Synthesis, Biological Evaluation and Docking Studies of 13-Epimeric 10-fluoro- and 10-Chloroestra-1,4-dien-3-ones as Potential Aromatase Inhibitors

 and
and
Abstract
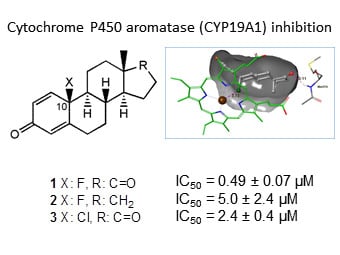
1. Introduction
2. Results and Discussion
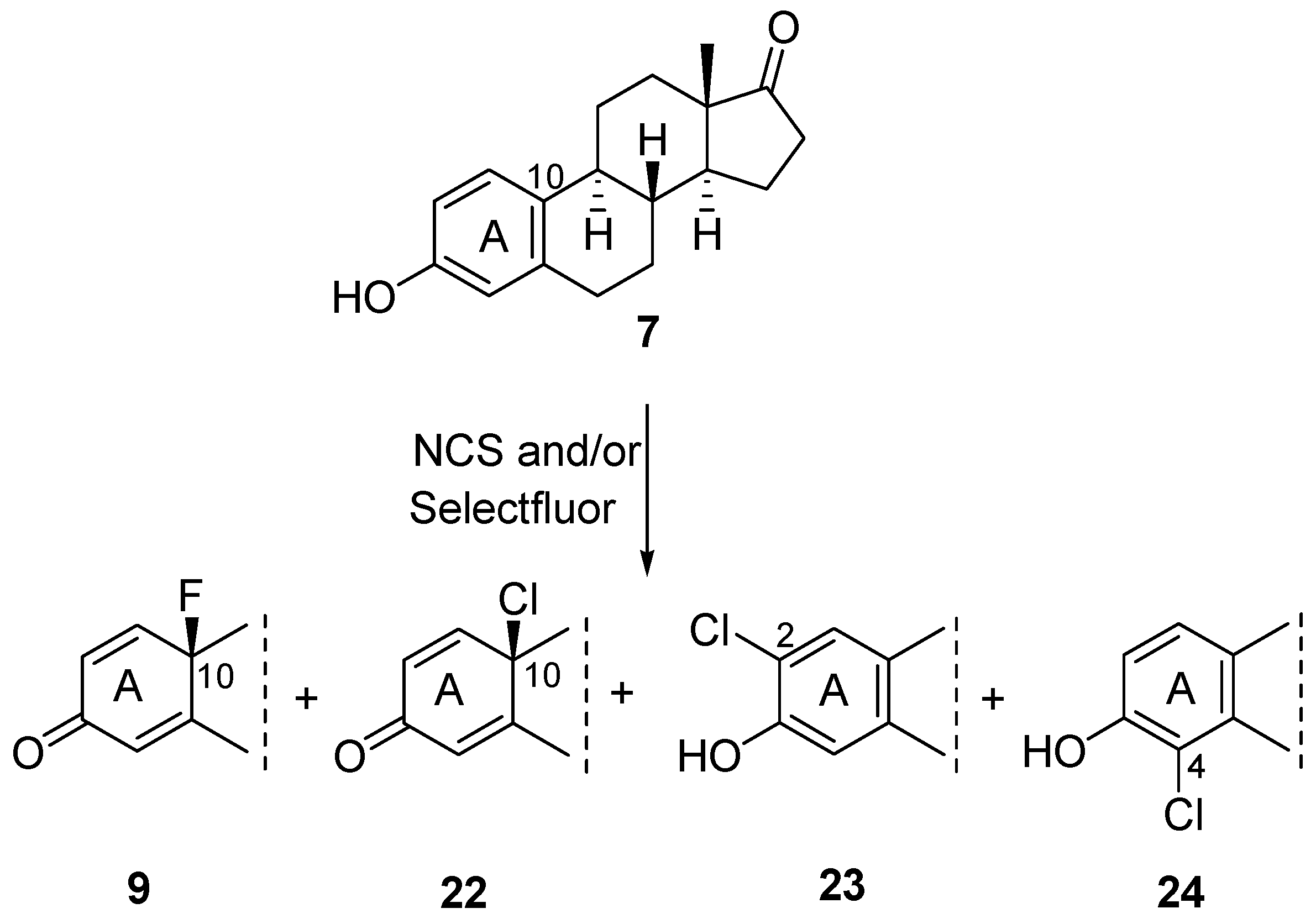
2.1. Chemistry
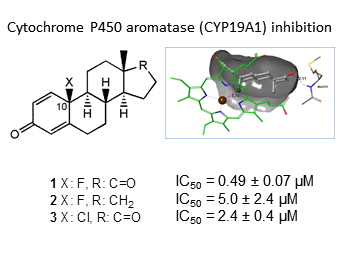
2.2. Aromatase Inhibition Studies
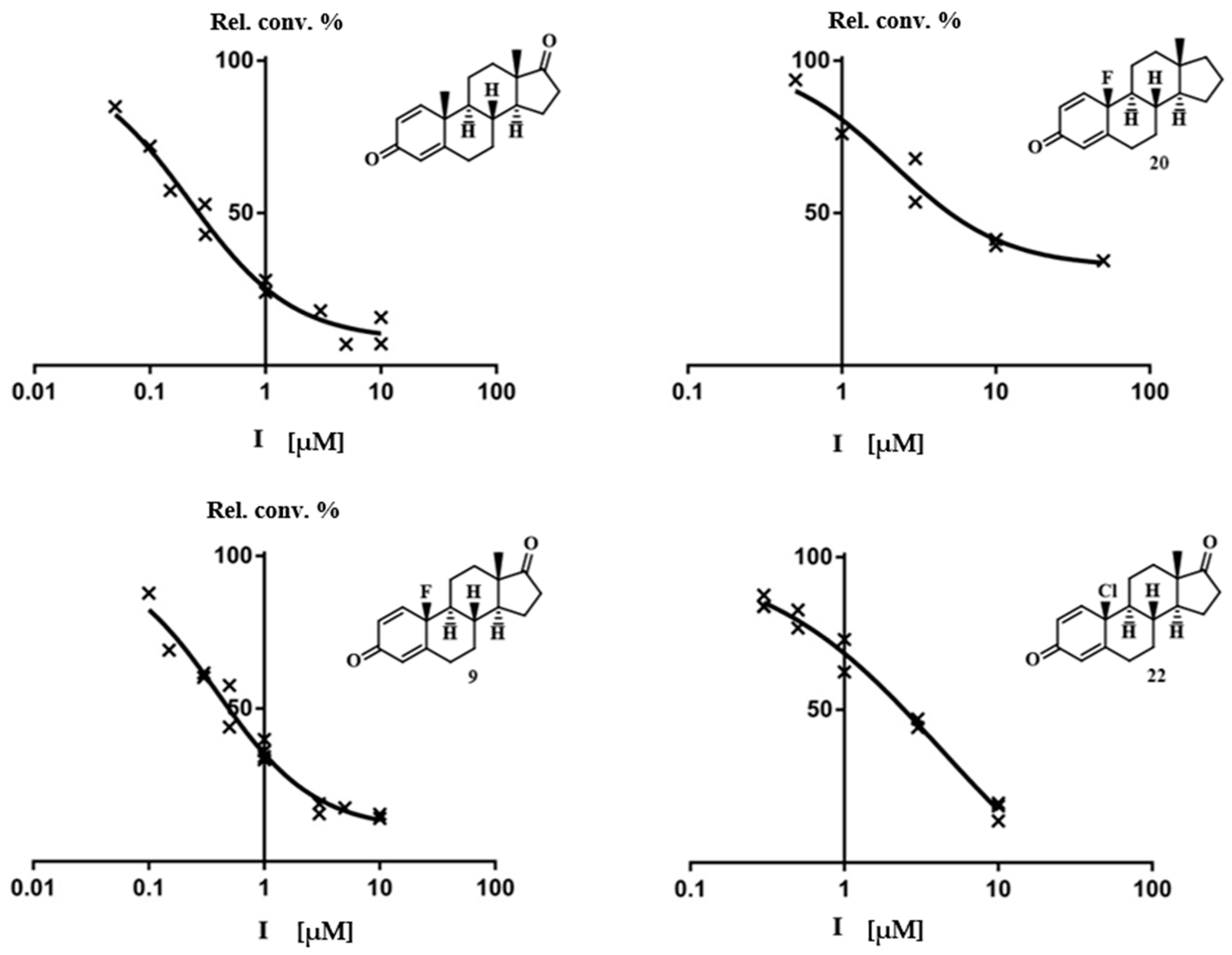
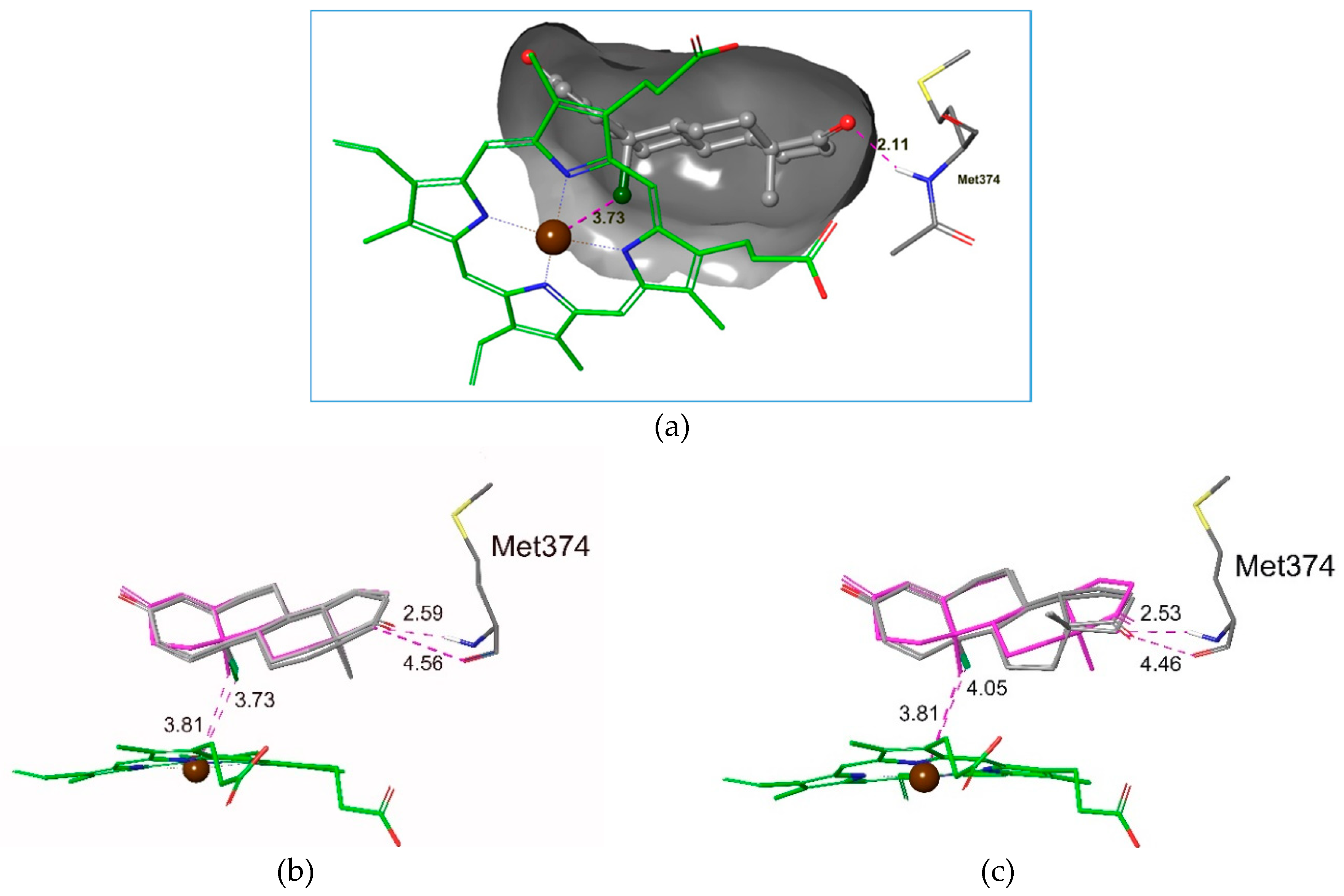
2.3. Molecular Docking Studies
3. Materials and Methods
3.1. Chemical Synthesis
3.1.1. General
3.1.2. Synthesis of 10β-Fluoroestra-1,4-dien-3-one (9) or 10β-Fluoro-13α-estra-1,4-dien-3-one (17) in acetonitrile
3.1.3. Synthesis of 10β-Fluoroestra-1,4-dien-3-one (9) or 10β-Fluoro-13α-estra-1,4-dien-3-one (17) in methanol
3.1.4. General Procedure for the Fluorination with Selectfluor in Acetonitrile or in Methanol by Adding TEMPO
3.1.5. Synthesis of 10β-Fluoro-17-deoxyestra-1,4-dien-3-one (20) or 10β-Fluoro-17-deoxy-13α-estra-1,4-dien-3-one (21)
3.1.6. Fluorination of estrone (7) with Selectfluor in TFA
3.1.7. Fluorination of Estrone (7) with Selectfluor in TFA by Adding TEMPO
3.1.8. Chlorination of Estrone (7) with NCS in Acetonitrile
3.1.9. Chlorination of Estrone (7) with NCS in TFA
3.1.10. Chlorination of estrone (7) with NCS in TFA by adding TEMPO
3.1.11. Fluorination and Chlorination of Estrone (7) with Selectfluor and NCS in Acetonitrile
3.1.12. Fluorination and Chlorination of Estrone (7) with Selectfluor and NCS in TFA
3.1.13. Fluorination and Chlorination of Estrone (7) with Selectfluor and NCS in TFA by Adding TEMPO
3.2. Aromatase Inhibition
3.2.1. General
3.2.2. Preparation of Enzyme Sources
3.2.3. Incubation Procedures
3.2.4. Inhibition Studies
3.3. Computational Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yadav, M.R.; Barmade, M.A.; Tamboli, R.S.; Murumkar, P.R. Developing steroidal aromatase inhibitors-an effective armament to win the battle against breast cancer. Eur. J. Med. Chem. 2015, 105, 1–38. [Google Scholar] [CrossRef]
- Miller, W.L.; Auchus, R.J. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 2011, 32, 81–151. [Google Scholar] [CrossRef] [PubMed]
- Brueggemeier, R.W. Update on the use of aromatase inhibitors in breast cancer. Expert Opin. Pharmacother 2006, 7, 1919–1930. [Google Scholar] [CrossRef] [PubMed]
