
Interactome Analysis and Docking Sites of MutS Homologs Reveal New Physiological Roles in Arabidopsis thaliana

Abstract
1. Introduction
2. Results
2.1. Multiple Sequence Alignment
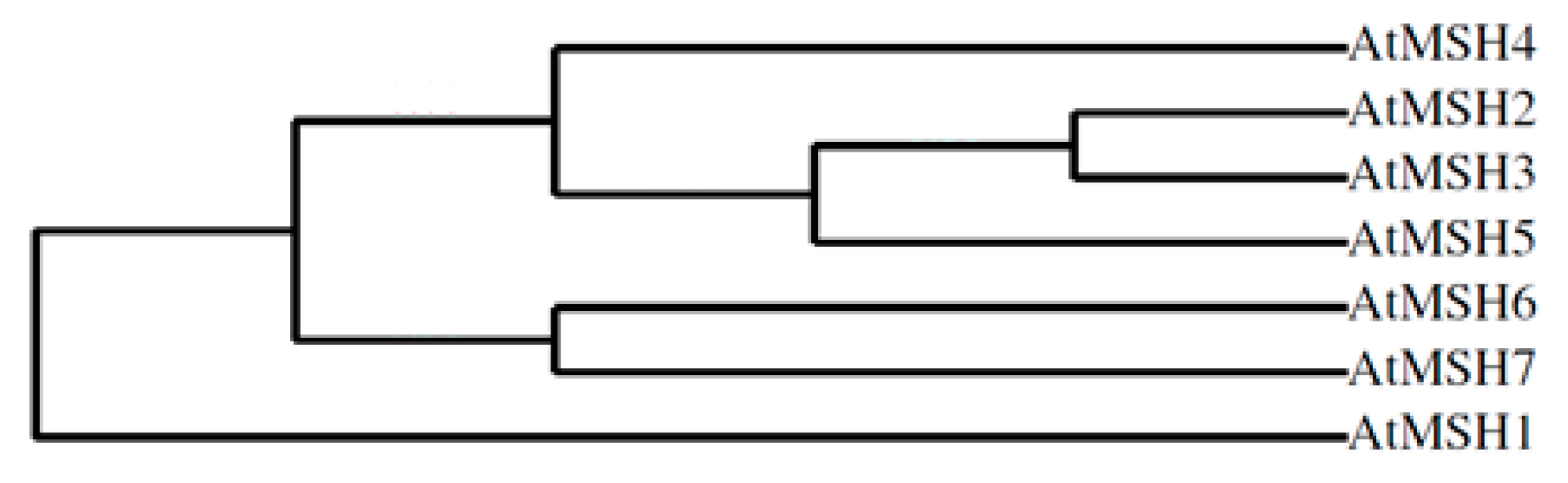
2.2. Phylogenetic Profile Rendering
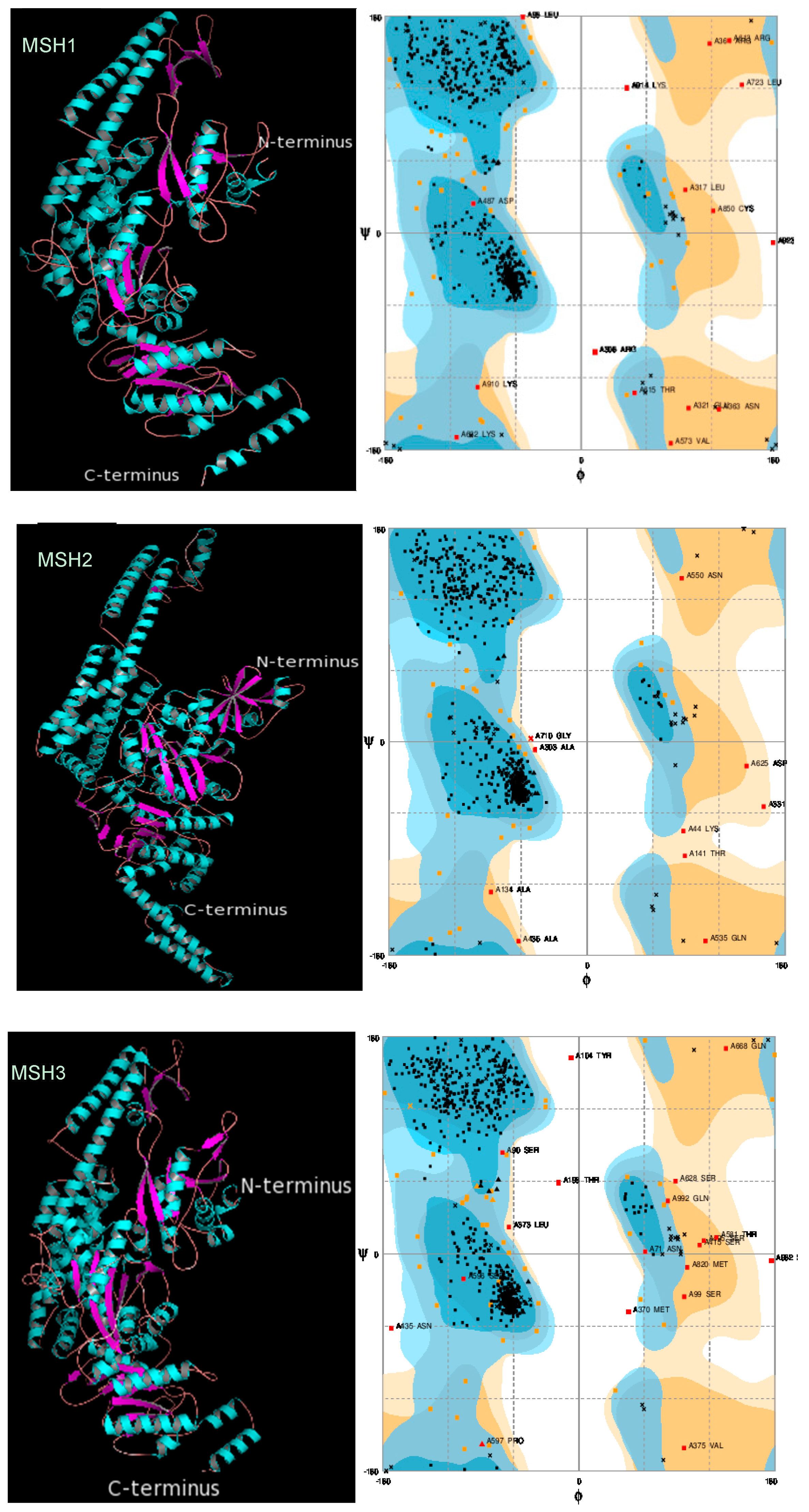
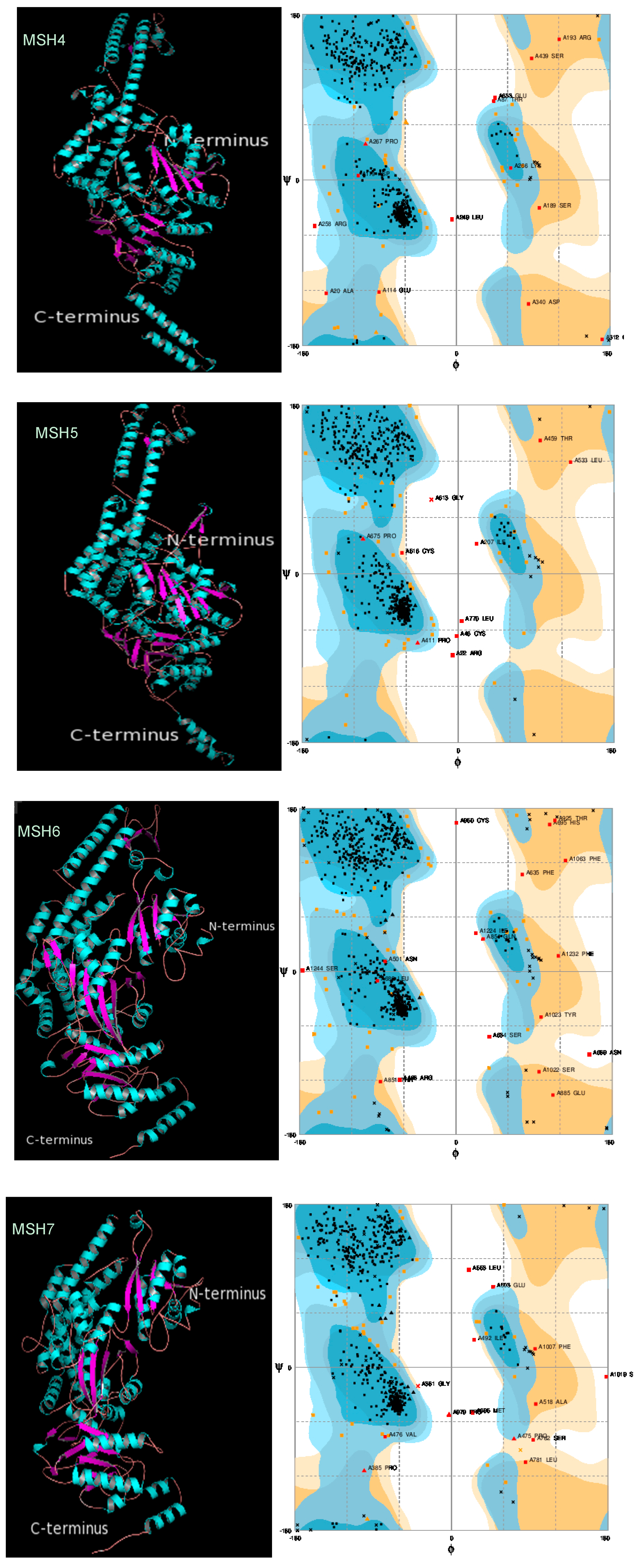
2.3. Protein 3D Structure Prediction and Refinement
2.4. Protein 3D Structure Validation
2.5. Protein Domain Identification
2.6. Interactome Analysis
2.7. Protein Subcellular Localization
2.8. Docking Site Prediction
3. Discussion
4. Materials and Methods
4.1. Sequences Retrieving and Multiple Sequence Alignment MSA
4.2. Phylogenetic Profile Rendering
4.3. Protein 3D Structure Prediction and Refinement
4.4. Protein 3D Structure Validation
4.5. Protein Domain Identification
4.6. Interactome Analysis
4.7. Protein Localization
4.8. Docking Site Prediction
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Helleday, T.; Eshtad, S.; Nik-Zainal, S. Mechanisms underlying mutational signatures in human cancers. Nat. Rev. Genet. 2014, 15, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Eker, A.P.M.; Quayle, C.; Chaves, I.; van der Horst, G.T.J. Direct DNA damage reversal: Elegant solutions for nasty problems. Cell. Mol. Life Sci. 2009, 66, 968–980. [Google Scholar] [CrossRef] [PubMed]
- Zharkov, D.O. Base excision DNA repair. Cell. Mol. Life Sci. 2008, 65, 1544–1565. [Google Scholar] [CrossRef] [PubMed]
- Schärer, O.D. Nucleotide excision repair in Eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef] [PubMed]
- Kolodner, R.D.; Marsischky, G.T. Eukaryotic DNA mismatch repair. Curr. Opin. Genet. Dev. 1991, 9, 89–96. [Google Scholar] [CrossRef]
- Gorbunova, V.; Levy, A. Non-homologous DNA end joining in plant cells is associated with deletions and filler DNA insertions. Nucl. Acids Res. 1997, 25, 4650–4657. [Google Scholar] [CrossRef] [PubMed]
- Hanin, M.; Paszkowski, J. Plant genome modification by homologous recombination. Curr. Opin. Plant Biol. 2003, 6, 157–162. [Google Scholar] [CrossRef]
- Ghosal, G.; Chen, J. DNA damage tolerance: A double-edged sword guarding the genome. Trans. Cancer Res. 2013, 2, 107–129. [Google Scholar]
- Jiricny, J. Postreplicative Mismatch Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012633. [Google Scholar] [CrossRef]
- Harfe, B.D.; Jinks-Robertson, S. DNA mismatch repair and genetic instability. Annu. Rev. Genet. 2000, 34, 359–399. [Google Scholar] [CrossRef]
- Rayssiguier, C.; Thaler, D.S.; Radman, M. The barrier to recotnbination between Escherichia coli and Salmonella typhimurium is disrupted in mismatch-repair mutants. Nature 1989, 342, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Petit, M.A.; Dimpfl, J.; Radman, M.; Echols, H. Control of large chromosomal duplications in Escherichia coli by the mismatch repair system. Genetics 1991, 129, 327–332. [Google Scholar] [PubMed]
