Lignin Modification Supported by DFT-Based Theoretical Study as a Way to Produce Competitive Natural Antioxidants

 , , ,
, , ,
Abstract
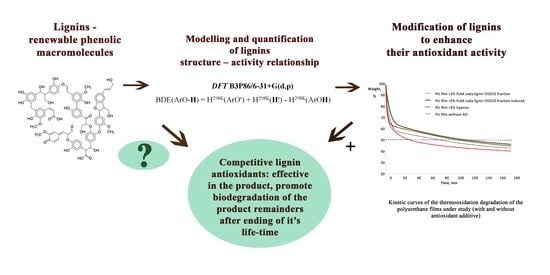
1. Introduction
2. Results and Discussion
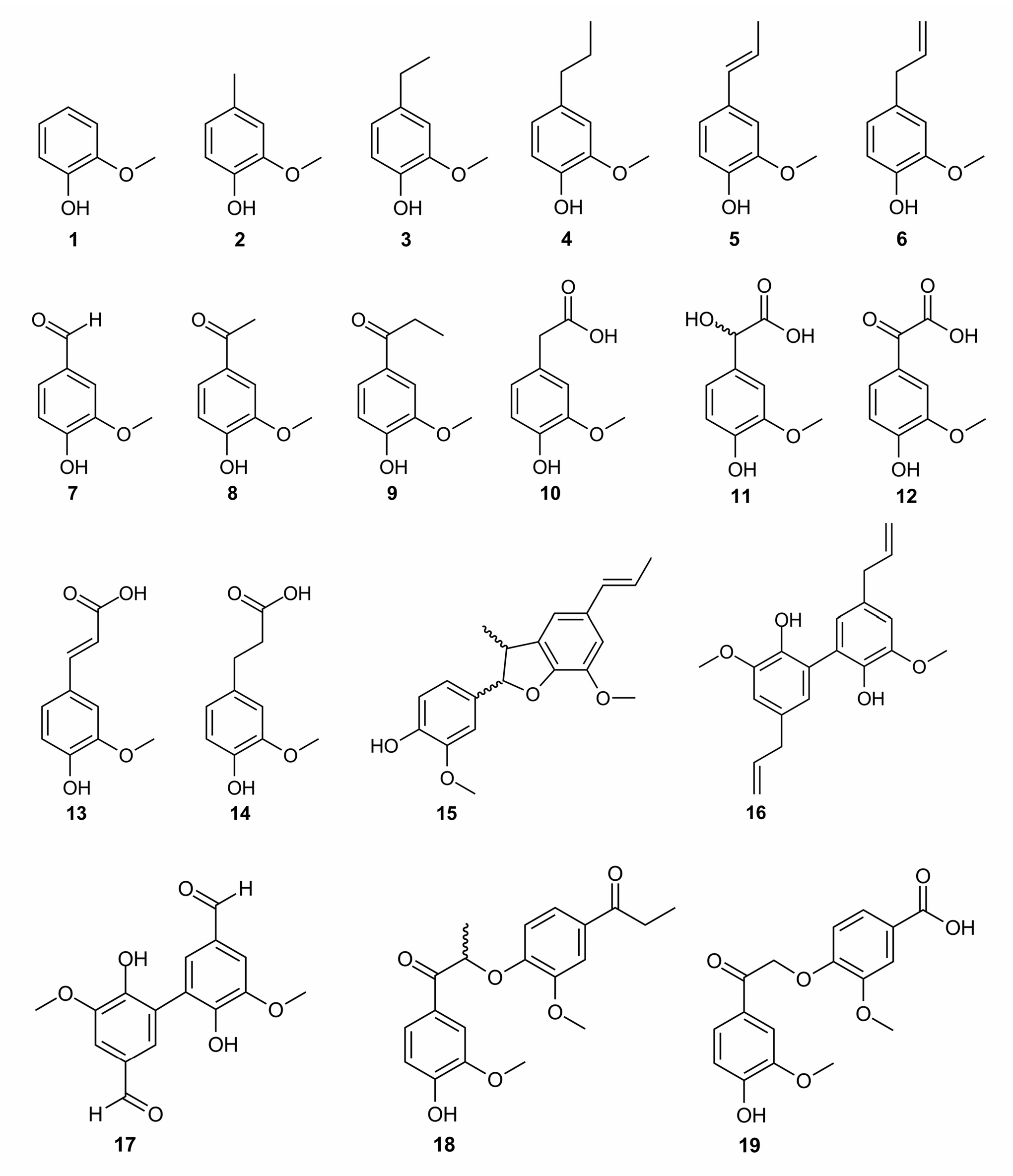
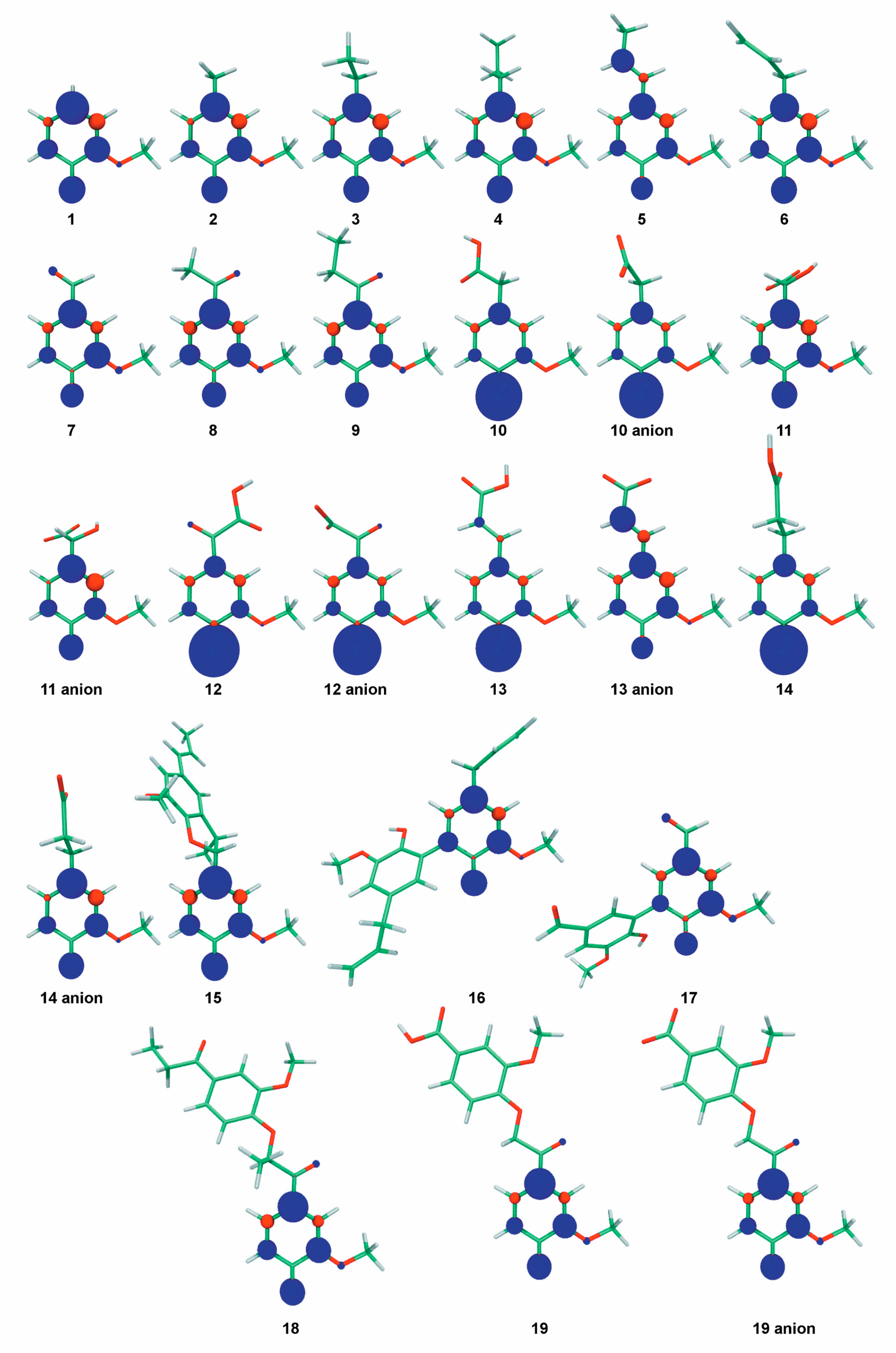
2.1. Role of the Alkyl Substituents
2.2. Role of π-Conjugation in the Side-Chain
2.3. Role of the Carbonyl Group in the α-Position of Guaiacyl Unit
2.4. Role of the Carboxylic Group in the β-Position of Guaiacyl Unit
2.5. Role of the Carboxylic Group in the γ-Position of Guaiacyl Unit
2.6. Effect of Dimerization
2.7. Quantification of the Direct Influence of the Each Structural Descriptor on the Antioxidant Activity of Lignins
2.8. Experimental Evaluation of Lignin Antioxidant Activity with Respect to Impact of Different Structural Descriptors
2.9. Rationalization and Enhancement of the Lignin Antioxidant Activity
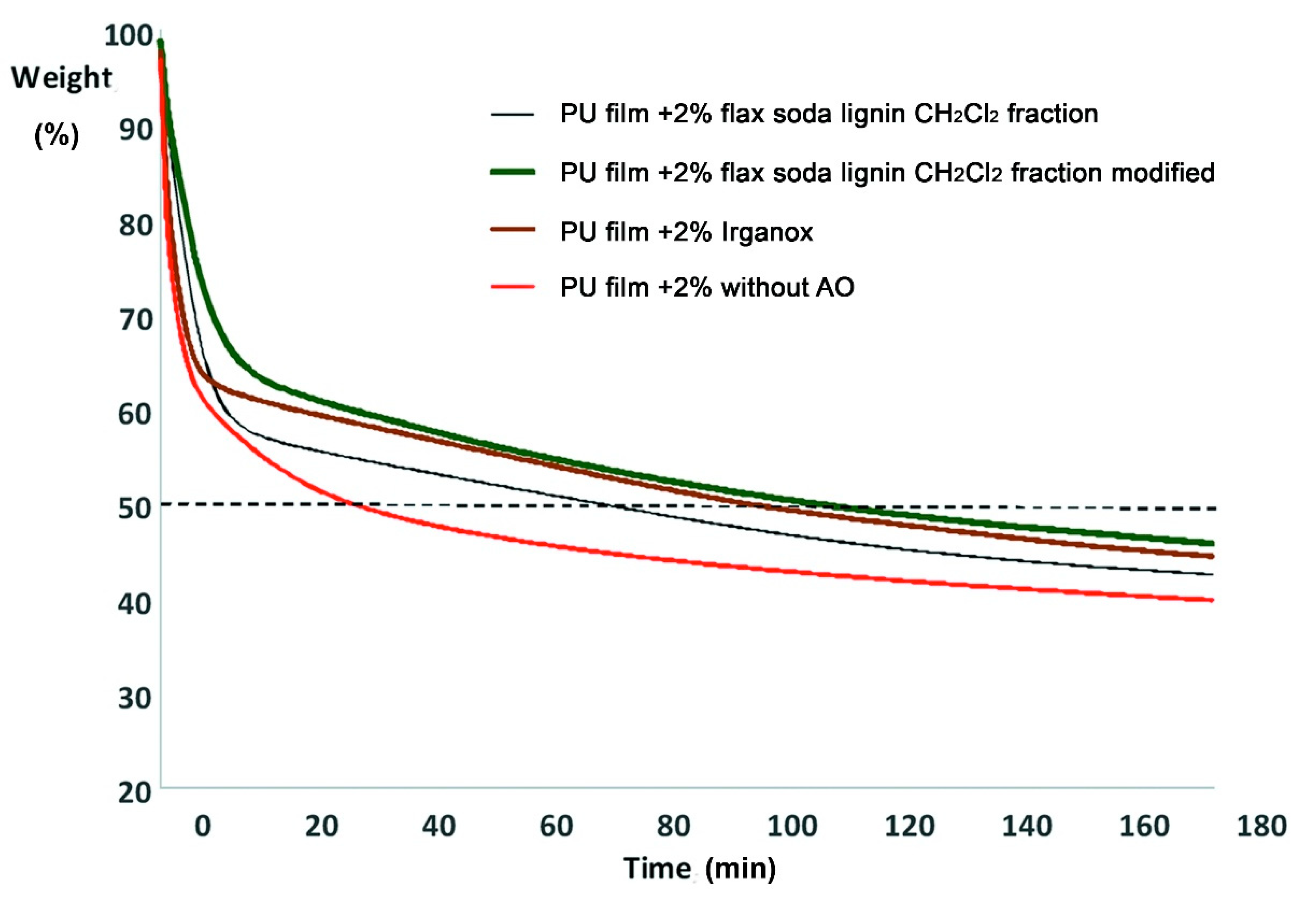
2.10. Modified Lignin Fractions as Alternative Antioxidants in Polyurethane Films
3. Materials and Methods
3.1. Compounds, Modeling Lignin Structural Units
3.2. High-Molecular Weight Lignins
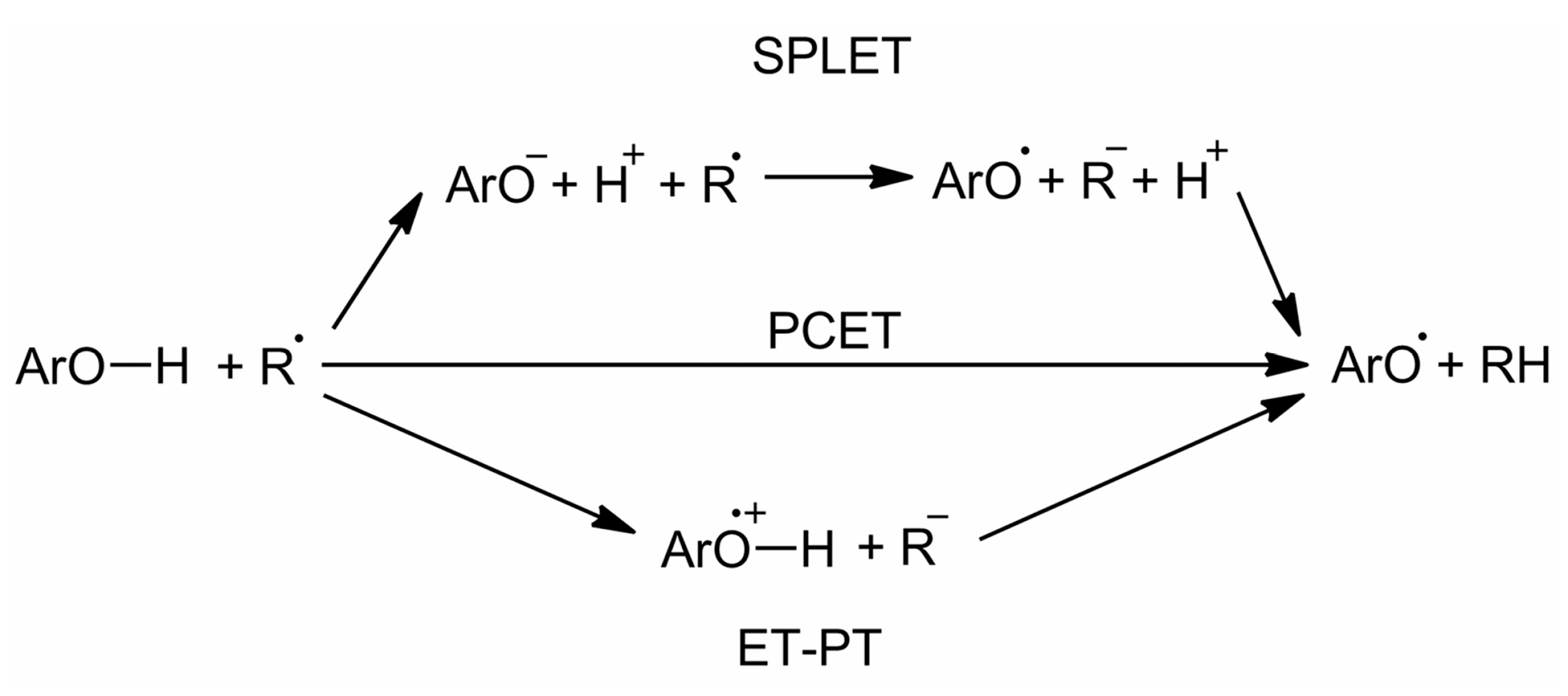
3.3. Computational Methods
3.4. Antioxidant Activity Assays (DPPH•, ABTS•+)
3.5. Characterization of the Polymeric and Oligomeric Lignin Products
3.5.1. Molecular Weight Distribution
3.5.2. Analytical Pyrolysis
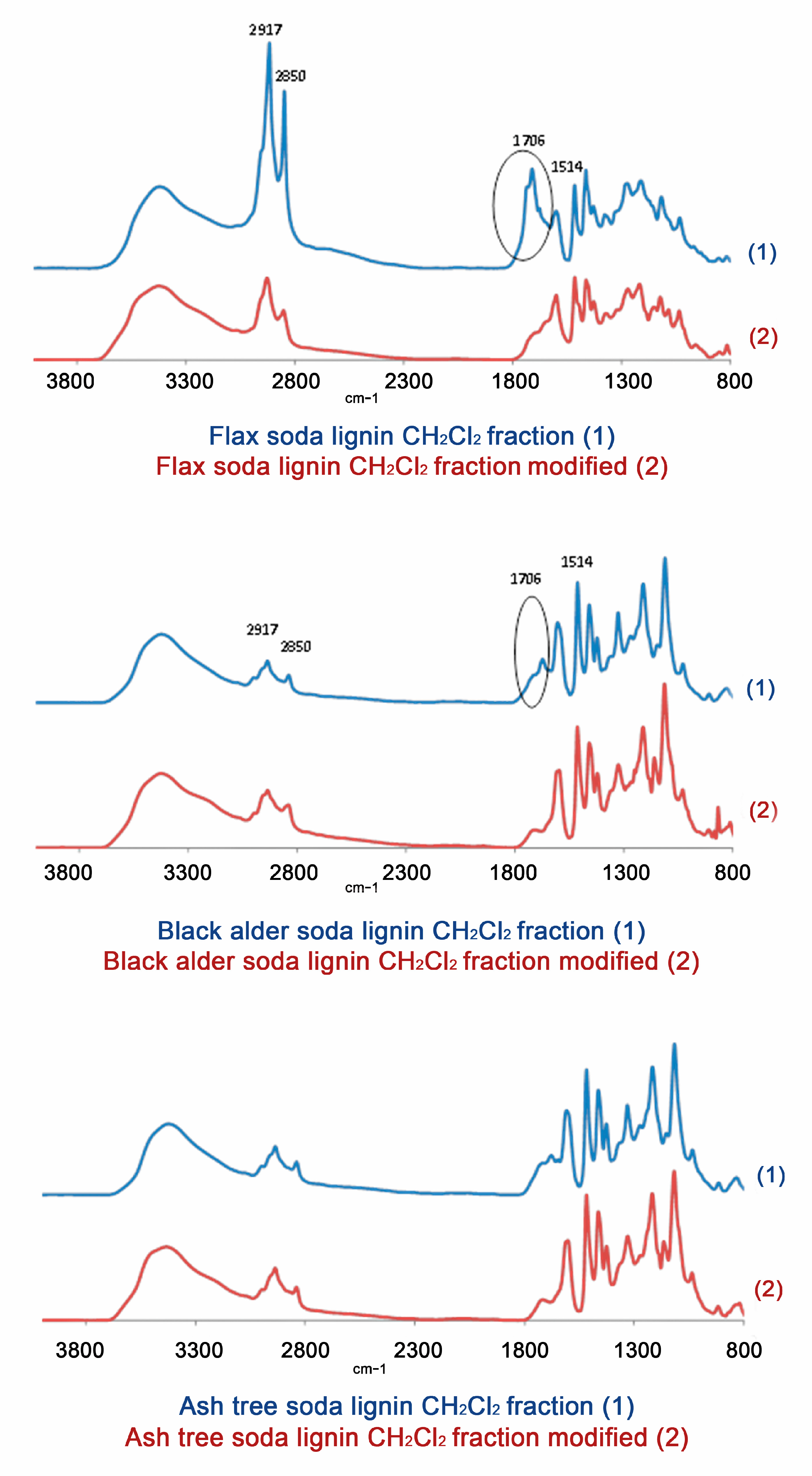
3.5.3. Spectroscopic Analyses
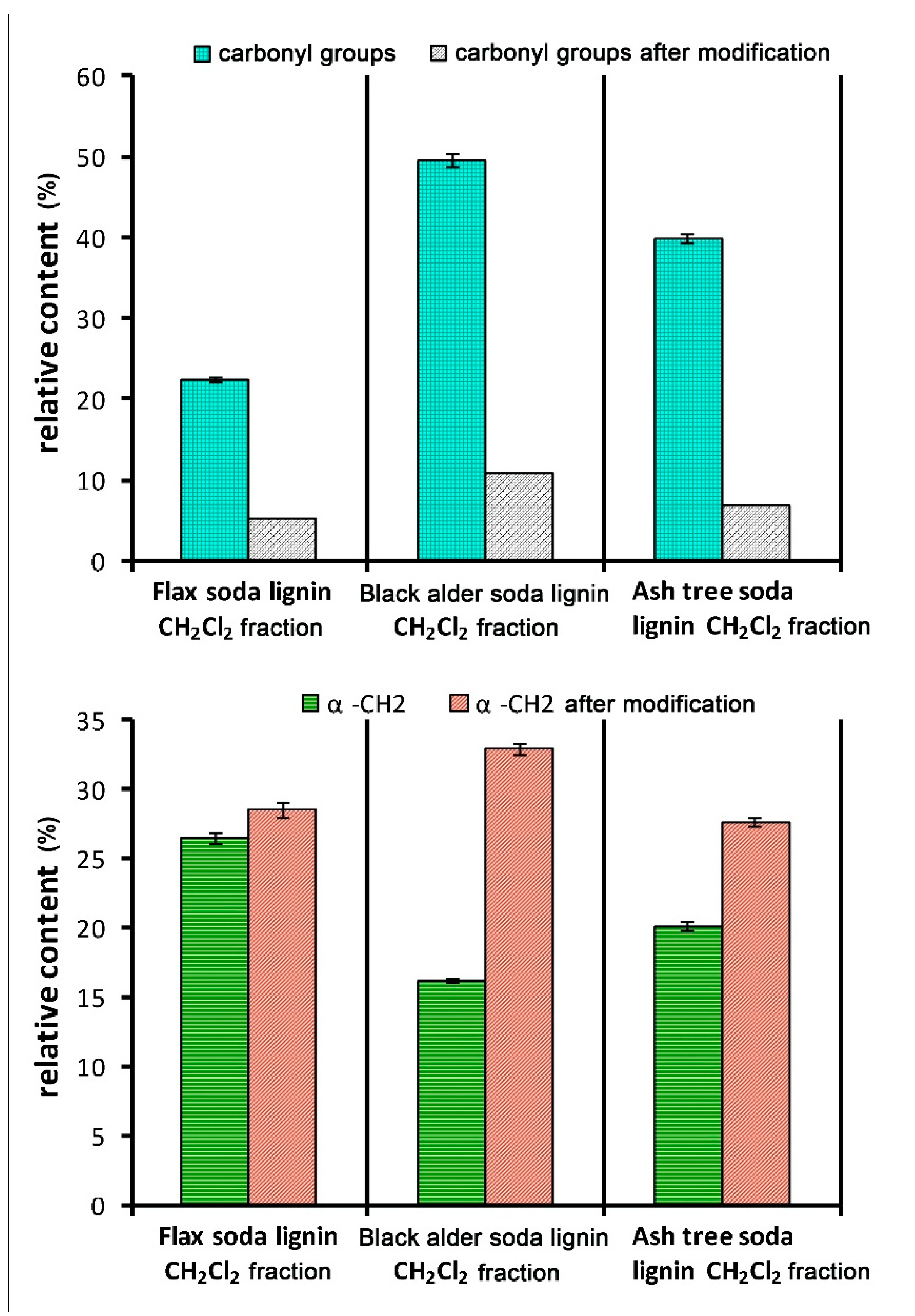
3.5.4. Functional Analysis
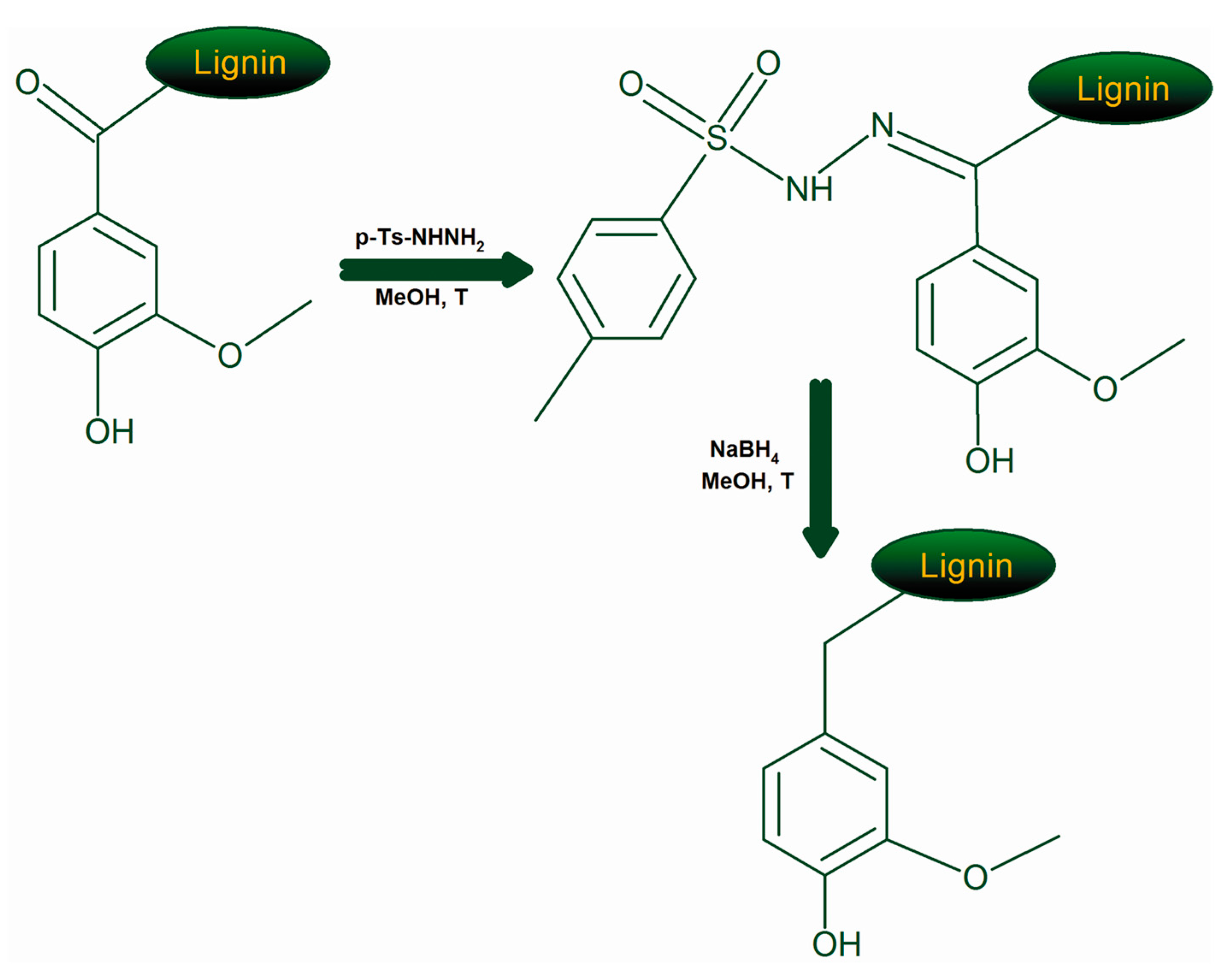
3.6. Modification of the Lignins by Reduction of Carbonyl Groups
3.7. The Effect of Lignin Products on Thermooxidative Destruction of Polyurethane Films
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cheng, F.; Brewer, C.E. Producing jet fuel from biomass lignin: Potential pathways to alkyl-benzenes and cycloalkanes. Renew. Sustain. Energy Rev. 2017, 72, 673–722. [Google Scholar] [CrossRef]
- Hou, Q.; Ju, M.; Li, W.; Liu, L.; Chen, Y.; Yang, Q.; Hou, Q.; Ju, M.; Li, W.; Liu, L.; et al. Pretreatment of Lignocellulosic Biomass with Ionic Liquids and Ionic Liquid-Based Solvent Systems. Molecules 2017, 22, 490. [Google Scholar] [CrossRef] [PubMed]
