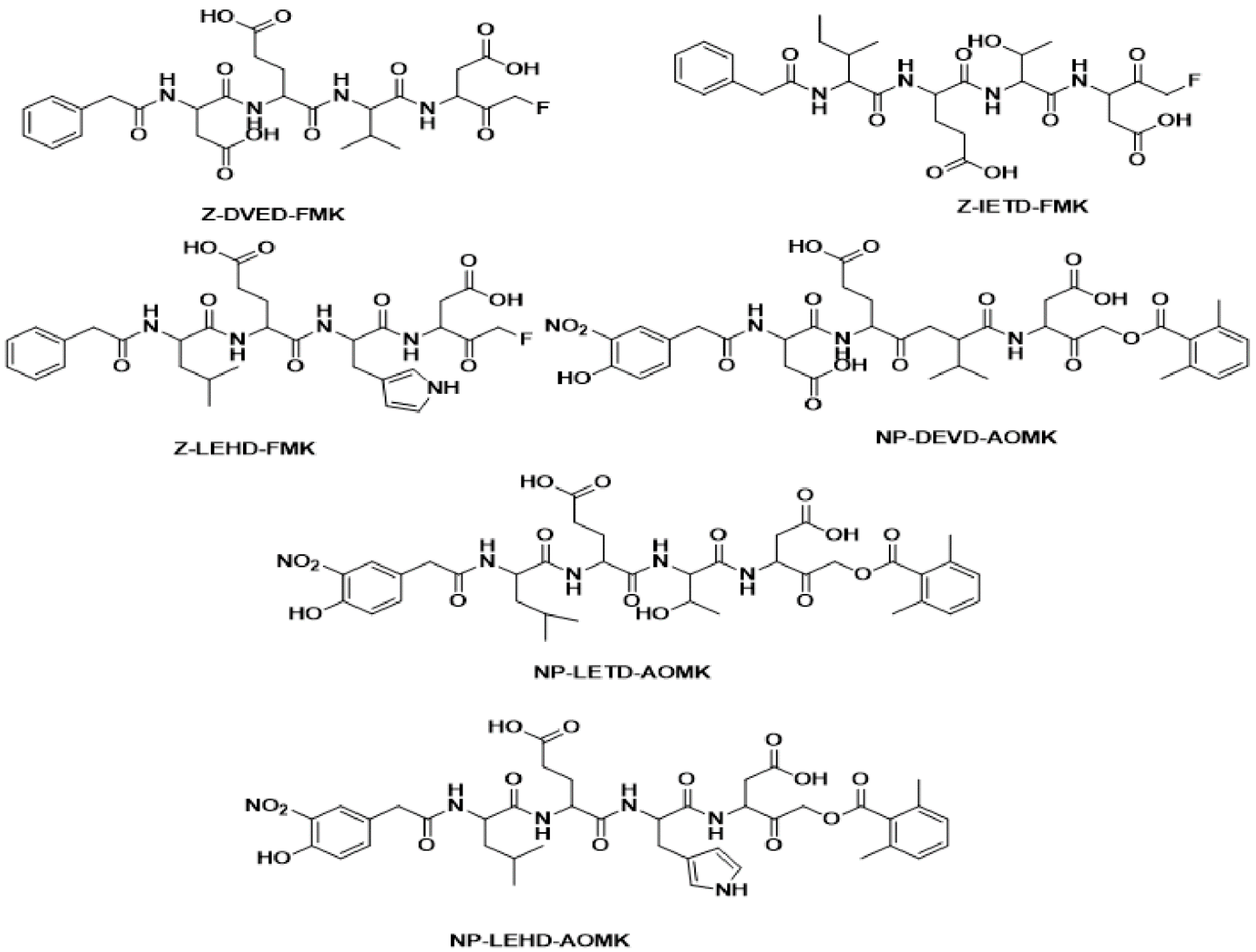{kind=link}
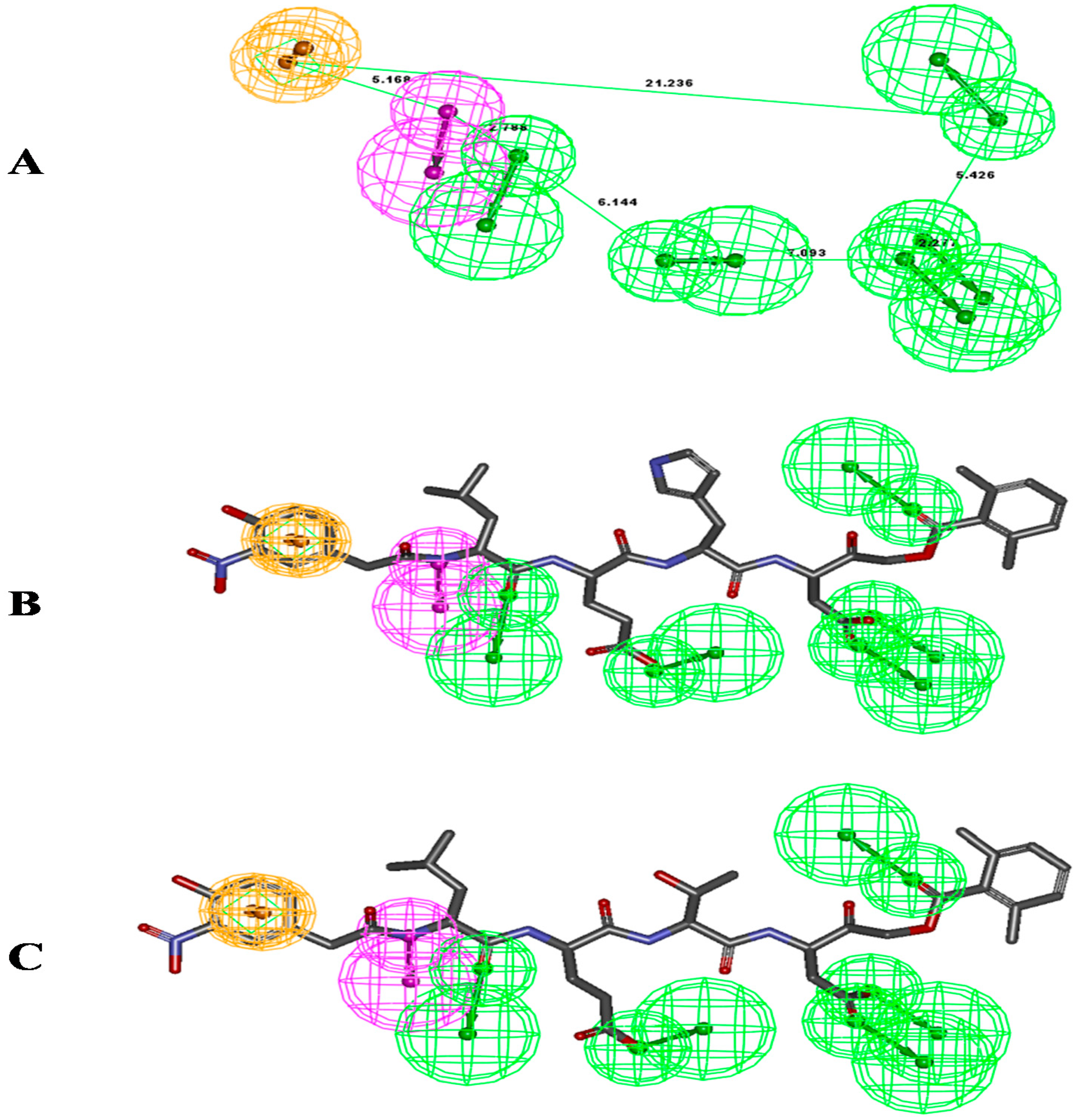{kind=link}
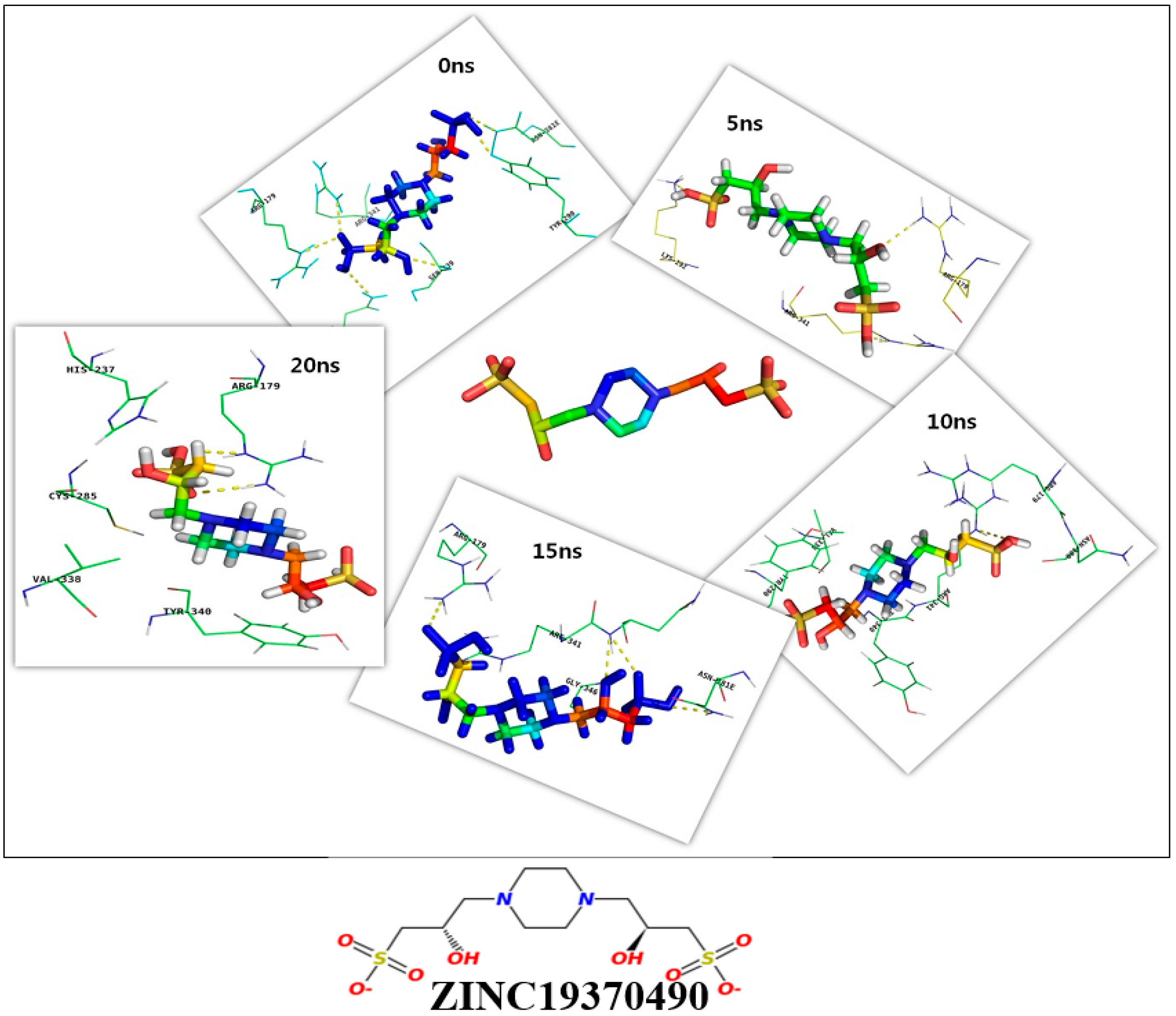{kind=link}
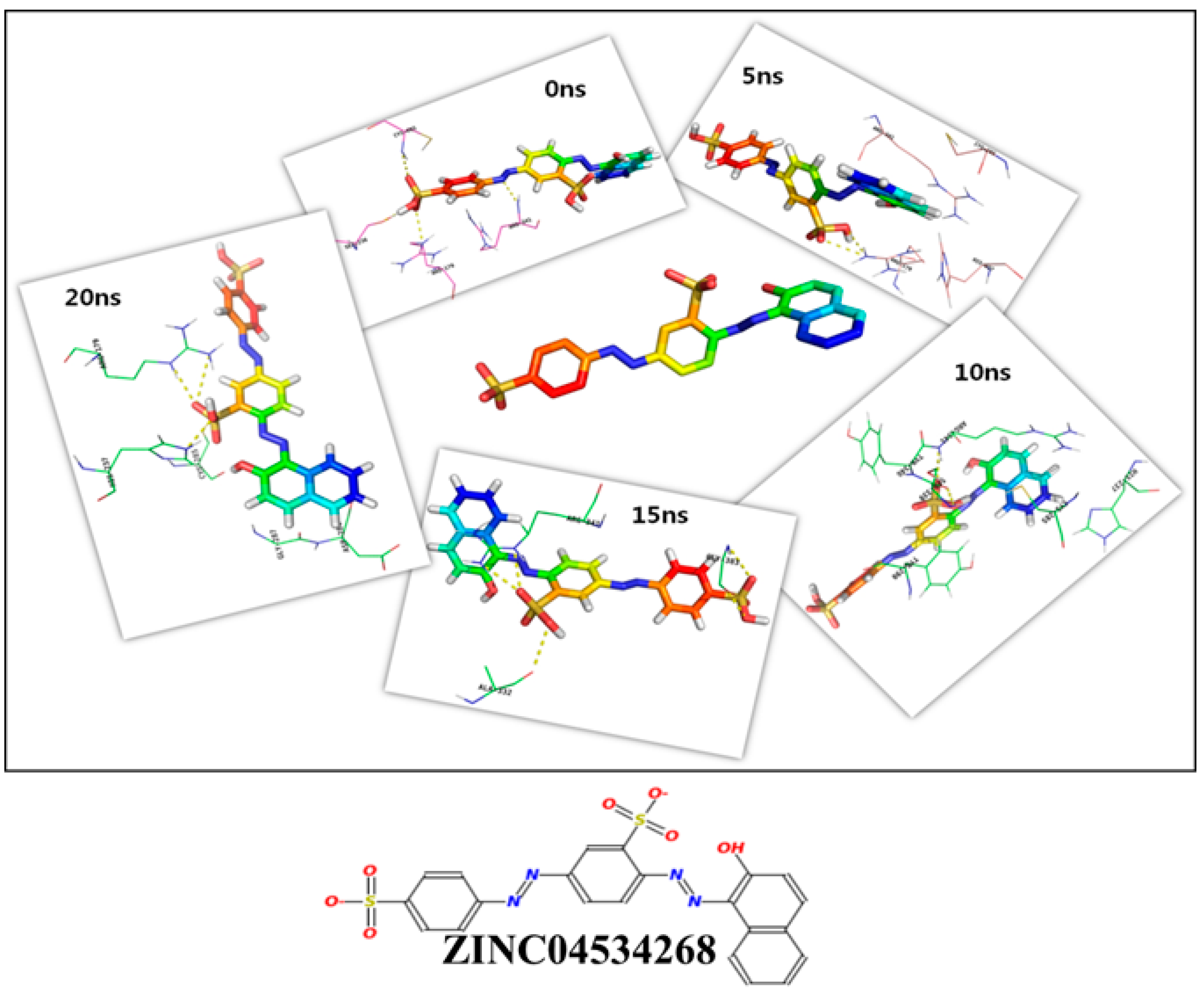{kind=link}
| 9 | Features | Rank | Direct Hit | Partial Hit | Max Fit |
|---|---|---|---|---|---|
| 01 | RDHHHHH | 152.441 | 111111 | 000000 | 7 |
| 02 | RZHHHHH | 152.286 | 111111 | 000000 | 7 |
| 03 | RZHHHHH | 152.086 | 111111 | 000000 | 7 |
| 04 | RZHHHHH | 152.058 | 111111 | 000000 | 7 |
| 05 | RZHHHHH | 151.770 | 111111 | 000000 | 7 |
| 06 | RZHHHHH | 151.719 | 111111 | 000000 | 7 |
| 07 | RZHHHHH | 151.511 | 111111 | 000000 | 7 |
| 08 | RZHHHHH | 151.147 | 111111 | 000000 | 7 |
| 09 | RZHHHHH | 151.017 | 111111 | 000000 | 7 |
| 10 | RZHHHHH | 150.863 | 111111 | 000000 | 7 |
| Ligand | MolDock Score | Rerank Score | Docking Score | Similarity | Fit Value |
|---|---|---|---|---|---|