- Umar, A.; Risinger, J.I.; Glaab, W.E.; Tindall, K.R.; Barrett, J.C.; Kunkel, T.A. Functional overlap in mismatch repair by human MSH3 and MSH6. Genetics 1998, 148, 1637–1646. [Google Scholar] [PubMed]
- Culligan, K.M.; Hays, J.B. Arabidopsis MutS Homologs—AtMSH2, AtMSH3, AtMSH6, and a Novel AtMSH7—Form Three Distinct Protein Heterodimers with Different Specificities for Mismatched DNA. Plant Cell 2000, 12, 991–1003. [Google Scholar] [PubMed]
- Hopfner, K.P.; Tainer, J.A. DNA Mismatch Repair: The Hands of a Genome Guardian. Structure 2000, 8, R237–R241. [Google Scholar] [CrossRef]
- Abdelnoor, R.V.; Yule, R.; Elo, A.; Christensen, A.C.; Meyer-Gauen, G.; Mackenzie, S.A. Substoichiometric shifting in the plant mitochondrial genome is influenced by a gene homologous to MutS. Proc. Natl. Acad. Sci. USA 2003, 100, 5968–5973. [Google Scholar] [CrossRef] [PubMed]
- Virdi, K.S.; Wamboldt, Y.; Kundariya, H.; Laurie, J.D.; Keren, I.; Kumar, K.R.S.; Block, A.; Mackenzie, S.A. MSH1 is a Plant Organellar DNA Binding and Thylakoid Protein under Precise Spatial Regulation to Alter Development. Mol. Plant 2016, 9, 245–260. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, A.P.; Abdelnoor, R.V.; Mackenzie, S.A. Transgenic induction of mitochondrial rearrangements for cytoplasmic male sterility in crop plants. Proc. Natl. Acad. Sci. USA 2007, 104, 1766–1770. [Google Scholar] [CrossRef] [PubMed]
- Lafleuriel, J.; Degroote, F.; Depeiges, A.; Picard, G. Impact of the loss of AtMSH2 on double-strand break-induced recombination between highly diverged homeologous sequences in Arabidopsis thaliana germinal tissues. Plant Mol. Biol. 2007, 63, 833–846. [Google Scholar] [CrossRef]
- Lario, L.D.; Ramirez-Parra, E.; Gutierrez, C.; Casati, P.; Spampinato, C.P. Regulation of plant MSH2 and MSH6 genes in the UV-B-induced DNA damage response. J. Exp. Bot. 2011, 62, 2925–2937. [Google Scholar] [CrossRef]
- Higgins, J.D.; Armstrong, S.J.; Franklin, F.C.; Jones, G.H. The Arabidopsis MutS homolog AtMSH4 functions at an early step in recombination: Evidence for two classes of recombination in Arabidopsis. Genes Dev. 2004, 18, 2557–2570. [Google Scholar] [CrossRef] [PubMed]
- Higgins, J.D.; Vignard, J.; Mercier, R.; Pugh, A.G.; Franklin, F.C.; Jones, G.H. AtMSH5 partners AtMSH4 in the class I meiotic crossover pathway in Arabidopsis thaliana, but is not required for synapsis. Plant J. 2008, 55, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Liu, X.; An, L.; Zhang, W.; Sun, J.; Pei, H.; Meng, H.; Fan, Y.; Zhang, C. The Arabidopsis MutS homolog AtMSH5 is required for normal meiosis. Cell Res. 2008, 18, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Culligan, K.M.; Meyer-Gauen, G.; Lyons-Weiler, J.; Hays, J.B. Evolutionary origin, diversification and specialization of eukaryotic MutS homolog mismatch repair proteins. Nucl. Acids Res. 2000, 28, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Kalman, M.; Ben-Tal, N. Quality assessment of protein model-structures using evolutionary conservation. Bioinformatics 2010, 26, 1299–1307. [Google Scholar] [CrossRef]