- Kai, D.; Tan, M.J.; Chee, P.L.; Chua, Y.K.; Yap, Y.L.; Loh, X.J. Towards lignin-based functional materials in a sustainable world. Green Chem. 2016, 18, 1175–1200. [Google Scholar] [CrossRef]
- Luterbacher, J.S.; Martin Alonso, D.; Dumesic, J.A. Targeted chemical upgrading of lignocellulosic biomass to platform molecules. Green Chem. 2014, 16, 4816–4838. [Google Scholar] [CrossRef]
- Ragauskas, A.J.; Beckham, G.T.; Biddy, M.J.; Chandra, R.; Chen, F.; Davis, M.F.; Davison, B.H.; Dixon, R.A.; Gilna, P.; Keller, M.; et al. Lignin valorization: improving lignin processing in the biorefinery. Science 2014, 344, 1246843. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Zhang, Q.; Lee, D.-J. Kraft lignin biorefinery: A perspective. Bioresour. Technol. 2018, 247, 1181–1183. [Google Scholar] [CrossRef]
- Boerjan, W.; Ralph, J.; Baucher, M. L IGNIN B IOSYNTHESIS. Annu. Rev. Plant Biol. 2003, 54, 519–546. [Google Scholar] [CrossRef]
- Boeriu, C.G.; Bravo, D.; Gosselink, R.J.A.; van Dam, J.E.G. Characterisation of structure-dependent functional properties of lignin with infrared spectroscopy. Ind. Crops Prod. 2004, 20, 205–218. [Google Scholar] [CrossRef]
- Vlaminck, L.; Lingier, S.; Hufendiek, A.; Du Prez, F.E. Lignin inspired phenolic polyethers synthesized via ADMET: Systematic structure-property investigation. Eur. Polym. J. 2017, 95, 503–513. [Google Scholar] [CrossRef]
- Vinardell, M.P.; Ugartondo, V.; Mitjans, M. Potential applications of antioxidant lignins from different sources. Ind. Crops Prod. 2008, 27, 220–223. [Google Scholar] [CrossRef]
- UGARTONDO, V.; MITJANS, M.; VINARDELL, M. Comparative antioxidant and cytotoxic effects of lignins from different sources. Bioresour. Technol. 2008, 99, 6683–6687. [Google Scholar] [CrossRef]
- Bertini, F.; Canetti, M.; Cacciamani, A.; Elegir, G.; Orlandi, M.; Zoia, L. Effect of ligno-derivatives on thermal properties and degradation behavior of poly(3-hydroxybutyrate)-based biocomposites. Polym. Degrad. Stab. 2012, 97, 1979–1987. [Google Scholar] [CrossRef]
- Domenek, S.; Louaifi, A.; Guinault, A.; Baumberger, S. Potential of Lignins as Antioxidant Additive in Active Biodegradable Packaging Materials. J. Polym. Environ. 2013, 21, 692–701. [Google Scholar] [CrossRef]
- CATIGNANI, G.L.; CARTER, M.E. Antioxidant Properties of Lignin. J. Food Sci. 1982, 47, 1745. [Google Scholar] [CrossRef]