- Lonning, P.E. Clinico-pharmacological aspects of different hormone treatments. Eur. J. Cancer 2000, 36, 81–82. [Google Scholar] [CrossRef]
- Marcotte, P.A.; Robinson, C.H. Synthesis and evaluation of 10β-substituted 4-estrene-3,17-diones as inhibitors of human placental microsomal aromatase. Steroids 1982, 39, 325–344. [Google Scholar] [CrossRef]
- Numazawa, M.; Nagaoka, M.; Handa, W.; Ogawa, Y.; Matsuoka, S. Studies directed towards a mechanistic evaluation of inactivation of aromatase by the suicide substrates androsta-1,4-diene-3,17-diones and its 6-ene derivatives Aromatase inactivation by the 19-substituted derivatives and their enzymic aromatization. J. Steroid Biochem. Mol. Biol. 2007, 107, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Sherwin, P.F.; McMullan, P.C.; Covey, D.F. Effects of steroid d-ring modification on suicide inactivation and competitive inhibition of aromatase by analogues of androsta-1,4-diene-3,17-dione. J. Med. Chem. 1989, 32, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Neumann, C.N.; Ritter, T. Introduction of Fluorine and Fluorine-Containing Functional Groups. Angew. Chem. Int. Ed. 2013, 52, 8214–8264. [Google Scholar] [CrossRef]
- Smart, B.E. Fluorine substituent effects (on bioactivity). J. Fluorine Chem. 2001, 109, 3–11. [Google Scholar] [CrossRef]
- Leo, A.; Hansch, C.; Elkins, D. Partition coefficients and their uses. Chem. Rev. 1971, 71, 525–616. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Unger, S.H.; Kim, K.H.; Nikaitan, D.; Lien, E.J. “Aromatic” substituent constants for structure-activity correlations. J. Med. Chem. 1973, 16, 1207–1216. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Taft, R.W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Pravst, I.; Papez Iskra, M.; Jereb, M.; Zupan, M.; Stavber, S. The role of F–N reagent and reaction conditions on fluorofunctionalisation of substituted phenols. Tetrahedron 2006, 62, 4474–4481. [Google Scholar] [CrossRef]
- Bogautdinov, R.P.; Fidarov, A.F.; Morozkina, S.N.; Zolotarev, A.A.; Starova, G.L.; Selivanov, S.I.; Shavva, A.G. Fluorination of steroid estrogens with Selectfluor: Elucidation of regio- and stereoselectivity. J. Fluorine Chem. 2014, 168, 218–222. [Google Scholar] [CrossRef]
