
Modeling the Tertiary Structure of the Rift Valley Fever Virus L Protein

 , , ,
, , , 
Abstract

1. Introduction
2. Results
2.1. Domain Identification
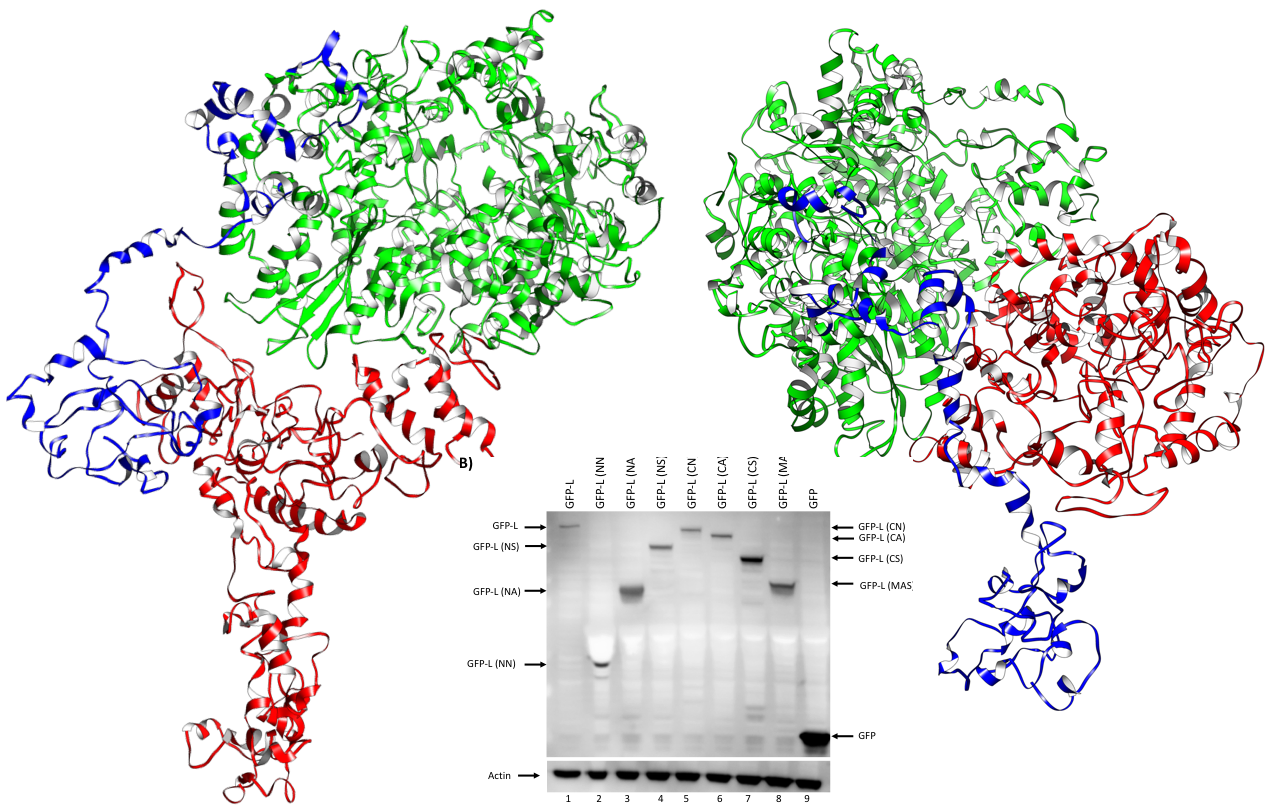
2.2. Modeling the Structure of RVFV L Protein Domains
2.3. Assembled L Protein Full-Length Structural Models
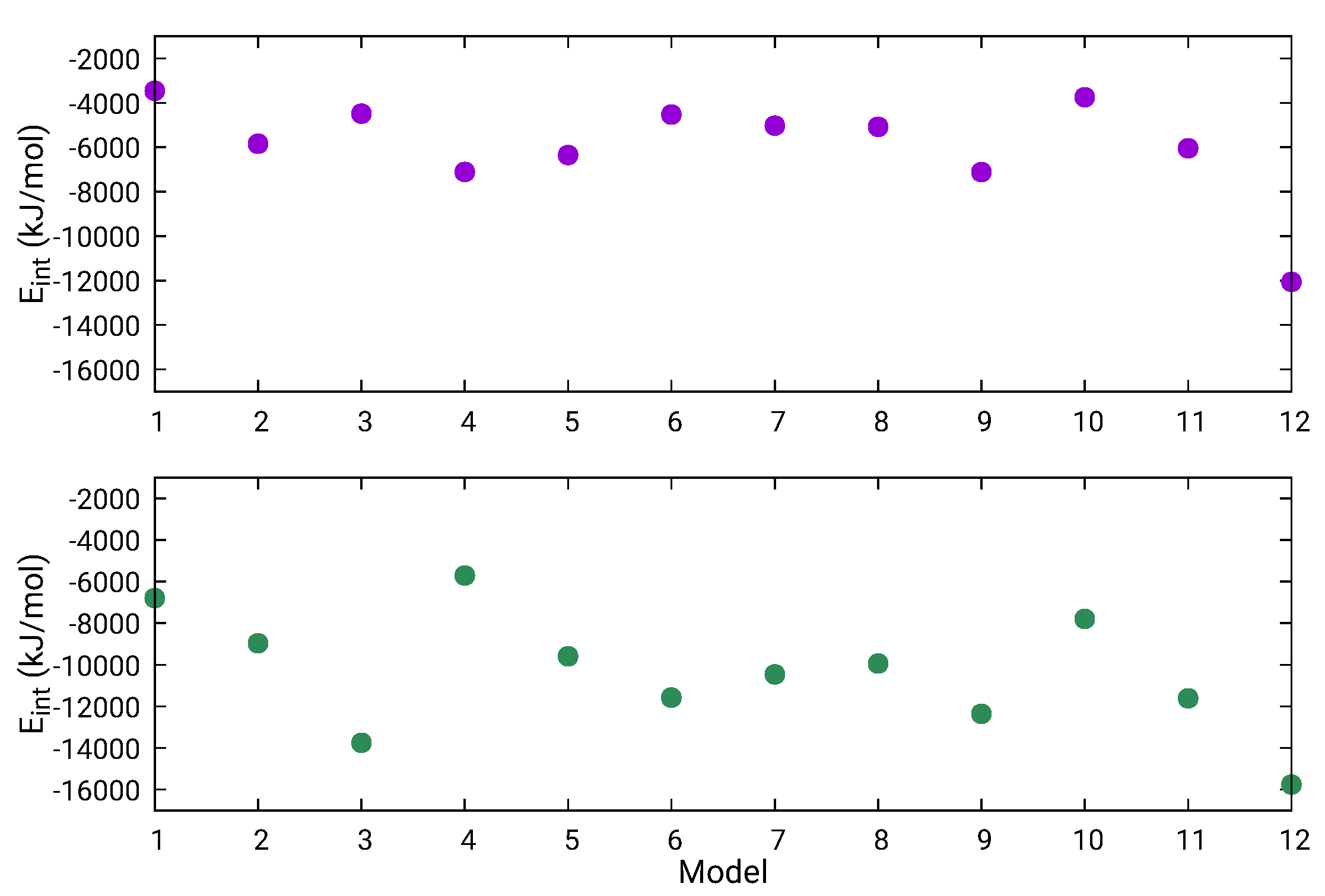
2.4. Energetic Refinement of Full-Length RVFV L-Protein Model Structures and Their Properties
3. Discussion
4. Materials and Methods
4.1. Domain Identification
4.2. Domain-Structure Modeling
4.3. All-Atom Molecular-Dynamics Investigation, Structural Relaxation, and Energetics Evaluation
4.4. Assembling Structural Models of Single Domains into a Full-Length Tertiary Structure
4.5. Tertiary-Structure Refinement
4.6. Tertiary-Structure Evaluation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| aa | amino acid |
| AZT | Azidothymidine |
| CASP | Critical Assessment of Protein Structure Prediction |
| CDC | Centers for Disease Control and Prevention |
| Cryo TEM | Transmission electron cryomicroscopy |
| FDA | U.S. Food and Drug Administration |
| HIV | Human immunodeficiency virus |
| lRMSD | least root-mean-squared-deviation |
| MD | Molecular Dynamics |
| NMR | Nuclear magnetic resonance |
| NP | Nucleoprotein |
| PAGE | Polyacrylamide gel electrophoresis |
| PDB | Protein Data Bank |
| PE | Potential energy |
| RdRp | RNA dependent RNA polymerase |
| RMSD | Root-mean-square deviation |
| RVFV | Rift Valley Fever Virus |
| SASA | Solvent Accesible Surface Area |
| SDS | Sodium dodecyl sulfate |
| USDA | U.S. Department of Agriculture |
References
- Bohr, D.D.; Wright, P.E. How do proteins interact? Science 2008, 320, 1429–1430. [Google Scholar] [CrossRef] [PubMed]
- Shehu, A. Conformational Search for the Protein Native State. In Protein Structure Prediction: Method and Algorithms; Rangwala, H., Karypis, G., Eds.; Book Series on Bioinformatics; Wiley: Hoboken, NJ, USA, 2010; Chapter 21. [Google Scholar]
- Bird, B.H.; Nichol, S.T. Breaking the chain: Rift Valley fever virus control via livestock vaccination. Curr. Opin. Virol. 2012, 2, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Hartman, A. Rift Valley Fever. Clin. Lab. Med. 2017, 37, 285–301. [Google Scholar] [CrossRef]
- Ikegami, T.; Makino, S. The pathogenesis of Rift Valley fever. Viruses 2011, 3, 493–519. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, K.L.; Banyard, A.C.; McElhinney, L.; Johnson, N.; Horton, D.L.; Hernandez-Triana, L.M.; Fooks, A.R. Rift Valley fever virus. Vaccine 2017, 33, 5520–5531. [Google Scholar] [CrossRef]