- Qazi, S.; Li, D.; Briens, C.; Berruti, F.; Abou-Zaid, M.; Qazi, S.S.; Li, D.; Briens, C.; Berruti, F.; Abou-Zaid, M.M. Antioxidant Activity of the Lignins Derived from Fluidized-Bed Fast Pyrolysis. Molecules 2017, 22, 372. [Google Scholar] [CrossRef] [PubMed]
- Morandim-Giannetti, A.A.; Agnelli, J.A.M.; Lanças, B.Z.; Magnabosco, R.; Casarin, S.A.; Bettini, S.H.P. Lignin as additive in polypropylene/coir composites: Thermal, mechanical and morphological properties. Carbohydr. Polym. 2012, 87, 2563–2568. [Google Scholar] [CrossRef]
- Fernandes, D.M.; Winkler Hechenleitner, A.A.; Job, A.E.; Radovanocic, E.; Gómez Pineda, E.A. Thermal and photochemical stability of poly(vinyl alcohol)/modified lignin blends. Polym. Degrad. Stab. 2006, 91, 1192–1201. [Google Scholar] [CrossRef]
- Canetti, M.; Bertini, F.; De Chirico, A.; Audisio, G. Thermal degradation behaviour of isotactic polypropylene blended with lignin. Polym. Degrad. Stab. 2006, 91, 494–498. [Google Scholar] [CrossRef]
- Schmidt, J.A.; Rye, C.S.; Gurnagul, N. Lignin inhibits autoxidative degradation of cellulose. Polym. Degrad. Stab. 1995, 49, 291–297. [Google Scholar] [CrossRef]
- Barclay, L.R.C.; Xi, F.; Norris, J.Q. Antioxidant Properties of Phenolic Lignin Model Compounds. J. Wood Chem. Technol. 1997, 17, 73–90. [Google Scholar] [CrossRef]
- Gregorová, A.; Cibulková, Z.; Košíková, B.; Šimon, P. Stabilization effect of lignin in polypropylene and recycled polypropylene. Polym. Degrad. Stab. 2005, 89, 553–558. [Google Scholar] [CrossRef]
- Domínguez-Robles, J.; Tamminen, T.; Liitiä, T.; Peresin, M.S.; Rodríguez, A.; Jääskeläinen, A.-S. Aqueous acetone fractionation of kraft, organosolv and soda lignins. Int. J. Biol. Macromol. 2018, 106, 979–987. [Google Scholar] [CrossRef]
- Park, S.Y.; Kim, J.-Y.; Youn, H.J.; Choi, J.W. Fractionation of lignin macromolecules by sequential organic solvents systems and their characterization for further valuable applications. Int. J. Biol. Macromol. 2018, 106, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Brodin, I.; Sjöholm, E.; Gellerstedt, G. Kraft lignin as feedstock for chemical products: The effects of membrane filtration. Holzforschung 2009, 63, 290–297. [Google Scholar] [CrossRef]