- Zaikin, P.A.; Dyan, O.T.; Evtushok, D.V.; Usoltsev, A.N.; Borodkin, G.I.; Karpova, E.V.; Shubin, V.G. Solvent-Free Fluorination of Electron-Rich Aromatic Compounds with F-TEDA-BF4: Toward “Dry” Processes. Eur. J. Org. Chem. 2017, 17, 2469–2474. [Google Scholar] [CrossRef]
- Stavber, G.; Zupan, M.; Jereb, M.; Stavber, S. Selective and Effective Fluorination of Organic Compounds in Water using SelectfluorTM F-TEDA-BF4. Org. Lett. 2004, 6, 4973–4976. [Google Scholar] [CrossRef]
- Stavber, S.; Jereb, M.; Zupan, M. Efficient Synthesis of 4-Fluorocyclohexa-2,5-dienone Derivatives Using N-Fluoro-1,4-diazoniabicyclo[2.2.2]octane Salt Analogues. Synlett 1999, 9, 1375–1378. [Google Scholar] [CrossRef]
- Butenandt, A.; Wolff, A.; Karlson, P. Über Lumi-oestron. Chem. Ber. 1941, 74, 1308–1312. [Google Scholar] [CrossRef]
- Schönecker, B.; Lange, C.; Kötteritzsch, M.; Günther, W.; Weston, J.; Anders, E.; Görls, H. Conformational design for 13α-steroids. J. Org. Chem. 2000, 65, 5487–5497. [Google Scholar] [CrossRef]
- Roy, A.J.; Maltais, R.; Poirier, D. Impact of estradiol structural modifications (18-methyl and/or 17-hydroxy inversion of configuration) on the in vitro and in vivo estrogenic activity. J. Steroid Biochem. Mol. Biol. 2011, 127, 324–330. [Google Scholar]
- Vincent, S.P.; Burkart, M.D.; Tsai, C.Y.; Zhang, Z.; Wong, C.H. Electrophilic Fluorination−Nucleophilic Addition Reaction Mediated by Selectfluor: Mechanistic Studies and New Applications. J. Org. Chem. 1999, 64, 5264–5279. [Google Scholar] [CrossRef]
- Differding, E.; Rüegg, G. Nucleophilic substitution versus electron transfer: 1. On the mechanism of electrophilic fluorinations. Tetrahedron Lett. 1991, 32, 3815–3818. [Google Scholar] [CrossRef]
- Ashby, E.C.; Pham, T.N. The question of the validity of using radical probes for determining SET. The reaction of alkyl halides with LiAlH4. Tetrahedron Lett. 1987, 28, 3197–3200. [Google Scholar] [CrossRef]
- Zhang, Y.; Wen, C.; Li, J. C5-Regioselective C–H fluorination of 8-aminoquinoline amides and sulfonamides with Selectfluor under metal-free conditions. Org. Biomol. Chem. 2018, 16, 1912–1920. [Google Scholar] [CrossRef]