- Bouloy, M.; Weber, F. Molecular biology of Rift Valley Fever virus. Open Virol. J. 2010, 4, 8–14. [Google Scholar] [CrossRef]
- Morin, B.; Coutard, B.; Lelke, M.; Ferron, F.; Kerber, R.; Jamal, S.; Frangeul, A.; Baronti, C.; Charrel, R.; de Lamballerie, X.; et al. The N-terminal domain of the arenavirus L protein is an RNA endonuclease essential in mRNA transcription. PLoS Pathog. 2010, 6, e1001038. [Google Scholar] [CrossRef] [PubMed]
- Patterson, J.L.; Holloway, B.; Kolakofsky, D. La Crosse virions contain a primer-stimulated RNA polymerase and a methylated cap-dependent endonuclease. J. Virol. 1984, 52, 215–222. [Google Scholar]
- Lopez, N.; Muller, R.; Prehaud, C.; Bouloy, M. The L-Protein of Rift Valley Fever Virus Can Rescue Viral Ribonucleoproteins and Transcribe Synthetic Genome-Like RNA Molecules. J. Virol. 1995, 69, 3972–3979. [Google Scholar]
- Venkataraman, S.; Prasad, B.V.L.S.; Selvarajan, R. RNA Dependent RNA Polymerases: Insights from Structure, Function and Evolution. Viruses 2018, 10, 76. [Google Scholar] [CrossRef] [PubMed]
- Müller, R.; Poch, O.; Delarue, M.; Bishop, D.H.; Bouloy, M. Rift-Valley Fever Virus L-Segment—Correction of the Sequence and Possible Functional-Role of Newly Identified Regions Conserved in RNA-Dependent Polymerases. J. Gen. Virol. 1994, 75, 1345–1352. [Google Scholar] [CrossRef]
- Zamoto-Niikura, A.; Terasaki, K.; Ikegami, T.; Peters, C.J.; Makino, S. Rift Valley Fever Virus L Protein Forms a Biologically Active Oligomer. J. Virol. 2009, 83, 12779–12789. [Google Scholar] [CrossRef]
- Littler, E.; Oberg, B. Achievements and challenges in antiviral drug discovery. Antivir. Chem. Chemother. 2005, 16, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Almsned, F.; Gogovi, G.; Bracci, N.; Kehn-Hall, K.; Blaisten-Barojas, E.; Shehu, A. Modeling the Tertiary Structure of a Multi-domain Protein. In Proceedings of the 2018 ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics, Washington, DC, USA, 29 August–1 September 2018; pp. 615–620. [Google Scholar]
- Berman, H.M.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Biol. 2003, 10, 980. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Choi, K.H. Viral Polymerases. Adv. Exp. Med. Biol. 2012, 726, 267–304. [Google Scholar]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef]