- New, L.; Liu, K.; Crouse, G.F. The yeast gene MSH3 defines a new class of eukaryotic MutS homologues. Mol. Gen. Genet. 1993, 239, 97–108. [Google Scholar]
- Lamers, M.H.; Perrakis, A.; Enzlin, J.H.; Winterwerp, H.H.; de Wind, N.; Sixma, T.K. The crystal structure of DNA mismatch repair protein MutS binding to a G x T mismatch. Nature 2000, 407, 711–717. [Google Scholar] [CrossRef]
- Obmolova, G.; Ban, C.; Hsieh, P.; Yang, W. Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature 2000, 407, 703–710. [Google Scholar] [CrossRef]
- Van Roey, P.; Meehan, L.; Kowalski, J.C.; Belfort, M.; Derbyshire, V. Catalytic domain structure and hypothesis for function of GIY-YIG intron endonuclease I-TevI. Nat. Struct. Biol. 2002, 9, 806–811. [Google Scholar] [CrossRef]
- Dunin-Horkawicz, S.; Feder, M.; Bujnicki, J.M. Phylogenomic analysis of the GIY-YIG nuclease superfamily. BMC Genom. 2006, 7, 98. [Google Scholar] [CrossRef]
- Raynaud, C.; Sozzani, R.; Glab, N.; Domenichini, S.; Perennes, C.; Cella, R.; Bergounioux, C. Two cell-cycle regulated SET-domain proteins interact with proliferating cell nuclear antigen (PCNA) in Arabidopsis. Plant J. 2006, 47, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Brasil, J.N.; Cabral, L.M.; Eloy, N.B.; Primo, L.M.F.; Barroso-Neto, I.L.; Grangeiro, L.P.P.; Hemerly, A.S. AIP1 is a novel Agenet/Tudor domain protein from Arabidopsis that interacts with regulators of DNA replication, transcription and chromatin remodeling. BMC Plant Biol. 2015, 15, 270. [Google Scholar] [CrossRef] [PubMed]
- Butler, G.S.; Overall, C.M. Proteomic identification of multitasking proteins in unexpected locations complicates drug targeting. Nat. Rev. Drug Discov. 2009, 8, 935–948. [Google Scholar] [CrossRef] [PubMed]
- Eisen, J.A. A phylogenomic study of the MutS family of proteins. Nucl. Acids Res. 1998, 26, 4291–4300. [Google Scholar] [CrossRef] [PubMed]
- Abdel Gawwad, M.R.; Alpdemir, S.; Eminagic, E. Interactome Analysis and Docking Sites of PCNA Subunits Reveal New Function in Arabidopsis thaliana. Curr. Proteom. 2014, 12, 152–167. [Google Scholar] [CrossRef]
- Hofmann, N.R. MutS HOMOLOG1 Stabilizes Plastid and Mitochondrial Genomes. Plant Cell 2011, 23, 3085. [Google Scholar] [CrossRef] [PubMed]
- Gueneau, E.; Dherin, C.; Legrand, P.; Tellier-Lebegue, C.; Gilquin, B.; Bonnesoeur, P.; Londino, F.; Charbonnier, J.B. Structure of the MutLα C-terminal domain reveals how Mlh1 contributes to Pms1 endonuclease site. Nat. Struct. Mol. Biol. 2013, 20, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Dion, E.; Li, L.; Jean, M.; Belzile, F. An Arabidopsis MLH1 mutant exhibits reproductive defects and reveals a dual role for this gene in mitotic recombination. Plant J. 2007, 51, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Hartwell, L.H.; Hood, L.; Goldberg, M.L.; Reynolds, A.E.; Silver, L.M. Genetics from Genes to Genomes, 4th ed.; McGraw-Hill: New York, NY, USA, 2011. [Google Scholar]