- Boeriu, C.G.; Fiţigău, F.I.; Gosselink, R.J.A.; Frissen, A.E.; Stoutjesdijk, J.; Peter, F. Fractionation of five technical lignins by selective extraction in green solvents and characterisation of isolated fractions. Ind. Crops Prod. 2014, 62, 481–490. [Google Scholar] [CrossRef]
- Thring, R.W.; Griffin, S.L. The heterogeneity of two Canadian kraft lignins. Can. J. Chem. 1995, 73, 629–634. [Google Scholar] [CrossRef]
- Ropponen, J.; Räsänen, L.; Rovio, S.; Ohra-aho, T.; Liitiä, T.; Mikkonen, H.; van de Pas, D.; Tamminen, T. Solvent extraction as a means of preparing homogeneous lignin fractions. Holzforschung 2011, 65, 543–549. [Google Scholar] [CrossRef]
- Gosselink, R.J.A.; van Dam, J.E.G.; de Jong, E.; Scott, E.L.; Sanders, J.P.M.; Li, J.; Gellerstedt, G. Fractionation, analysis, and PCA modeling of properties of four technical lignins for prediction of their application potential in binders. Holzforschung 2010, 64, 193–200. [Google Scholar] [CrossRef]
- Li, H.; McDonald, A.G. Fractionation and characterization of industrial lignins. Ind. Crops Prod. 2014, 62, 67–76. [Google Scholar] [CrossRef]
- Pouteau, C.; Dole, P.; Cathala, B.; Averous, L.; Boquillon, N. Antioxidant properties of lignin in polypropylene. Polym. Degrad. Stab. 2003, 81, 9–18. [Google Scholar] [CrossRef]
- Pouteau, C.; Cathala, B.; Dole, P.; Kurek, B.; Monties, B. Structural modification of Kraft lignin after acid treatment: characterisation of the apolar extracts and influence on the antioxidant properties in polypropylene. Ind. Crops Prod. 2005, 21, 101–108. [Google Scholar] [CrossRef]
- Jääskeläinen, A.-S.; Liitiä, T.; Mikkelson, A.; Tamminen, T. Aqueous organic solvent fractionation as means to improve lignin homogeneity and purity. Ind. Crops Prod. 2017, 103, 51–58. [Google Scholar] [CrossRef]
- Arshanitsa, A.; Ponomarenko, J.; Dizhbite, T.; Andersone, A.; Gosselink, R.J.A.; van der Putten, J.; Lauberts, M.; Telysheva, G. Fractionation of technical lignins as a tool for improvement of their antioxidant properties. J. Anal. Appl. Pyrolysis 2013, 103, 78–85. [Google Scholar] [CrossRef]
- Ponomarenko, J.; Dizhbite, T.; Lauberts, M.; Viksna, A.; Dobele, G.; Bikovens, O.; Telysheva, G. Characterization of Softwood and Hardwood LignoBoost Kraft Lignins with Emphasis on their Antioxidant Activity. BioResources 2014, 9, 2051–2068. [Google Scholar] [CrossRef]