- Reguera, J.; Gerlach, P.; Cusack, S. Towards a structural understanding of RNA synthesis by negative strand RNA viral polymerases. Curr. Opin. Struct. Biol. 2009, 36, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Jaroszewski, L.; Li, Z.; Godzik, A. AIDA: Ab initio domain assembly for automated multi-domain protein structure prediction and domain-domain interaction prediction. Bioinformatics 2015, 31, 2098–2105. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera: A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Ye, Y.; Godzik, A. Flexible structure alignment by chaining aligned fragment pairs allowing twists. Bioinformatics 2003, 19, ii246–ii255. [Google Scholar] [CrossRef]
- Burchard, A.; Schmidt, M.; Stockmayer, W.H. Information on Polydispersity and Branching from Combined Quasi-Elastic and Intergrated Scattering. Macromolecules 1980, 13, 1265–1272. [Google Scholar] [CrossRef]
- Wilkins, D.K.; Grimshaw, S.B.; Receveur, V.; Dobson, C.M.; Jones, J.A.; Smith, L.J. Hydrodynamic Radii of Native and Denatured Proteins Measured by Pulse Field Gradient NMR Techniques. Biochemistry 1999, 38, 16424–16431. [Google Scholar] [CrossRef]
- Tanford, C. Physical Chemistry of Macromolecules; John Wiley & Sons, Inc.: New York, NY, USA, 1961; Chapter IV. [Google Scholar]
- Atkins, C.; Freiberg, A. Recent advances in the development of antiviral therapeutics for Rift Valley fever virus infection. Future Virol. 2017, 12, 651–665. [Google Scholar] [CrossRef]
- Georgiou, D.; Karakasidis, T.; Nieto, J.; Torres, A. Use of fuzzy clustering technique and matrices to classify amino acids and its impact to Chou’s pseudo amino acid composition. J. Theor. Biol. 2009, 257, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Chothia, C.; Lesk, A.M. The relation between the divergence of sequence and structure in proteins. EMBO J. 1986, 5, 823–826. [Google Scholar] [CrossRef]
- Altschul, S.; Gish, F.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Schultz, J.; Milpetz, F.; Bork, P.; Ponting, C.P. SMART, a simple modular architecture research tool: Identification of signaling domains. Proc. Natl. Acad. Sci. USA 1998, 95, 5857–5864. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Xu, J. Raptorx: Exploiting structure information for protein alignment by statistical inference. Proteins 2011, 79, 161–171. [Google Scholar] [CrossRef]
- Källberg, M.; Wang, H.; Wang, S.; Peng, J.; Wang, Z.; Lu, H.; Xu, J. Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 2012, 7, 1511–1522. [Google Scholar] [CrossRef]