- Numazawa, M.; Ando, M.; Watari, Y.; Tominaga, T.; Hayata, Y.; Yoshimura, A. Structure-activity relationships of 2-, 4-, or 6-substituted estrogens as aromatase inhibitors. J. Steroid Biochem. Mol. Biol. 2005, 96, 51–58. [Google Scholar] [CrossRef]
- Kellis, J.T., Jr.; Vickery, L.E. Purification and characterization of human placental aromatase cytochrome P-450. J. Biol. Chem. 1987, 262, 4413–4420. [Google Scholar]
- Yoshida, N.; Osawa, Y. Purification of human placental aromatase cytochrome P-450 with monoclonal antibody and its characterization. Biochemistry 1991, 30, 3003–3010. [Google Scholar] [CrossRef] [PubMed]
- Numazawa, M.; Kamiyama, T.; Tachibana, M.; Oshibe, M. Synthesis and structure−activity relationships of 6-substituted androst-4-ene analogs as aromatase inhibitors. J. Med. Chem. 1996, 39, 2245–2252. [Google Scholar] [CrossRef] [PubMed]
- Bacsa, I.; Herman, B.E.; Jójárt, R.; Herman, K.S.; Wölfling, J.; Schneider, G.; Varga, M.; Tömböly, C.; Lanišnik Rižner, T.; Szécsi, M.; et al. Synthesis and structure–activity relationships of 2- and/or 4-halogenated 13β- and 13α-estrone derivatives as enzyme inhibitors of estrogen biosynthesis. J. Enzyme Inhib. Med. Chem. 2018, 33, 1271–1282. [Google Scholar] [CrossRef] [PubMed]
- Flynn, G.A.; Johnston, J.O.; Wright, C.L.; Metcalf, B.W. The time-dependent inactivation of aromatase by 17beta-hydroxy-10-methylthioestra-1,4-dien-3-one. Biochem. Biophys. Res. Commun. 1998, 103, 913–918. [Google Scholar] [CrossRef]
- Lovett, J.A.; Darby, M.V.; Counsell, R.E. Synthesis and evaluation of 19-aza- and 19-aminoandrostenedione analogues as potential aromatase inhibitors. J. Med. Chem. 1984, 27, 734–740. [Google Scholar] [CrossRef]
- Bednarski, P.J.; Porubek, D.J.; Nelson, S.D. Thiol-containing androgens as suicide substrates of aromatase. J. Med. Chem. 1985, 28, 775–779. [Google Scholar] [CrossRef]
- Bednarski, P.J.; Nelson, S.D. Interactions of thiol-containing androgens with human placental aromatase. J. Med. Chem. 1989, 32, 203–213. [Google Scholar] [CrossRef]
- Nakamura, H.; Shiozawa, T.; Terao, Y.; Shiraishi, F.; Fukazawa, H. By-products Produced by the Reaction of Estrogens with Hypochlorous Acid and their Estrogen Activities. J. Health. Sci. 2006, 52, 124–131. [Google Scholar] [CrossRef]
- Schrödinger Release 2018-4: Glide; Schrödinger, LLC: New York, NY, USA, 2018.