- Sandhu, A.P.S. Evaluation of a Mitochondrial Mutator System in Higher Plants; University of Nebraska: Lincoln, NE, USA, 2008. [Google Scholar]
- Miller-Messmer, M.; Kühn, K.; Bichara, M.; Le Ret, M.; Imbault, P.; Gualberto, J.M. RecA-Dependent DNA Repair Results in Increased Heteroplasmy of the Arabidopsis Mitochondrial Genome. Plant Physiol. 2012, 159, 211–226. [Google Scholar] [CrossRef] [PubMed]
- del Olmo, I.; Lopez-Gonzalez, L.; Martin-Trillo, M.M.; Martinez-Zapater, J.M.; Pineiro, M.; Jarillo, J.A. EARLY IN SHORT DAYS 7 (ESD7) encodes the catalytic subunit of DNA polymerase epsilon and is required for flowering repression through a mechanism involving epigenetic gene silencing. Plant J. 2010, 61, 623–636. [Google Scholar] [CrossRef]
- Bertrand, P.; Tishkoff, D.X.; Filosi, N.; Dasgupta, R.; Kolodner, R.D. Physical interaction between components of DNA mismatch repair and nucleotide excision repair. Proc. Natl. Acad. Sci. USA 1998, 95, 14278–14283. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Zhang, X.; Liu, J.; Wang, Y.; He, J.; Yang, T.; Hong, X.; Yang, Q.; Gong, Z. Epigenetic regulation, somatic homologous recombination, and abscisic acid signaling are influenced by DNA polymerase epsilon mutation in Arabidopsis. Plant Cell 2009, 21, 386–402. [Google Scholar] [CrossRef] [PubMed]
- Hartung, F.; Wurz-Wildersinn, R.; Fuchs, J.; Schubert, I.; Suer, S.; Puchta, H. The catalytically active tyrosine residues of both SPO11-1 and SPO11-2 are required for meiotic double-strand break induction in Arabidopsis. Plant Cell 2007, 19, 3090–3099. [Google Scholar] [CrossRef] [PubMed]
- Bagherieh-Najjar, M.B.; de Vries, O.M.H.; Kroon, J.T.M.; Wright, E.L.; Elborough, K.M.; Hille, J.; Dijkwel, P.P. Arabidopsis RecQsim, a plant-specific member of the RecQ helicase family, can suppress the MMS hypersensitivity of the yeast sgs1 mutant. Plant Mol. Biol. 2003, 52, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Dubest, S.; Gallego, M.E.; White, C.I. Role of the AtRad1p endonuclease in homologous recombination in plants. EMBO Rep. 2002, 3, 1049–1054. [Google Scholar] [CrossRef] [PubMed]
- Dubest, S.; Gallego, M.E.; White, C.I. Roles of the AtErcc1 protein in recombination. Plant J. 2004, 39, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Saparbaev, M.; Prakash, L.; Prakash, S. Requirement of mismatch repair genes MSH2 and MSH3 in the RAD1–RAD10 pathway of mitotic recombination in Saccharomyces cerevisiae. Genetics 1996, 142, 727–736. [Google Scholar]
- Stacey, N.J.; Kuromori, T.; Azumi, Y.; Roberts, G.; Breuer, C.; Wada, T.; Maxwell, A.; Roberts, K.; Sugimoto-Shirasu, K. Arabidopsis SPO11-2 functions with SPO11-1 in meiotic recombination. Plant J. 2006, 48, 206–216. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. Available online: http://www.ncbi.nlm.nih.gov/ (accessed on 19 February 2016).