- Boratyn, G.; Schäffer, A.A.; Agarwala, R.; Altschul, S.F.; Lipman, D.J.; Madden, T.L. Domain enhanced lookup time accelerated BLAST. Biol. Direct. 2012, 7, 1–14. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, J.; Li, R.; Shi, Q.; Xue, Z.; Zhang, Y. ThreaDomEx: A unified platform for predicting continuous and discontinuous protein domains by multiple-threading and segment assembly. Nucleic Acids Res. 2017, 45, W400–W407. [Google Scholar] [CrossRef] [PubMed]
- Eickholt, J.; Deng, X.; Cheng, J. DoBo: Protein domain boundary prediction by integrating evolutionary signals and machine learning. BMC Bioinf. 2011, 12, 43. [Google Scholar] [CrossRef]
- Suyama, M.; Ohara, O. DomCut: Prediction of inter-domain linker regions in amino acid sequences. Bioinformatics 2003, 19, 673–674. [Google Scholar] [CrossRef]
- Marsden, R.L.; McGuffin, L.J.; Jones, D.T. Rapid protein domain assignment from amino acid sequence using predicted secondary structure. Protein Sci. 2009, 11, 2814–2824. [Google Scholar] [CrossRef]
- Mongan, J.; Case, D.A.; McCammon, J.A. Constant pH molecular dynamics in generalized Born implicit solvent. J. Comput. Chem. 2004, 25, 2038–2048. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. WIREs Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Ponder, J.W.; Case, D.A. Force fields for protein simulations. Adv. Prot. Chem. 2003, 66, 27–85. [Google Scholar]
- Maier, J.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.; Simmerling, C. f14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Tsui, V.; Case, D.A. Theory and applications of the generalized Born solvation model in macromolecular simulations. Biopolymers 2001, 56, 275–291. [Google Scholar] [CrossRef]
- Miao, G.; Zander, J.; Sung, K.W.; Slimane, B. Fundamentals of Mobile Data Networks; Cambridge University Press: Cambridge, UK, 2016. [Google Scholar]
- Bhattacharya, D.; Cheng, J. 3Drefine: Consistent protein structure refinement by optimizing hydrogen bonding network and atomic-level energy minimization. Proteins Struct. Funct. Bioinf. 2013, 81, 119–131. [Google Scholar] [CrossRef]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef]
- Shuid, A.N.; Kempster, R.; McGuffin, L.J. ReFOLD: A server for the refinement of 3D protein models guided by accurate quality estimates. Nucleic Acids Res. 2017, 45, W422–W428. [Google Scholar] [CrossRef]
- Heo, L.; Feig, M. PREFMD: A web server for protein structure refinement via molecular dynamics simulations. Bioinformatics 2018, 34, 1063–1065. [Google Scholar] [CrossRef]
- Rodrigues, J.P.; Levitt, M.; Chopra, G. KoBaMIN: A knowledge-based minimization web server for protein structure refinement. Nucleic Acids Res. 2012, 40, W323–W328. [Google Scholar] [CrossRef]