| ZINC00959167 | −79.5965 | −68.8523 | −328.308 | −247.343 | 4.999 |
| ZINC01234548 | −109.655 | −94.2736 | −362.447 | −248.796 | 4.79982 |
| ZINC09212678 | −80.7085 | −69.2156 | −328.066 | −246.039 | 4.89982 |
| ZINC01301026 | −72.0231 | −58.011 | −342.572 | −265.101 | 4.99982 |
| ZINC03830398 | −76.8326 | −60.6093 | −345.951 | −270.038 | 4.922 |
| ZINC06143162 | −88.6888 | −65.8386 | −339.951 | −250.383 | 4.4982 |
| ZINC14671560 | −97.5435 | −77.8008 | −377.247 | −280.961 | 4.482 |
| ZINC04534268 | −116.417 | −94.1023 | −440.055 | −309.038 | 4.9482 |
| ZINC19370490 | −138.402 | −117.46 | −480.995 | −329.518 | 4.682 |
| ZINC02775438 | −91.4008 | −67.6985 | −406.978 | −311.933 | 4.621 |
| Time Period | Gold Fitness Score | Residues | |
|---|---|---|---|
| Hydrogen Bond | Hydrophobic Interaction | ||
| 0 ns | 53.78 | R179, Q283, Y290, S339, R341, N381 | R179, C285, S339, Y340, R341 |