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Ghosh, D.; Lo, J.; Morton, D.; Valette, D.; Xi, J.; Griswold, J.; Hubbell, S.; Egbuta, C.; Jiang, W.; An, J.; et al. Novel Aromatase Inhibitors by Structure-Guided Design. J. Med. Chem. 2012, 55, 8464–8476. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
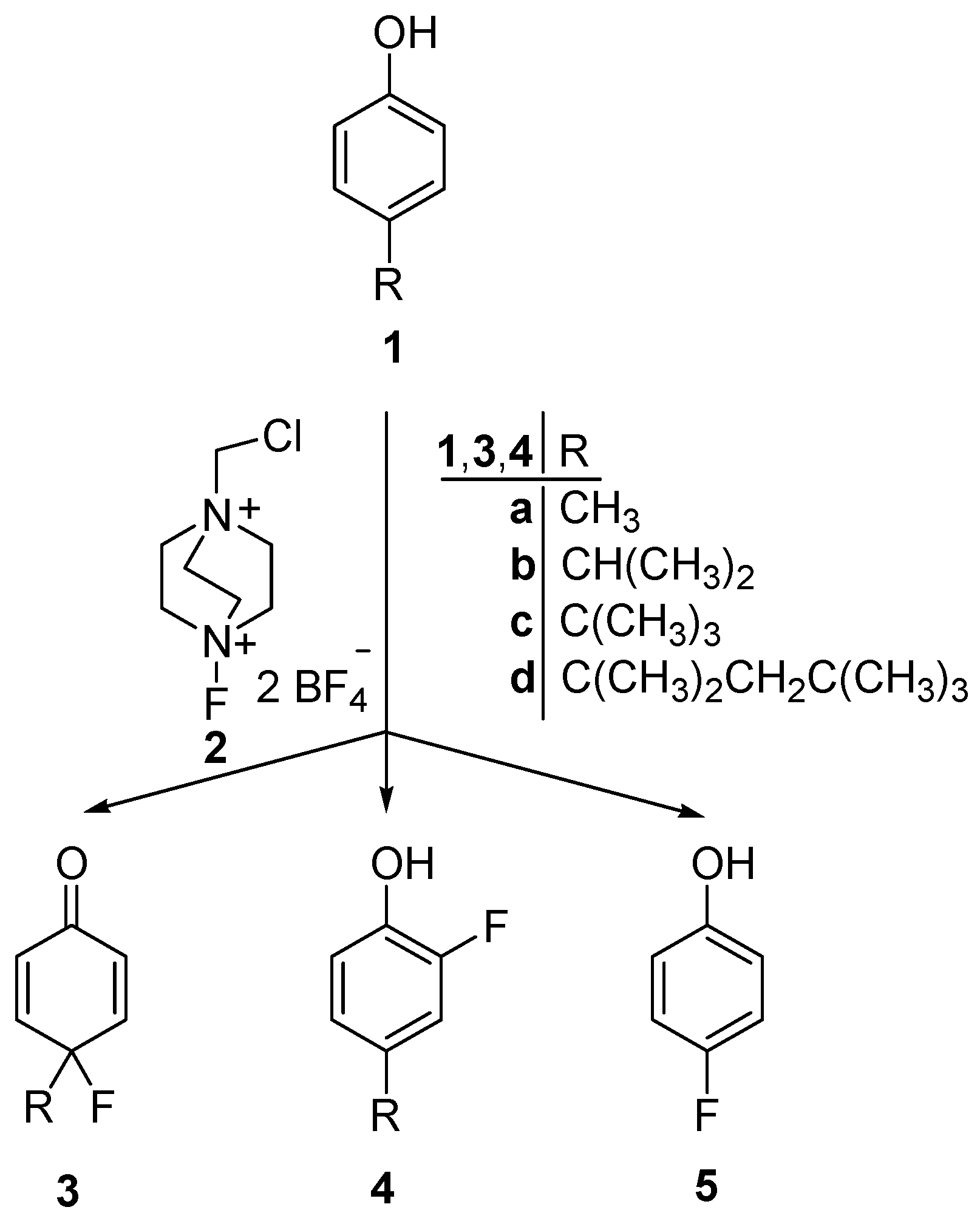{kind=link}
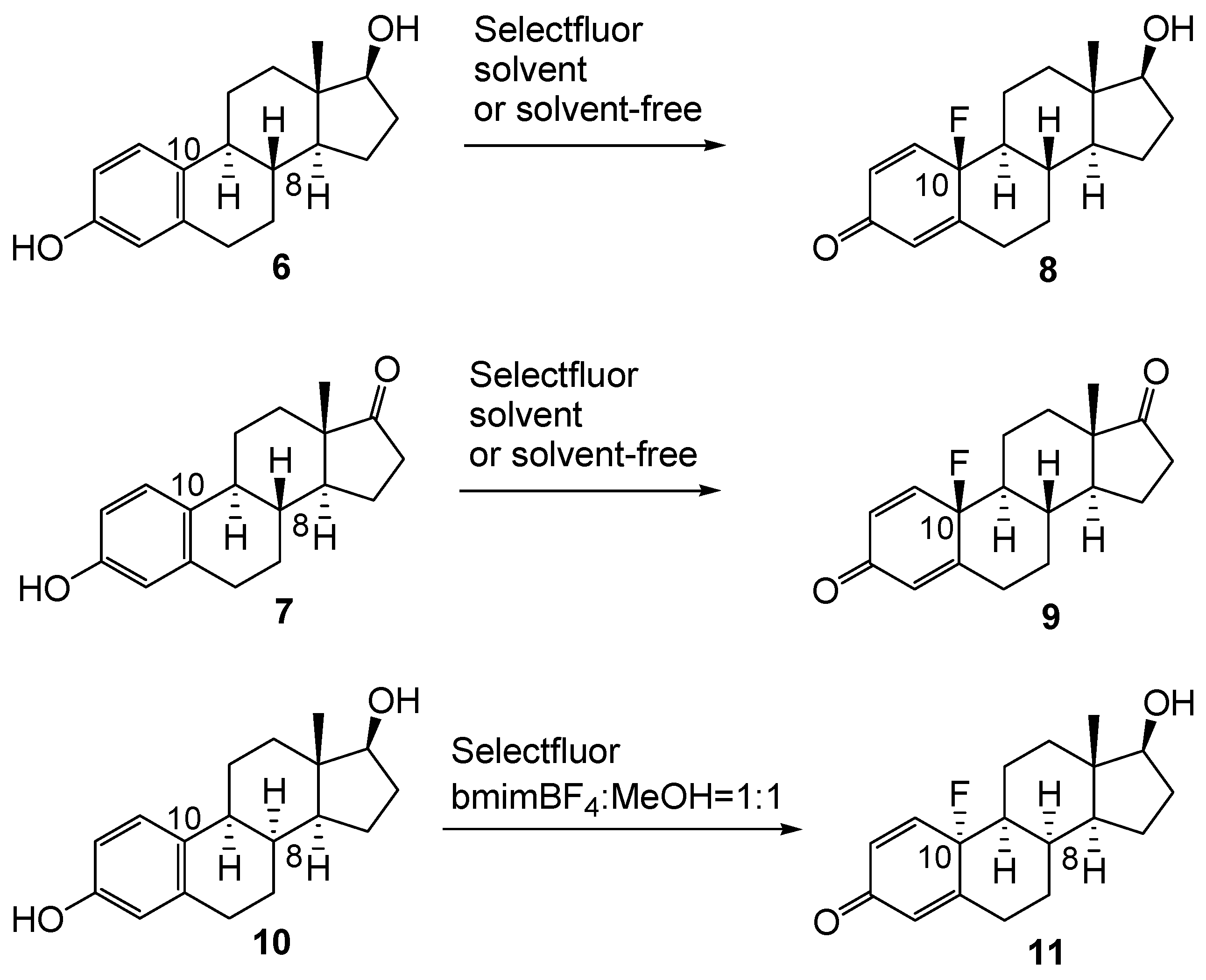{kind=link}
{kind=link}
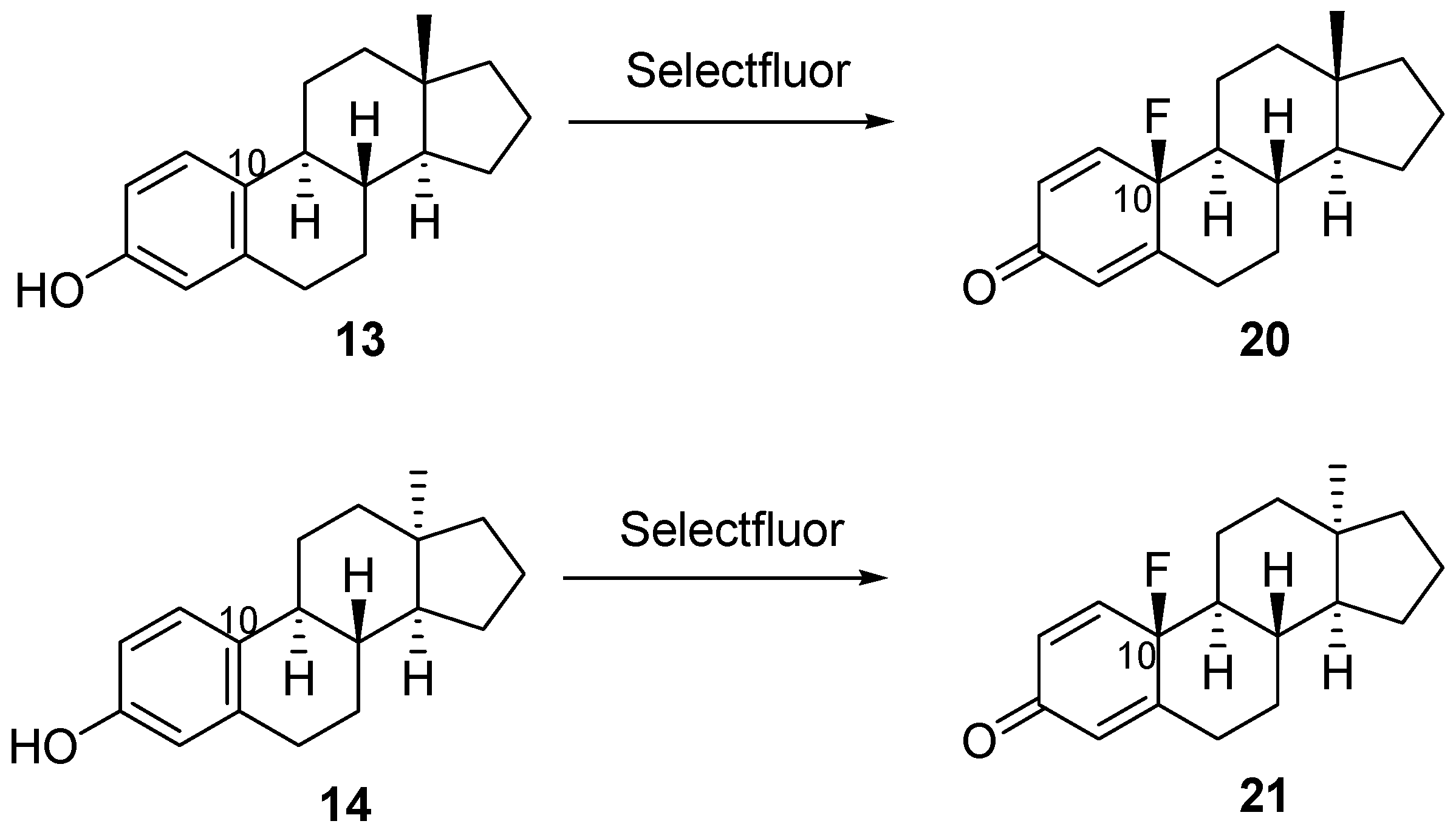{kind=link}
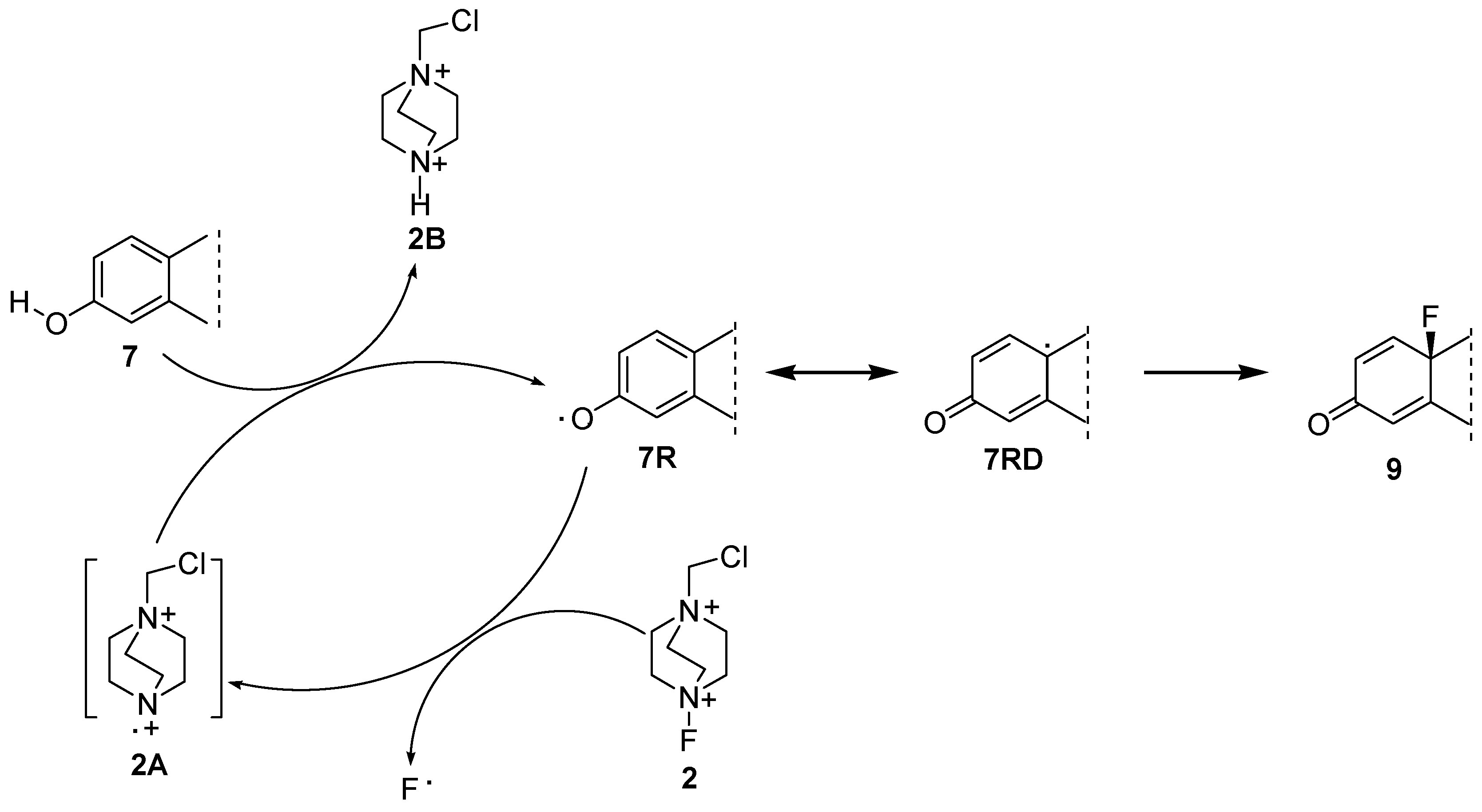{kind=link}
{kind=link}
| Entry | Substrate | Solvent | Temperature | Reaction Time | Product | Yield (%) |
|---|---|---|---|---|---|---|
| 1 | 7 | acetonitrile | rt | 24 h | 9 | 95 |
| 2 | 7 | acetonitrile | 80 °C | 1 h | 9 | 97 |
| 3 a | 7 | acetonitrile | rt | 24 h | 9 | 3 |
| 4 | 7 | methanol | rt | 24 h | 9 + (15 + 16) b | 76 + (16) |
| 5 | 7 | methanol | reflux | 1 h | 9 + (15 + 16) | 78 + (15) |
| 6 a | 7 | methanol | rt | 24 h | 9 | 2 |
| 7 | 12 | acetonitrile | rt | 24 h | 17 | 97 |
| 8 | 12 | acetonitrile | 80 °C | 1 h | 17 | 98 |