- Davis, I.W.; Leaver-Fay, A.; Chen, V.B.; Block, J.N.; Kapral, G.J.; Wang, X.; Murray, L.W.; Arendall, W.B., III; Snoeyink, J.; Richardson, J.S.; et al. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007, 35, W375–W383. [Google Scholar] [CrossRef]
Sample Availability: The coordinates of the 12 predicted tertiary structure models are enclosed in the
Supplementary Materials. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB id | Organism | C-Score | PDB id | Organism | C-Score | Model | Description | C-Score |
|---|---|---|---|---|---|---|---|---|
| L1-nt | - | −4.32 | L2-nt | - | −0.09 | L3-nt | I-TASSER model | −1.68 |
| L1-4miw | Lassa virus | −1.55 | L2-5amq | La Crosse | 0.07 | L3-nt-MD | L3-nt with MD aa 1861–2092 | −1.95 |
| L1-5ize | Hantaan virus | −0.97 | L2-5amr | La Crosse | −0.09 | L3-AIDA | L3-nt-MD, AIDA | - |
| L1-5hsb | Andes virus | −0.74 | L2-1yuy | Hepatitis C | −0.05 | L3-Chimera | L3-nt-MD, Chimera | - |
| L1-5j1n | Lassa virus | −1.45 | L2-4xhi | Thosea Asigna | 0.04 | |||
| L1-MD | - | - | L2-4ucy | Metapneu- movirus | 0.17 |
| Domain Segment | PE (kJ/mol) | (nm) | (nm) | (nm) |
|---|---|---|---|---|
| L1-MD | −8.62 | 6.66 | 3.05 | 6.03 |
| L3 | −7.57 | 7.23 | 2.45 | 5.47 |
| L3-MD | −8.70 | 3.89 | 2.97 | 5.11 |
| MP-Score | Clash-Score | Rot-Out | Ram-Out | Ram-fv | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Model | Before min | After min | Before min | After min | Before min | After min | Before min | After min | Before min | After min |
| L1-5ize + L2-4xhi + L3-nt | 3.75 | 2.48 | 74.1 | 1.89 | 7.91 | 8.52 | 8.42 | 7.22 | 80.53 | 72.16 |
| L1-5ize + L2-4xhi + L3-MD | 3.70 | 2.52 | 73.34 | 1.95 | 7.53 | 9.68 | 7.85 | 6.96 | 82.54 | 78.54 |
| L1-5ize + L2-4xhi + L3-AIDA | 3.79 | 2.49 | 73.36 | 1.71 | 8.82 | 9.18 | 9.76 | 8.40 | 80.05 | 71.19 |
| L1-5ize + L2-4xhi + L3-Chimera | 3.91 | 2.60 | 88.81 | 2.04 | 9.84 | 10.95 | 10.07 | 7.47 | 79.58 | 70.82 |
| L1-5hsb + L2-4xhi + L3-nt | 3.76 | 2.64 | 77.25 | 2.61 | 8.02 | 10.12 | 8.18 | 7.27 | 81.15 | 72.94 |
| L1-5hsb + L2-4xhi + L3-MD | 3.73 | 2.50 | 74.62 | 1.98 | 8.23 | 9.07 | 7.70 | 7.22 | 83.16 | 73.20 |
| L1-5hsb + L2-4xhi + L3-AIDA | 3.78 | 2.56 | 77.23 | 2.16 | 8.23 | 9.40 | 9.33 | 6.96 | 80.19 | 71.39 |
| L1-5hsb + L2-4xhi + L3-Chimera | 3.90 | 2.56 | 89.62 | 2.13 | 9.36 | 9.13 | 9.73 | 7.89 | 79.29 | 69.43 |
| L1-MD + L2-4xhi + L3-nt | 3.73 | 2.60 | 72.28 | 2.16 | 8.02 | 10.90 | 7.94 | 6.24 | 81.53 | 72.37 |