- An, L.; Wang, G.; Jia, H.; Liu, C.; Sui, W.; Si, C. Fractionation of enzymatic hydrolysis lignin by sequential extraction for enhancing antioxidant performance. Int. J. Biol. Macromol. 2017, 99, 674–681. [Google Scholar] [CrossRef]
- Brunow, G.; Kilpeläinen, I.; Sipilä, J.; Syrjänen, K.; Karhunen, P.; Setälä, H.; Rummakko, P. Oxidative Coupling of Phenols and the Biosynthesis of Lignin. In Lignin and Lignan Biosynthesis; American Chemical Society: Washington, DC, USA, 1998; pp. 131–147. [Google Scholar]
- Önnerud, H.; Gellerstedt, G. Inhomogeneities in the Chemical Structure of Hardwood Lignins. Holzforschung 2003, 57, 255–265. [Google Scholar] [CrossRef]
- Pan, X.; Kadla, J.F.; Ehara, K.; Gilkes, N.; Saddler, J.N. Organosolv Ethanol Lignin from Hybrid Poplar as a Radical Scavenger: Relationship between Lignin Structure, Extraction Conditions, and Antioxidant Activity. J. Agric. Food Chem. 2006, 54, 5806–5813. [Google Scholar] [CrossRef] [PubMed]
- Anouar, E.; Košinová, P.; Kozlowski, D.; Mokrini, R.; Duroux, J.L.; Trouillas, P. New aspects of the antioxidant properties of phenolic acids: a combined theoretical and experimental approach. Phys. Chem. Chem. Phys. 2009, 11, 7659. [Google Scholar] [CrossRef] [PubMed]
- Anouar, E.; Calliste, C.A.; Košinová, P.; Di Meo, F.; Duroux, J.L.; Champavier, Y.; Marakchi, K.; Trouillas, P. Free Radical Scavenging Properties of Guaiacol Oligomers: A Combined Experimental and Quantum Study of the Guaiacyl-Moiety Role. J. Phys. Chem. A 2009, 113, 13881–13891. [Google Scholar] [CrossRef]
- Di Meo, F.; Lemaur, V.; Cornil, J.; Lazzaroni, R.; Duroux, J.-L.; Olivier, Y.; Trouillas, P. Free Radical Scavenging by Natural Polyphenols: Atom versus Electron Transfer. J. Phys. Chem. A 2013, 117, 2082–2092. [Google Scholar] [CrossRef]
- Setzer, W.N. Lignin-derived oak phenolics: a theoretical examination of additional potential health benefits of red wine. J. Mol. Model. 2011, 17, 1841–1845. [Google Scholar] [CrossRef]
- Kjällstrand, J.; Petersson, G. Phenolic antioxidants in wood smoke. Sci. Total Environ. 2001, 277, 69–75. [Google Scholar] [CrossRef]