| 9 a | 12 | acetonitrile | rt | 24 h | 17 | 4 |
| 10 | 12 | methanol | rt | 24 h | 17 + (18 + 19) c | 71 + (12) |
| 11 | 12 | methanol | reflux | 1 h | 17 + (18 + 19) | 73 + (13) |
| 12 a | 12 | methanol | rt | 24 h | 17 | 94 |
| 13 | 13 | acetonitrile | rt | 24 h | 20 | 94 |
| 14 | 14 | acetonitrile | rt | 24 h | 21 | 92 |
| Entry | Substrate | NCS and or Selectfluor (1.1 equiv.) | Solvent | Temp. | Reaction Time | Yield Products 9 + 22 + 23 + 24 (%) |
|---|---|---|---|---|---|---|
| 1 a | 7 | NCS | acetonitrile | rt | 24 h | 0 + 0 + 30 + 45 |
| 2 a | 7 | NCS | acetonitrile | 80 °C | 1 h | 0 + 0 + 30 + 45 |
| 3 | 7 | NCS | TFA | rt | 24 h | 0 + 55 + 10 + 20 |
| 4 | 7 | NCS | TFA | 80 °C | 1 h | 0 + 55 + 10 + 20 |
| 5 b | 7 | NCS | TFA | 80 °C | 1 h | 0 + 54 + 11 + 21 |
| 6 | 7 | Selectfluor | TFA | rt | 24 h | 96 + 0 + 0 + 0 |
| 7 | 7 | Selectfluor | TFA | 80 °C | 1 h | 95 + 0 + 0 + 0 |
| 8 b | 7 | Selectfluor | TFA | rt | 24 h | 95 + 0 + 0 + 0 |
| 9 a | 7 | NCS, Selectfluor | acetonitrile | rt | 24 h | 62 + 0 + 15 + 20 |
| 10 a | 7 | NCS, Selectfluor | acetonitrile | 80 °C | 1 h | 62 + 0 + 15 + 20 |