- Swarbreck, D.; Wilks, C.; Lamesch, P.; Berardini, T.Z.; Garcia-Hernandez, M.; Foerster, H.; Li, D.; Huala, E. The Arabidopsis Information Resource (TAIR): Gene structure and function annotation. Nucl. Acids Res. 2008, 36, D1009–D1014. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.G.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; Higgins, D. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Goujon, M.; McWilliam, H.; Li, W.; Valentin, F.; Squizzato, S.; Paern, J.; Lopez, R. A new bioinformatics analysis tools framework at EMBL-EBI. Nucl. Acids Res. 2010, 38, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cowley, A.; Uludag, M.; Gur, T.; McWilliam, H.; Squizzato, S.; Park, Y.M.; Lopez, R. The EMBL-EBI bioinformatics web and programmatic tools framework. Nucl. Acids Res. 2015, 43, W580–W584. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cowley, A.; Uludag, M.; Gur, T.; McWilliam, H.; Squizzato, S.; Park, Y.M.; Lopez, R. Analysis Tool Web Services from the EMBL-EBI. Nucl. Acids Res. 2013, 41, W597–W600. [Google Scholar]
- Dereeper, A.; Audic, S.; Claverie, J.M.; Blanc, G. BLAST-EXPLORER helps you building datasets for phylogenetic analysis. BMC Evol. Biol. 2010, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny.fr: Robust phylogenetic analysis for the non-specialist. Nucl. Acids Res. 2008, 36 (Suppl. 2), W465–W469. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, D.; Cheng, J. 3Drefine software for protein 3D structure refinement and its assessment in CASP10. PLoS ONE 2013, 8, e69648. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, D.; Cheng, J. 3Drefine: Consistent Protein Structure Refinement by Optimizing Hydrogen Bonding Network and Atomic Level Energy Minimization. Proteins Struct. Funct. Bioinform. 2012, 81, 119–131. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System; Version 1.8; CRC Press LLC: Boca Raton, FL, USA, 2014.
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- DeepView-Swiss-PdbViewer. Available online: http://www.expasy.org/spdbv/ (accessed on 3 May 2016).
- Lovell, S.C.; Davis, I.W.; Arendall, W.B.; de Bakker, P.I.W.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Calpha geometry: Phi, psi and Cbeta deviation. Proteins Struct. Funct. Genet. 2002, 50, 437–450. [Google Scholar] [CrossRef]
- Ramachandran, G.N.; Ramakrishnan, C.; Sasisekharan, V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 1963, 7, 95–99. [Google Scholar] [CrossRef]
- Schultz, J.; Milpetz, F.; Bork, P.; Ponting, C.P. SMART, a simple modular architecture research tool: Identification of signaling domains. Proc. Natl. Acad. Sci. USA 1998, 95, 5857–5864. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Doerks, T.; Bork, P. SMART: Recent updates, new developments and status in 2015. Nucl. Acids Res. 2015, 43, D257–D260. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; von Mering, C. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucl. Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, Z.; Mei, Q.; Chen, M. PSI: A Comprehensive and Integrative Approach for Accurate Plant Subcellular Localization Prediction. PLoS ONE 2013, 8, e75826. [Google Scholar] [CrossRef]
- Shen, H.B.; Chou, K.C. Nuc-PLoc: A new web-server for predicting protein subnuclear localization by fusing PseAA composition and PsePSSM. Protein Eng. Des. Sel. 2007, 20, 561–567. [Google Scholar] [CrossRef]
- Kozakov, D.; Beglov, D.; Bohnuud, T.; Mottarella, S.; Xia, B.; Hall, D.R.; Vajda, S. How good is automated protein docking? Proteins Struct. Funct. Bioinform. 2013, 81, 2159–2166. [Google Scholar] [CrossRef]
- Kozakov, D.; Brenke, R.; Comeau, S.R.; Vajda, S. PIPER: An FFT-based protein docking program with pairwise potentials. Proteins Struct. Funct. Bioinform. 2006, 65, 392–406. [Google Scholar] [CrossRef]
- Comeau, S.R.; Gatchell, D.W.; Vajda, S.; Camacho, C.J. ClusPro: An automated docking and discrimination method for the prediction of protein complexes. Bioinformatics 2004, 20, 45–50. [Google Scholar] [CrossRef]
- Comeau, S.R.; Gatchell, D.W.; Vajda, S.; Camacho, C.J. ClusPro: A fully automated algorithm for protein-protein docking. Nucl. Acids Res. 2004, 32 (Suppl. 2), W96–W99. [Google Scholar] [CrossRef]
- Porollo, A.; Meller, J. Prediction-based Fingerprints of Protein-Protein Interactions. Proteins Struct. Funct. Bioinform. 2007, 66, 630–645. [Google Scholar] [CrossRef] [PubMed]
- Janin, J.; Henrick, K.; Moult, J.; Ten Eyck, L.; Sternberg, M.J.; Vajda, S.; Vakser, I.; Wodak, S.J. CAPRI: A critical assessment of predicted interactions. Proteins 2003, 52, 2–9. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
| Protein | Percent Identity | ||||||
|---|---|---|---|---|---|---|---|
| MSH1 | MSH4 | MSH5 | MSH6 | MSH7 | MSH2 | MSH3 | |
| MSH1 | 100.00 | 19.08 | 18.03 | 22.86 | 21.49 | 19.09 | 20.36 |