- Kozlowski, D.; Trouillas, P.; Calliste, C.; Marsal, P.; Lazzaroni, R.; Duroux, J.-L. Density Functional Theory Study of the Conformational, Electronic, and Antioxidant Properties of Natural Chalcones. J. Phys. Chem. A 2007, 111, 1138–1145. [Google Scholar] [CrossRef]
- Kozlowski, D.; Marsal, P.; Steel, M.; Mokrini, R.; Duroux, J.-L.; Lazzaroni, R.; Trouillas, P. Theoretical Investigation of the Formation of a New Series of Antioxidant Depsides from the Radiolysis of Flavonoid Compounds. Radiat. Res. 2007, 168, 243–252. [Google Scholar] [CrossRef]
- Calliste, C.A.; Kozlowski, D.; Duroux, J.L.; Champavier, Y.; Chulia, A.J.; Trouillas, P. A new antioxidant from wild nutmeg. Food Chem. 2010, 118, 489–496. [Google Scholar] [CrossRef]
- Bortolomeazzi, R.; Verardo, G.; Liessi, A.; Callea, A. Formation of dehydrodiisoeugenol and dehydrodieugenol from the reaction of isoeugenol and eugenol with DPPH radical and their role in the radical scavenging activity. Food Chem. 2010, 118, 256–265. [Google Scholar] [CrossRef]
- Ragnar, M.; Lindgren, C.T.; Nilvebrant, N.-O. pKa-Values of Guaiacyl and Syringyl Phenols Related to Lignin. J. Wood Chem. Technol. 2000, 20, 277–305. [Google Scholar] [CrossRef]
- Rived, F.; Rosés, M.; Bosch, E. Dissociation constants of neutral and charged acids in methyl alcohol. The acid strength resolution. Anal. Chim. Acta 1998, 374, 309–324. [Google Scholar] [CrossRef]
- Stella, A.O.; Maria, Z.T.; Anastasios, P.V.; Evangelos, G.B. Structure−DPPH• Scavenging Activity Relationships: Parallel Study of Catechol and Guaiacol Acid Derivatives. J. Agric. Food Chem. 2006, 54, 5763–5768. [Google Scholar]
- Villaño, D.; Fernández-Pachón, M.S.; Moyá, M.L.; Troncoso, A.M.; García-Parrilla, M.C. Radical scavenging ability of polyphenolic compounds towards DPPH free radical. Talanta 2007, 71, 230–235. [Google Scholar] [CrossRef]
- Kanitskaja, L.V.; Medvedeva, S.A.; Ivanova, S.Z.; Kushnarev, D.F.; Ri, B. 1H NMR spectroscopy as a method for the identification of hydroxyl-containing lignin fragments. Khimija Drev. 1987, 6, 3–10. [Google Scholar]
- Li, S.; Lundquist, K. A new method for the analysis of phenolic groups in lignins by ʹH NMR spectrometry. Nord. Pulp Pap. Res. J. 1994, 9, 191–195. [Google Scholar] [CrossRef]
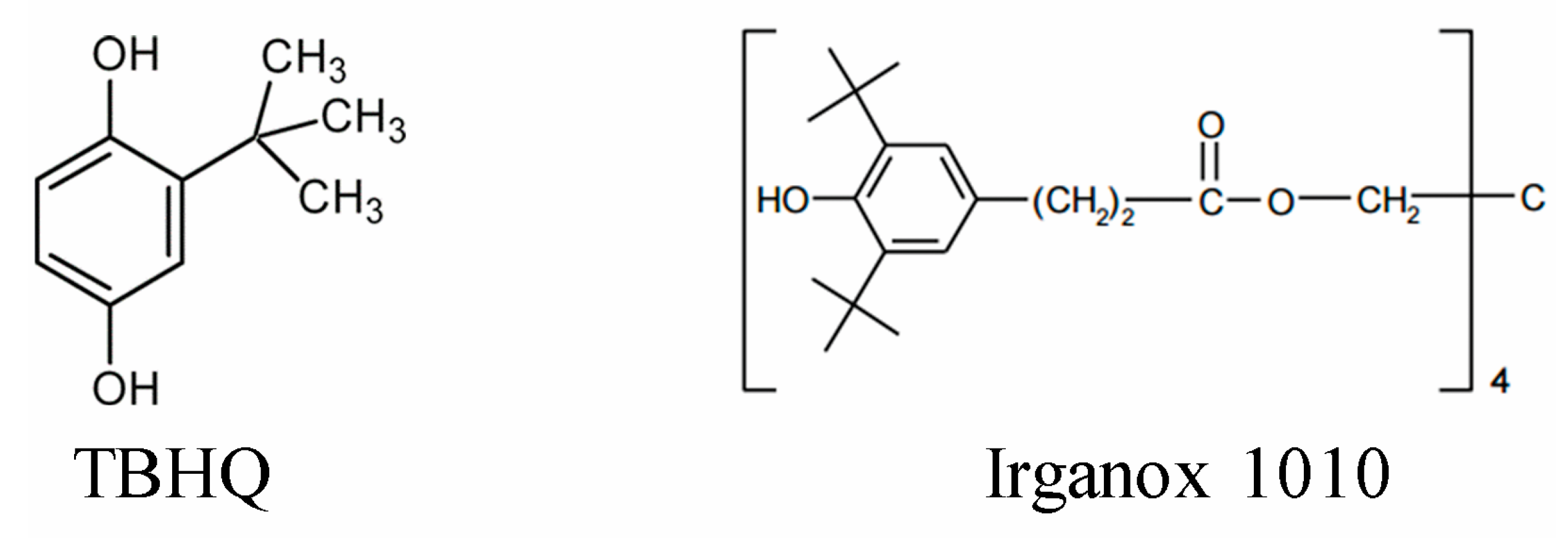
- Irganox® 1010. Available online: https://worldaccount.basf.com/en_GB/welcome.html (accessed on 5 April 2019).