| 5 ns | 53.30 | R179, K292, R341 | R179, Y290, K292, Y340, R341, P343 |
| 10 ns | 54.06 | R341 | R179, N180, Y290, V338, Y340, R341 |
| 15 ns | 58.12 | R179, R341, N342, G346, N381E | C285, T333, V338, S339, Y340, R341, E345, G346, T347, W348, N381E, Q385 |
| 20 ns | 51.50 | R179 | H237, C285, V338, Y340 |
| Time Period | Gold Fitness Score | Residues | |
|---|---|---|---|
| Hydrogen Bond | Hydrophobic Interaction | ||
| 0 ns | 60.48 | R179, S236, C285, R341 | H237, C285, S339, Y340, R341, D381B, K381D, |
| 5 ns | 52.37 | R179 | R179, H237, C285, V338, R341, P343, N381E, |
| 10 ns | 55.96 | S339, R341 | R179, H237, C285, Y290, K292, S339, Y340, R341 |
| 15 ns | 57.52 | A332, R341, G383 | R179, N180, G181, D185, Q283, Y340, R341, G346, T347, D381A, N381E, G383, Q385, |
| 20 ns | 58.68 | R179, H237 | S175J, H237, C285, G287, D288, Y290, V338, Y340, R341, P343, |
| Ligand | Electrostatic | Polar Solvation | Van der Waal | SASA | SAV | Binding Free Energy (KJ/mol) |
|---|---|---|---|---|---|---|
| Z-IETD-FMK | −81.44 (33.4) | −4.98 (4.5) | −121.622 (15.2) | −14.52 (1.25) | −159.2 (9.5) | −382.524 (29.45) |
| Z-IETD-FMK | −155.45 (30.5) | 58.326 (10.5) | −140.4 (8.45) | −17.90 (2.58) | −198.5 (6.47) | −453.924 (45.98) |
| Z-LEHD-FMK | −58.45 (21.25) | 212.670 (12.36) | −108.45 (19.585) | −19.07 (2.04) | −169.7 (7.8) | −143.08 (29.704) |
| NP-DEVD_AOMK | −53.11 (48.90) | 80.37 (4.80) | −109. 48 (5.08) | −13.09 (1.08) | −117.9 (6.24) | −106.99 (34.19) |
| NP-LETD-AOMK | −145.48 (46.2) | 254.051 (15.66) | −314. 94 (18.56) | −17.6 (1.69) | −178.56 (9.4) | −401.93 (26.78) |
| NP-LEHD AOMK | −58.67 (45.09) | 198.07 (12.360) | −265.98 (14.8) | −11.04 (2.05) | −164.5 (2.63) | −302.12 (41.29) |
| ZINC19370490 | −154.45 (4.22) | 109.45 (29.07) | −190.07 (26.6) | −19.05 (2.44) | −180.56 (4.5) | −434.68 (39.45) |