| 11 | 7 | NCS, Selectfluor | TFA | rt | 24 h | 36 + 26 + 11 + 16 |
| 12 | 7 | NCS, Selectfluor | TFA | 80 °C | 1 h | 36 + 26 + 11 + 16 |
| 13 b | 7 | NCS, Selectfluor | TFA | 80 °C | 1 h | 36 + 25 + 11 + 17 |
| Compound | Structure | IC50 ± SD (µM) or Rel. conv. ± SD (%) |
|---|---|---|
| 9 |  | IC50 = 0.49 ± 0.07 |
| 20 |  | IC50 = 5.0 ± 2.4 |
| 17 |  | IC50 > 10 93 ± 11 |
| 21 |  | IC50 > 10 100 ± 6 |
| 22 |  | IC50 = 2.4 ± 0.4 |
| Androst-4-ene-3,17-dione |  | IC50 = 0.22 ± 0.2 |
| Androst-1,4-diene-3,17-dione |  | IC50 = 0.26 ± 0.06 |
| Compound | Emodel Score | Glide Score |
|---|---|---|
| 22 | −80.929 | −4.973 |
| 9 | −79.415 | −4.626 |
| 20 | −72.055 | −4.192 |
| 21 | −64.406 | −3.642 |
| 17 | −61.448 | −3.642 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jójárt, R.; Traj, P.; Kovács, É.; Horváth, Á.; Schneider, G.; Szécsi, M.; Pál, A.; Paragi, G.; Mernyák, E. Synthesis, Biological Evaluation and Docking Studies of 13-Epimeric 10-fluoro- and 10-Chloroestra-1,4-dien-3-ones as Potential Aromatase Inhibitors. Molecules 2019, 24, 1783. https://doi.org/10.3390/molecules24091783