| MSH4 | 19.08 | 100.00 | 22.51 | 24.53 | 25.29 | 24.56 | 22.88 |
| MSH5 | 18.03 | 22.51 | 100.00 | 23.60 | 22.33 | 22.93 | 24.16 |
| MSH6 | 22.86 | 24.53 | 23.60 | 100.00 | 33.30 | 23.95 | 26.30 |
| MSH7 | 21.49 | 25.29 | 22.33 | 33.30 | 100.00 | 25.37 | 25.56 |
| MSH2 | 19.09 | 24.56 | 22.93 | 23.95 | 25.37 | 100.00 | 27.93 |
| MSH3 | 20.36 | 22.88 | 24.16 | 26.30 | 25.56 | 27.93 | 100.00 |
| Protein | RAMPAGE (Residues in Allowed Region) | PROCHECK (G-Factor) | dDFIRE |
|---|---|---|---|
| MSH1 | 98.1% | −0.28 | −1755.42 |
| MSH2 | 98.9% | −0.05 | −2105.67 |
| MSH3 | 98.0% | −0.18 | −2052.20 |
| MSH4 | 98.1% | −0.16 | −1753.06 |
| MSH5 | 98.7% | −0.23 | −1797.04 |
| MSH6 | 98.1% | −0.18 | −2093.36 |
| MSH7 | 98.3% | −0.19 | −1841.97 |
| Domain and Accession | Protein | |||||||
|---|---|---|---|---|---|---|---|---|
| AtMSH1 | AtMSH2 | AtMSH3 | AtMSH4 | AtMSH5 | AtMSH6 | AtMSH7 | ||
| MUTSac (SM000534) | Start | 761 | 659 | 810 | 546 | 562 | 1076 | 846 |
| End | 947 | 855 | 1006 | 733 | 757 | 1268 | 1043 | |
| MUTSd (SM000533) | Start | - | 314 | 440 | 190 | 211 | 716 | 573 |
| End | - | 642 | 793 | 531 | 547 | 1056 | 822 | |
| Pfam:MutS_I (PF01624) | Start | 125 | 22 | 105 | - | - | 380 | 268 |
| End | 228 | 129 | 218 | - | - | 496 | 382 | |
| Pfam:MutS_II (PF05188) | Start | - | 142 | 235 | - | - | 505 | 388 |
| End | - | 284 | 361 | - | - | 676 | 542 | |
| Pfam:GIY-YIG (PF01541) | Start | 1024 | - | - | - | - | - | - |
| End | 1091 | - | - | - | - | - | - | |
| TUDOR (PF00567) | Start | - | - | - | - | - | 121 | - |
| End | - | - | - | - | - | 179 | - | |
| MSH | Interactor | CV | ||
|---|---|---|---|---|
| Name | Accession | Function | ||
| AtMSH1 | MLH1 | AT4G09140.1 | MUTL-homologue 1; correcting IDLs in MMR coming from DNA replication, DNA damage or heterologous recombination in meiosis | 0.999 |
| MLH3 | AT4G35520.1 | MUTL protein homolog 3; correcting IDLs in MMR coming from DNA replication, DNA damage or heterologous recombination in meiosis | 0.996 | |
| PMS1 | AT4G02460.1 | Postmeiotic segregation 1; correcting non-Watson–Crick base pairing and IDLs in MMR; coming from DNA replication, DNA damage or heterologous recombination in meiosis | 0.999 | |
| PCNA1 | AT1G07370.1 | Proliferating cellular nuclear antigen 1; auxiliary protein of DNA polδ; controls eukaryotic DNA replication | 0.992 | |
| PCNA2 | AT2G29570.1 | proliferating cell nuclear antigen 2; auxiliary protein of DNA polδ; controls eukaryotic DNA replication | 0.991 | |
| MSH2 | AT3G18524.1 | MUTS homolog 2; (see Introduction for detailed description) | 0.981 | |
| MSH5 | AT3G20475.1 | MUTS-homolog 5; (see Introduction for detailed description) | 0.981 | |
| RECA3 | AT3G10140.1 | RECA homolog 3; plays role in recombination ability DNA strand transfer | 0.980 | |
| TIL1 | AT1G08260.1 | TILTED 1; DNA polymerase II; involved in DNA replication. Important physiological role (timing and determination of cell fate during plant embryogenesis and root pole development; required for proper shoot (SAM) and root apical meristem (RAM) function; required for flowering repression | 0.974 | |
| TIL2 | AT2G27120.1 | TILTED 2; DNA polymerase II; involved in DNA replication, promotes cell cycle and cell type patterning. Contributes to flowering time repression | 0.974 | |
| AtMSH2 | MLH1 | AT4G09140.1 | MUTL-homologue 1; correcting IDLs in MMR coming from DNA replication, DNA damage or heterologous recombination in meiosis | 0.999 |
| MLH3 | AT4G35520.1 | MUTL protein homolog 3; correcting IDLs in MMR coming from DNA replication, DNA damage or heterologous recombination in meiosis | 0.997 | |
| PMS1 | AT4G02460.1 | Postmeiotic segregation 1; correcting non-Watson–Crick base pairing and IDLs in MMR; coming from DNA replication, DNA damage or heterologous recombination in meiosis. | 0.999 | |
| PCNA1 | AT1G07370.1 | proliferating cellular nuclear antigen 1; auxiliary protein of DNA polδ; controls eukaryotic DNA replication | 0.997 | |
| PCNA2 | AT2G29570.1 | proliferating cell nuclear antigen 2; auxiliary protein of DNA polδ; controls eukaryotic DNA replication | 0.998 | |
| MSH7 | AT3G24495.1 | MUTS homolog 7; (see Introduction for detailed description) | 0.997 | |
| MSH6 | AT4G02070.1 | MUTS homolog 6; (see Introduction for detailed description) | 0.983 | |
| UVH1 | AT5G41150.1 | DNA repair endonuclease UVH1; probably involved in NER and repair of UV light damage, and oxidative damage. In vitro, repairs DSBs and is required for homologous recombination | 0.991 | |
| TIL1 | AT1G08260.1 | TILTED 1; DNA polymerase II; involved in DNA replication. Important physiological role (timing and determination of cell fate during plant embryogenesis and root pole development; required for proper shoot (SAM) and root apical meristem (RAM) function; required for flowering repression | 0.974 | |