- Chattopadhyay, D.K.; Webster, D.C. Thermal stability and flame retardancy of polyurethanes. Prog. Polym. Sci. 2009, 34, 1068–1133. [Google Scholar] [CrossRef]
- Mottern, H.O. A New Vanillin Synthesis. J. Am. Chem. Soc. 1934, 56, 2107–2108. [Google Scholar] [CrossRef]
- Rab, M.Z. The synthesis of lignin models, containing.- β-ether bond. For. J. 1968, 6, 120–123. (In Russian) [Google Scholar]
- Erdtman, H.; Leopold, B.; Linderholm, H.; Kainulainen, A.; Halonen, A.; Pulkkinen, E. Aromatic Keto- and Hydroxy-polyethers as Lignin Models. II. Acta Chem. Scand. 1949, 3, 1358–1374. [Google Scholar] [CrossRef]
- Zakis, G.F. Functional Analysis of Lignins and Their Derivatives; TAPPI Press: Atlanta, GA, USA, 1994; ISBN 0898522587. [Google Scholar]
- Abächerli Alfred, D.F. Method for Preparing Alkaline Solutions Containing Aromatic Polymers 1998. U.S. Patent 6,239,198, 20 March 1998. [Google Scholar]
- Arshanitsa, A.; Krumina, L.; Telysheva, G.; Dizhbite, T. Exploring the application potential of incompletely soluble organosolv lignin as a macromonomer for polyurethane synthesis. Ind. Crops Prod. 2016, 92, 1–12. [Google Scholar] [CrossRef]
- Trouillas, P.; Marsal, P.; Siri, D.; Lazzaroni, R.; Duroux, J.-L. A DFT study of the reactivity of OH groups in quercetin and taxifolin antioxidants: The specificity of the 3-OH site. Food Chem. 2006, 97, 679–688. [Google Scholar] [CrossRef]
- Trouillas, P.; Marsal, P.; Svobodová, A.; Vostálová, J.; Gažák, R.; Hrbáč, J.; Sedmera, P.; Křen, V.; Lazzaroni, R.; Duroux, J.-L.; et al. Mechanism of the Antioxidant Action of Silybin and 2,3-Dehydrosilybin Flavonolignans: A Joint Experimental and Theoretical Study. J. Phys. Chem. A 2008, 112, 1054–1063. [Google Scholar] [CrossRef]
- Gaussian 09; Gausian, Inc.: Wallingford, CT, USA, 2010.
- Baltrušaitytė, V.; Venskutonis, P.R.; Čeksterytė, V. Radical scavenging activity of different floral origin honey and beebread phenolic extracts. Food Chem. 2007, 101, 502–514. [Google Scholar] [CrossRef]
- Ringena, O.; Lebioda, S.; Lehnen, R.; Saake, B. Size-exclusion chromatography of technical lignins in dimethyl sulfoxide/water and dimethylacetamide. J. Chromatogr. A 2006, 1102, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Evtuguin, D.V.; Neto, C.P.; Silva, A.M.; Domingues, P.M.; Amado, F.M.; Robert, D.; Faix, O. Comprehensive study on the chemical structure of dioxane lignin from plantation Eucalyptus globulus wood. J. Agric. Food Chem. 2001, 49, 4252–4261. [Google Scholar] [CrossRef] [PubMed]
- Caglioti, L. Reduction of Ketones by Use of the Tosylhydrazone Derivatives: Androstan-17 b-ol. Org. Synth. 1972, 52, 122. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of the Compound | Lignin Modeling Compound | BDE (kcal mol−1) | DPPH• RDI | ETE (kcal mol−1) | ABTS•+ RDI | pKa in Water b |
|---|---|---|---|---|---|---|
| 1 | guaiacol | 82.4 | 1.00 ± 0.03 | 109.0 | - | 9.93 |
| 2 | methylguaiacol | 80.3 | 1.38 ± 0.03 | 105.7 | 0.92 ± 0.03 | 10.27 |
| 3 | ethylguaiacol | 80.5 | 1.35 ± 0.04 | 106.0 | - | - |
| 4 | propylguaiacol | 80.4 | 1.53 ± 0.02 | 106.0 | 1.58 ± 0.07 | 9.85 |
| 5 | isoeugenol | 77.7 | 0.90 ± 0.09 | 105.7 | 0.67 ± 0.05 | 9.89 |
| 6 | eugenol | 81.0 | 1.72 ± 0.07 | 107.3 | 0.96 ± 0.04 | 10.15 |
| 7 | vanillin | 85.3 | 0.02 ± 0.01 | 121.4 | 0.57 ± 0.03 | 7.40 |
| 8 | acetovanillone | 85.2 | 0.03 ± 0.01 | 119.5 | 0.54 ± 0.03 | 7.81 |
| 9 | propiovanillone | 85.0 | 0.05 ± 0.01 | 118.9 | 0.45 ± 0.04 | 7.98 |
| 10 | homovanillic acid | 82.0/78.7 | 1.18 ± 0.03 | 109.1/102.0 | 1.67 ± 0.02 | 4.41/ 10.52 |
| 11 | vanillylmandelic acid | 82.5/79.0 | 0.98 ± 0.02 | 111.3/103.3 | 0.92 ± 0.02 | 3.43/ 9.93 |
| 12 | vanilglycolic acid | 86.9/83.7 | 0.08 ± 0.01 | 125.7/116.7 | 0.26 ± 0.01 | 1.60/ 7.54 |
| 13 | ferulic acid | 81.8/77.6 | 1.23 ± 0.03 | 116.8/106.0 | 2.39 ± 0.07 | 4.56/ 9.39 |
| 14 | dihydroferulic acid | 81.2/79.5 | 1.23 ± 0.06 | 107.4/103.5 | 1.39 ± 0.04 | - |
| 15 | dehydrodiisoeugenol | 81.9 | 0.67 ± 0.02 a | 109.5 | - | - |