| ZINC04534268 | −225.64 (79.82) | 76.697 (9.14) | −217.56 (78.2) | −17.89 (1.99) | −88.67 (2.6) | −484.063 (31.08) |
| Properties | ZINC04534268 | ZINC19370490 |
|---|---|---|
| GI absorption | Low | Low |
| BBB permeant | No | No |
| P-gp substrate | No | Yes |
| CYP1A2 inhibitor | No | No |
| CYP2C19 inhibitor | No | No |
| CYP2C9 inhibitor | No | No |
| CYP2D6 inhibitor | No | No |
| CYP3A4 inhibitor | No | No |
| LogKp(skin permeation) | −6.67 cm/s | −14.17 cm/s |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, K.; Balaramnavar, V.M.; Chaturvedi, N.; Khan, S.; Haque, S.; Lee, Y.-H.; Choi, I. Targeting Caspase 8: Using Structural and Ligand-Based Approaches to Identify Potential Leads for the Treatment of Multi-Neurodegenerative Diseases. Molecules 2019, 24, 1827. https://doi.org/10.3390/molecules24091827
Ahmad K, Balaramnavar VM, Chaturvedi N, Khan S, Haque S, Lee Y-H, Choi I. Targeting Caspase 8: Using Structural and Ligand-Based Approaches to Identify Potential Leads for the Treatment of Multi-Neurodegenerative Diseases. Molecules. 2019; 24(9):1827. https://doi.org/10.3390/molecules24091827
Chicago/Turabian StyleAhmad, Khurshid, Vishal M. Balaramnavar, Navaneet Chaturvedi, Saif Khan, Shafiul Haque, Yong-Ho Lee, and Inho Choi. 2019. "Targeting Caspase 8: Using Structural and Ligand-Based Approaches to Identify Potential Leads for the Treatment of Multi-Neurodegenerative Diseases" Molecules 24, no. 9: 1827. https://doi.org/10.3390/molecules24091827
APA StyleAhmad, K., Balaramnavar, V. M., Chaturvedi, N., Khan, S., Haque, S., Lee, Y.-H., & Choi, I. (2019). Targeting Caspase 8: Using Structural and Ligand-Based Approaches to Identify Potential Leads for the Treatment of Multi-Neurodegenerative Diseases. Molecules, 24(9), 1827. https://doi.org/10.3390/molecules24091827