| RECQSIM | AT5G27680.1 | RECQ helicase SIM; Involved in DNA repair; 3′-5′ helicase specific for plants | 0.991 | |
| ERCC1 | AT3G05210.1 | DNA excision repair protein ERCC-1; involved in NER. In vitro, repairs DSBs and is required for homologous recombination. UVH1/RAD1-ERCC1/RAD10 complex acts as endonuclease | 0.990 | |
| AtMSH3 | MLH1 | AT4G09140.1 | MUTL-homologue 1; correcting IDLs in MMR coming from DNA replication, DNA damage or heterologous recombination in meiosis | 0.999 |
| MLH3 | AT4G35520.1 | MUTL protein homolog 3; correcting IDLs in MMR coming from DNA replication, DNA damage or heterologous recombination in meiosis | 0.997 | |
| PMS1 | AT4G02460.1 | Postmeiotic segregation 1; correcting non-Watson-Crick base pairing and IDLs in MMR; coming from DNA replication, DNA damage or heterologous recombination in meiosis | 0.999 | |
| PCNA1 | AT1G07370.1 | proliferating cellular nuclear antigen 1; auxiliary protein of DNA polδ; controls eukaryotic DNA replication | 0.955 | |
| PCNA2 | AT2G29570.1 | proliferating cell nuclear antigen 2; auxiliary protein of DNA polδ; controls eukaryotic DNA replication | 0.968 | |
| AT2G02550 | AT2G02550.2 | PIN domain-containing protein; nuclease | 0.965 | |
| AT1G29630 | AT1G29630.2 | exonuclease 1; dsDNAexonuclease. May be involved in DNA mismatch repair (MMR) | 0.965 | |
| AT1G18090 | AT1G18090.1 | 5′-3′ exonuclease family protein | 0.965 | |
| ERCC1 | AT3G05210.1 | DNA excision repair protein ERCC-1; involved in NER. In vitro, repairs DSBs and is required for homologous recombination. UVH1/RAD1-ERCC1/RAD10 complex acts as endonuclease | 0.953 | |
| RECQSIM | AT5G27680.1 | RECQ helicase SIM; Involved in DNA repair; 3′-5′ helicase specific for plants | 0.863 | |
| AtMSH4 | MLH1 | AT4G09140.1 | MUTL-homologue 1; correcting IDLs in MMR coming from DNA replication, DNA damage or heterologous recombination in meiosis | 0.999 |
| MLH3 | AT4G35520.1 | MUTL protein homolog 3; correcting IDLs in MMR coming from DNA replication, DNA damage or heterologous recombination in meiosis | 0.999 | |
| PMS1 | AT4G02460.1 | Postmeiotic segregation 1; correcting non-Watson–Crick base pairing and IDLs in MMR; coming from DNA replication, DNA damage or heterologous recombination in meiosis | 0.998 | |
| MSH5 | AT3G20475.1 | MUTS-homologue 5; (see Introduction for detailed description) | 0.987 | |
| RAD51 | AT5G20850.1 | DNA repair protein RAD51-like 1; binds ss- and dsDNA; DNA-dependent ATPase; repair of meiotic DBSs generated by AtSPO11-1 and in homologous recombination. Important for vegetative growth and root mitosis | 0.984 | |
| MUS81 | AT4G30870.1 | MMS andUV sensitive 81; part of endonuclease complex. Involved in DNA repair and homologous recombination (HR) in somatic cells. | 0.980 | |
| ATSPO11-1 | AT3G13170.1 | Meiotic recombination protein SPO11-1; part of meiotic recombination. Cleaves DNA to make DSB and start meiotic recombination | 0.970 | |
| RCK | AT3G27730.1 | ROCK-N-ROLLERS; DNA helicase important for meiosis | 0.964 | |
| DMC1 | AT3G22880.1 | Disruption of meiotic control 1; May participate in meiotic recombination | 0.958 | |
| SPO11-2 | AT1G63990.1 | sporulation 11-2; involved in meiotic recombination. Cleaves DNA to make DSB and start meiotic recombination | 0.931 | |
| AtMSH5 | MLH1 | AT4G09140.1 | MUTL-homologue 1; correcting IDLs in MMR coming from DNA replication, DNA damage or heterologous recombination in meiosis | 0.999 |
| MLH3 | AT4G35520.1 | MUTL protein homolog 3; correcting IDLs in MMR coming from DNA replication, DNA damage or heterologous recombination in meiosis | 0.999 | |
| PMS1 | AT4G02460.1 | Postmeiotic segregation 1; correcting non-Watson-Crick base pairing and IDLs in MMR; coming from DNA replication, DNA damage or heterologous recombination in meiosis | 0.999 | |
| PCNA1 | AT1G07370.1 | proliferating cellular nuclear antigen 1; auxiliary protein of DNA polδ; controls eukaryotic DNA replication | 0.993 | |
| PCNA2 | AT2G29570.1 | proliferating cell nuclear antigen 2; auxiliary protein of DNA polδ; controls eukaryotic DNA replication | 0.993 | |
| RECQ4A | AT1G10930.1 | ATP-dependent DNA helicase Q-like 4A; DNA helicase possibly involved in repair of DNA | 0.994 | |
| RECQSIM | AT5G27680.1 | RECQ helicase SIM; Involved in DNA repair; 3′-5′ helicase specific for plants | 0.991 | |
| RecQI3 | AT4G35740.1 | ATP-dependent DNA helicase Q-like 3; DNA helicase; possible role in DNA repair. Mediates DNA strand annealing | 0.991 | |
| RECQI1 | AT3G05740.1 | RECQ helicase l1; DNA helicase; possible role in DNA repair | 0.991 | |
| RECQ4B | AT1G60930.1 | RECQ helicase L4B; DNA helicase; possible role in DNA repair; promotes crossovers | 0.991 | |
| AtMSH6 | MLH1 | AT4G09140.1 | MUTL-homologue 1; correcting IDLs in MMR coming from DNA replication, DNA damage or heterologous recombination in meiosis | 0.999 |