| 16 | dehydrodieugenol | 80.1 | 2.72 ± 0.03 a | 106.3 | - | - |
| 17 | divanillin | 84.8 | n.d. c | 121.6 | 0.16 ± 0.03 | 6.16/ 10.07 |
| 18 | dipropiovanillone | 86.0 | 0.014 ± 0.01 | 122.0 | - | - |
| 19 | acetovanillonylvanillic acid | 86.5/85.6 | 0.02 ± 0.01 | 121.5/120.0 | 0.26 | - |
| Structural Descriptor | Compounds with and without the Indicated Chemical Feature * | Average ΔBDE of Phenolic OH Group between Compounds Bearing or not the Corresponding Chemical Substituent | Normalized Negative Impact of the Structural Descriptor, % | Normalized Positive Impact of the Structural Descriptor, % |
|---|---|---|---|---|
| α-CH2 | 2 (1) | −2.1 | - | 43 |
| α-CH2-CH3 | 3 (1) | −1.9 | - | 39 |
| α-CH2-CH2-CH3 | 4 (1) | −2.0 | - | 41 |
| α-C=C | 5(4), 13(14) | −2.3 | - | 47 |
| β-C=C | 6(4) | +0.6 | 12 | - |
| α-C=O | 7(2), 8 (3), 9(4), 12(10) | +4.8 | 98 | - |
| β-COOH | 10 (3), 12 (8) | +1.6 | 33 | - |
| γ-COOH | 14 (4) | +0.8 | 16 | - |
| α-OH | 11 (10) | +0.5 | 10 | - |
| α-O-4/β-5 | 15 (4) | +1.5 | 31 | - |
| β-O-4 ether linked | 18 (9), 19 (8) | +1.4 | 29 | - |
| biphenyl (5-5) | 16 (6), 17 (7) | −0.7 | - | 14 |
| Sample | Lignin Content, % * | OCH3, % | Mw, Da | Phenolic OH/100 Phenyl Propane Units (PPU) | Relative Content of α-Carbonyl group in the Phenyl-Propane Units, % ** | Number of Scavenged DPPH• Radicals Per OHphen (RDI) |
|---|---|---|---|---|---|---|
| Flax soda lignin | 94.5 ± 0.5 | 8.1 ± 0.1 | 8358 | 26 | 9.1 ± 0.1 | 1.33 ± 0.07 |
| Flax soda lignin CH2Cl2 fraction | 95.4 ± 0.4 | 6.5 ± 0.1 | 847 | 33 | 22.3 ± 0.1 | 0.71 ± 0.04 |
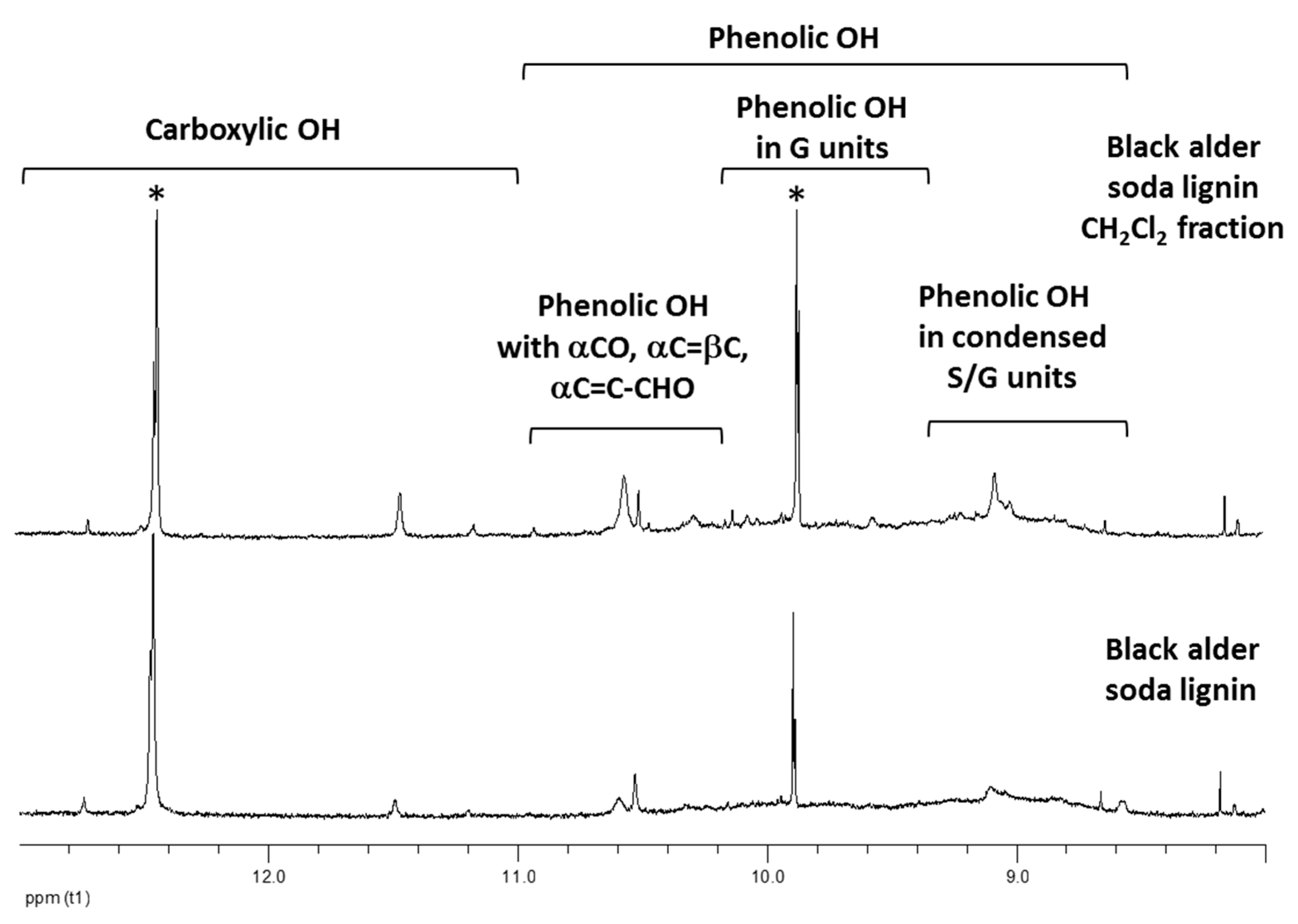
| Black alder soda lignin | 74.0 ± 0.7 | 11.2 ± 0.1 | 7617 | 48 | 32.5 ± 0.1 | 0.77 ± 0.03 |
| Black alder soda lignin, CH2Cl2 fraction | 99.2 ± 0.7 | 24.0 ± 0.2 | 638 | 88 | 49.5 ± 0.1 | 0.69 ± 0.03 |
| Ash-tree soda lignin | 92.0 ± 0.9 | 16.3 ± 0.1 | 4505 | 47 | 21.7 ± 0.1 | 1.01 ± 0.05 |
| Ash-tree soda lignin CH2Cl2 fraction | 98.2 ± 0.5 | 13.3 ± 0.1 | 818 | 85 | 39.9 ± 0.1 | 0.72 ± 0.04 |
| TBHQ | - | - | - | 200 | - | 1.31 ± 0.05 |
| Irganox | - | - | - | 100 | - | 1.31 ± 0.07 |
| Sample | Relative Content of the Phenolic Units Conjugated with α-Carbonyl Group, vs. Total Phenolic Groups, % | Number of Scavenged DPPH• Radicals Per One Phenolic OH Group |
|---|---|---|
| Flax soda lignin CH2Cl2 fraction | 14.0 | 0.71 ± 0.04 |
| Flax soda lignin CH2Cl2 fraction modified | 6.0 | 2.05 ± 0.8 |
| Black alder soda lignin CH2Cl2fraction | 36.5 | 0.69 ± 0.03 |
| Black alder soda lignin CH2Cl2fraction, modified | 13.9 | 1.80 ± 0.02 |
| Ash-tree soda lignin CH2Cl2 fraction | 25.6 | 0.72 ± 0.04 |
| Ash-tree soda lignin CH2Cl2fraction, modified | 11.3 | 1.26 ± 0.07 |
| TBHQ | - | 1.31 ± 0.05 |
| Irganox 1010 | - | 1.31 ± 0.07 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lauberte, L.; Fabre, G.; Ponomarenko, J.; Dizhbite, T.; Evtuguin, D.V.; Telysheva, G.; Trouillas, P. Lignin Modification Supported by DFT-Based Theoretical Study as a Way to Produce Competitive Natural Antioxidants. Molecules 2019, 24, 1794. https://doi.org/10.3390/molecules24091794
Lauberte L, Fabre G, Ponomarenko J, Dizhbite T, Evtuguin DV, Telysheva G, Trouillas P. Lignin Modification Supported by DFT-Based Theoretical Study as a Way to Produce Competitive Natural Antioxidants. Molecules. 2019; 24(9):1794. https://doi.org/10.3390/molecules24091794
Chicago/Turabian StyleLauberte, Liga, Gabin Fabre, Jevgenija Ponomarenko, Tatiana Dizhbite, Dmitry V. Evtuguin, Galina Telysheva, and Patrick Trouillas. 2019. "Lignin Modification Supported by DFT-Based Theoretical Study as a Way to Produce Competitive Natural Antioxidants" Molecules 24, no. 9: 1794. https://doi.org/10.3390/molecules24091794
APA StyleLauberte, L., Fabre, G., Ponomarenko, J., Dizhbite, T., Evtuguin, D. V., Telysheva, G., & Trouillas, P. (2019). Lignin Modification Supported by DFT-Based Theoretical Study as a Way to Produce Competitive Natural Antioxidants. Molecules, 24(9), 1794. https://doi.org/10.3390/molecules24091794