| L1-MD + L2-4xhi + L3-MD | 3.70 | 2.49 | 76.02 | 1.86 | 7.42 | 9.18 | 7.42 | 6.86 | 83.43 | 72.89 |
| L1-MD + L2-4xhi + L3-AIDA | 3.77 | 2.53 | 72.81 | 1.89 | 8.66 | 9.79 | 9.04 | 7.68 | 80.57 | 71.75 |
| L1-MD + L2-4xhi + L3-Chimera | 3.90 | 2.57 | 90.22 | 2.28 | 9.63 | 9.07 | 10.16 | 8.45 | 80.30 | 70.36 |
| Model | PE (kJ/mol) | (nm) | (nm) | (nm) | SASA (nm) | RMSD (nm) | ||
|---|---|---|---|---|---|---|---|---|
| 1 | L1-MD + L2-4xhi + L3-nt | −6.664 | 5.10 | 9.49 | 0.05 ± 0.01 | 2.58 | 845.0 | 3.15 |
| 2 | L1-5ize + L2-4xhi + L3-nt | −6.710 | 5.29 | 9.51 | 2.05 ± 0.23 | 16.27 | 832.4 | 3.55 |
| 3 | L1-5hsb + L2-4xhi + L3-nt | −6.751 | 4.34 | 8.67 | 0.25 ± 0.03 | 5.64 | 816.0 | 3.37 |
| 4 | L1-5ize + L2-4xhi + L3-AIDA | −6.762 | 4.91 | 9.27 | 1.61 ± 0.18 | 14.33 | 802.8 | 3.74 |
| 5 | L1-MD + L2-4xhi + L3-MD | −6.781 | 4.78 | 9.15 | 0.17 ± 0.02 | 4.71 | 826.7 | 3.14 |
| 6 | L1-5ize + L2-4xhi + L3-MD | −6.791 | 5.11 | 9.44 | 0.48 ± 0.05 | 7.88 | 840.0 | 3.49 |
| 7 | L1-5hsb + L2-4xhi + L3-MD | −6.792 | 4.87 | 9.12 | 1.78 ± 0.19 | 15.04 | 801.5 | 3.55 |
| 8 | L1-5ize + L2-4xhi + L3-Chimera | −6.799 | 4.66 | 8.84 | 0.24 ± 0.03 | 5.60 | 759.8 | 1.85 |
| 9 | L1-5hsb + L2-4xhi + L3-Chimera | −6.820 | 4.71 | 8.99 | 0.54 ± 0.06 | 8.41 | 742.8 | 1.50 |
| 10 | L1-5hsb + L2-4xhi + L3-AIDA | −6.833 | 4.72 | 8.89 | 1.72 ± 0.18 | 14.80 | 771.0 | 3.31 |
| 11 | L1-MD + L2-4xhi + L3-AIDA | −6.901 | 4.43 | 8.71 | 0.66 ± 0.08 | 9.26 | 787.3 | 2.54 |
| 12 | L1-MD + L2-4xhi + L3-Chimera | −6.919 | 4.81 | 8.91 | 0.22 ± 0.02 | 5.28 | 747.6 | 0.00 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gogovi, G.K.; Almsned, F.; Bracci, N.; Kehn-Hall, K.; Shehu, A.; Blaisten-Barojas, E. Modeling the Tertiary Structure of the Rift Valley Fever Virus L Protein. Molecules 2019, 24, 1768. https://doi.org/10.3390/molecules24091768
Gogovi GK, Almsned F, Bracci N, Kehn-Hall K, Shehu A, Blaisten-Barojas E. Modeling the Tertiary Structure of the Rift Valley Fever Virus L Protein. Molecules. 2019; 24(9):1768. https://doi.org/10.3390/molecules24091768
Chicago/Turabian StyleGogovi, Gideon K., Fahad Almsned, Nicole Bracci, Kylene Kehn-Hall, Amarda Shehu, and Estela Blaisten-Barojas. 2019. "Modeling the Tertiary Structure of the Rift Valley Fever Virus L Protein" Molecules 24, no. 9: 1768. https://doi.org/10.3390/molecules24091768
APA StyleGogovi, G. K., Almsned, F., Bracci, N., Kehn-Hall, K., Shehu, A., & Blaisten-Barojas, E. (2019). Modeling the Tertiary Structure of the Rift Valley Fever Virus L Protein. Molecules, 24(9), 1768. https://doi.org/10.3390/molecules24091768