Jójárt R, Traj P, Kovács É, Horváth Á, Schneider G, Szécsi M, Pál A, Paragi G, Mernyák E. Synthesis, Biological Evaluation and Docking Studies of 13-Epimeric 10-fluoro- and 10-Chloroestra-1,4-dien-3-ones as Potential Aromatase Inhibitors. Molecules. 2019; 24(9):1783. https://doi.org/10.3390/molecules24091783
Chicago/Turabian StyleJójárt, Rebeka, Péter Traj, Édua Kovács, Ágnes Horváth, Gyula Schneider, Mihály Szécsi, Attila Pál, Gábor Paragi, and Erzsébet Mernyák. 2019. "Synthesis, Biological Evaluation and Docking Studies of 13-Epimeric 10-fluoro- and 10-Chloroestra-1,4-dien-3-ones as Potential Aromatase Inhibitors" Molecules 24, no. 9: 1783. https://doi.org/10.3390/molecules24091783
APA StyleJójárt, R., Traj, P., Kovács, É., Horváth, Á., Schneider, G., Szécsi, M., Pál, A., Paragi, G., & Mernyák, E. (2019). Synthesis, Biological Evaluation and Docking Studies of 13-Epimeric 10-fluoro- and 10-Chloroestra-1,4-dien-3-ones as Potential Aromatase Inhibitors. Molecules, 24(9), 1783. https://doi.org/10.3390/molecules24091783