| MLH3 | AT4G35520.1 | MUTL protein homolog 3; correcting IDLs in MMR coming from DNA replication, DNA damage or heterologous recombination in meiosis | 0.998 | |
| PMS1 | AT4G02460.1 | Postmeiotic segregation 1; correcting non-Watson–Crick base pairing and IDLs in MMR; coming from DNA replication, DNA damage or heterologous recombination in meiosis | 0.999 | |
| PCNA1 | AT1G07370.1 | proliferating cellular nuclear antigen 1; auxiliary protein of DNA polδ; controls eukaryotic DNA replication | 0.995 | |
| PCNA2 | AT2G29570.1 | proliferating cell nuclear antigen 2; auxiliary protein of DNA polδ; controls eukaryotic DNA replication | 0.996 | |
| MSH5 | AT3G20475.1 | MUTS-homologue 5; (see Introduction for detailed description) | 0.984 | |
| MSH2 | AT3G18524.1 | MUTS homolog 2; (see Introduction for detailed description) | 0.983 | |
| TIL1 | AT1G08260.1 | TILTED 1; DNA polymerase II; involved in DNA replication. Important physiological role (timing and determination of cell fate during plant embryogenesis and root pole development; required for proper shoot (SAM) and root apical meristem (RAM) function; required for flowering repression | 0.974 | |
| TIL2 | AT2G27120.1 | TILTED 2; DNA polymerase II; involved in DNA replication, promotes cell cycle and cell type patterning. Contributes to flowering time repression | 0.974 | |
| RECQSIM | AT5G27680.1 | RECQ helicase SIM; Involved in DNA repair; 3′-5′ helicase specific for plants | 0.970 | |
| AtMSH7 | MLH1 | AT4G09140.1 | MUTL-homologue 1; correcting IDLs in MMR coming from DNA replication, DNA damage or heterologous recombination in meiosis. | 0.999 |
| MLH3 | AT4G35520.1 | MUTL protein homolog 3; correcting IDLs in MMR coming from DNA replication, DNA damage or heterologous recombination in meiosis | 0.997 | |
| PMS1 | AT4G02460.1 | Postmeiotic segregation 1; correcting non-Watson–Crick base pairing and IDLs in MMR; coming from DNA replication, DNA damage or heterologous recombination in meiosis | 0.999 | |
| PCNA1 | AT1G07370.1 | proliferating cellular nuclear antigen 1; auxiliary protein of DNA polδ; controls eukaryotic DNA replication | 0.996 | |
| PCNA2 | AT2G29570.1 | proliferating cell nuclear antigen 2; auxiliary protein of DNA polδ; controls eukaryotic DNA replication | 0.995 | |
| MSH5 | AT3G20475.1 | MUTS-homologue 5; (see Introduction for detailed description) | 0.984 | |
| MSH2 | AT3G18524.1 | MUTS homolog 2; (see Introduction for detailed description) | 0.997 | |
| TIL1 | AT1G08260.1 | TILTED 1; DNA polymerase II; involved in DNA replication Important physiological role (timing and determination of cell fate during plant embryogenesis and root pole development; required for proper shoot (SAM) and root apical meristem (RAM) function; required for flowering repression | 0.976 | |
| TIL2 | AT2G27120.1 | TILTED 2; DNA polymerase II; involved in DNA replication, promotes cell cycle and cell type patterning. Contributes to flowering time repression | 0.976 | |
| RECQSIM | AT5G27680.1 | RECQ helicase SIM; Involved in DNA repair; 3′-5′ helicase specific for plants | 0.968 | |
| Protein | Subcellular Localization | Subnuclear Localization |
|---|---|---|
| MSH1 | Mitochondrion Chloroplast | - - |
| MSH2 | Nucleus | Nucleolus |
| MSH3 | Nucleus | Nucleolus |
| MSH4 | Nucleus | Nucleolus |
| MSH5 | Nucleus | Nucleolus |
| MSH6 | Nucleus | Nucleolus |
| MSH7 | Nucleus | Nucleolus |
| Protein Name | Accession Number | Sequence Length | |
|---|---|---|---|
| NCBI | TAIR | ||
| AtMSH1 | Q84LK0.1 | AT3G24320.1 | 1118 aa |
| AtMSH2 | O24617.1 | AT3G18524.1 | 937 aa |
| AtMSH3 | O65607.2 | AT4G25540.1 | 1081 aa |
| AtMSH4 | F4JP48.1 | AT4G17380.1 | 792 aa |
| AtMSH5 | F4JEP5.1 | AT3G20475.1 | 807 aa |
| AtMSH6 | O04716.2 | AT4G02070.1 | 1324 aa |
| AtMSH7 | Q9SMV7.1 | AT3G24495.1 | 1109 aa |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
AbdelGawwad, M.R.; Marić, A.; Al-Ghamdi, A.A.; Hatamleh, A.A. Interactome Analysis and Docking Sites of MutS Homologs Reveal New Physiological Roles in Arabidopsis thaliana. Molecules 2019, 24, 2493. https://doi.org/10.3390/molecules24132493
AbdelGawwad MR, Marić A, Al-Ghamdi AA, Hatamleh AA. Interactome Analysis and Docking Sites of MutS Homologs Reveal New Physiological Roles in Arabidopsis thaliana. Molecules. 2019; 24(13):2493. https://doi.org/10.3390/molecules24132493
Chicago/Turabian StyleAbdelGawwad, Mohamed Ragab, Aida Marić, Abdullah Ahmed Al-Ghamdi, and Ashraf A. Hatamleh. 2019. "Interactome Analysis and Docking Sites of MutS Homologs Reveal New Physiological Roles in Arabidopsis thaliana" Molecules 24, no. 13: 2493. https://doi.org/10.3390/molecules24132493
APA StyleAbdelGawwad, M. R., Marić, A., Al-Ghamdi, A. A., & Hatamleh, A. A. (2019). Interactome Analysis and Docking Sites of MutS Homologs Reveal New Physiological Roles in Arabidopsis thaliana. Molecules, 24(13), 2493. https://doi.org/10.3390/molecules24132493