
Triazolopeptides Inhibiting the Interaction between Neuropilin-1 and Vascular Endothelial Growth Factor-165

 , , , , and
, , , , and 
Abstract
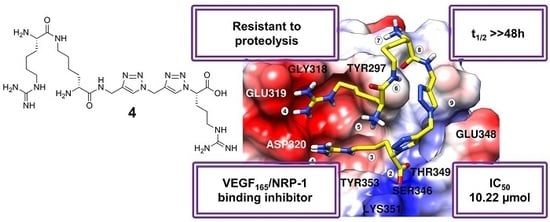
:
1. Introduction
2. Results and Discussion
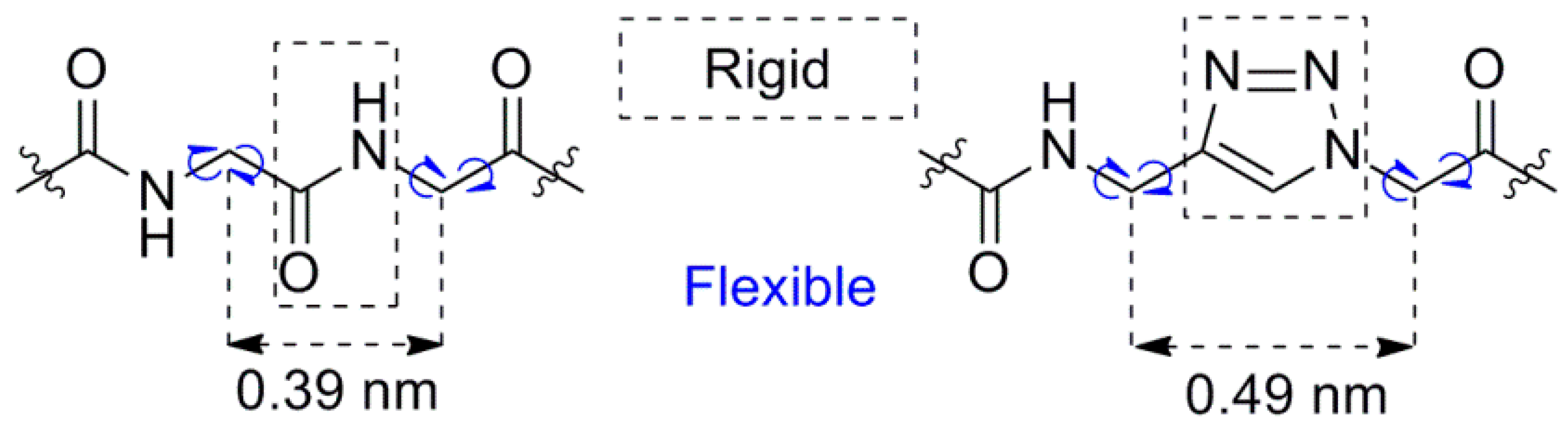
2.1. Synthesis of Triazolopeptides on Solid Support
2.1.1. Conversion of Primary Amine Group into Azide on Solid Support
2.1.2. Click Reaction on Solid Support
2.1.3. Homoarginine (Har) Synthesis on Solid Support
2.1.4. Final Cleavage from Solid Support and Triazolopeptides Isolation
2.2. Inhibition of NRP-1/VEGF-A
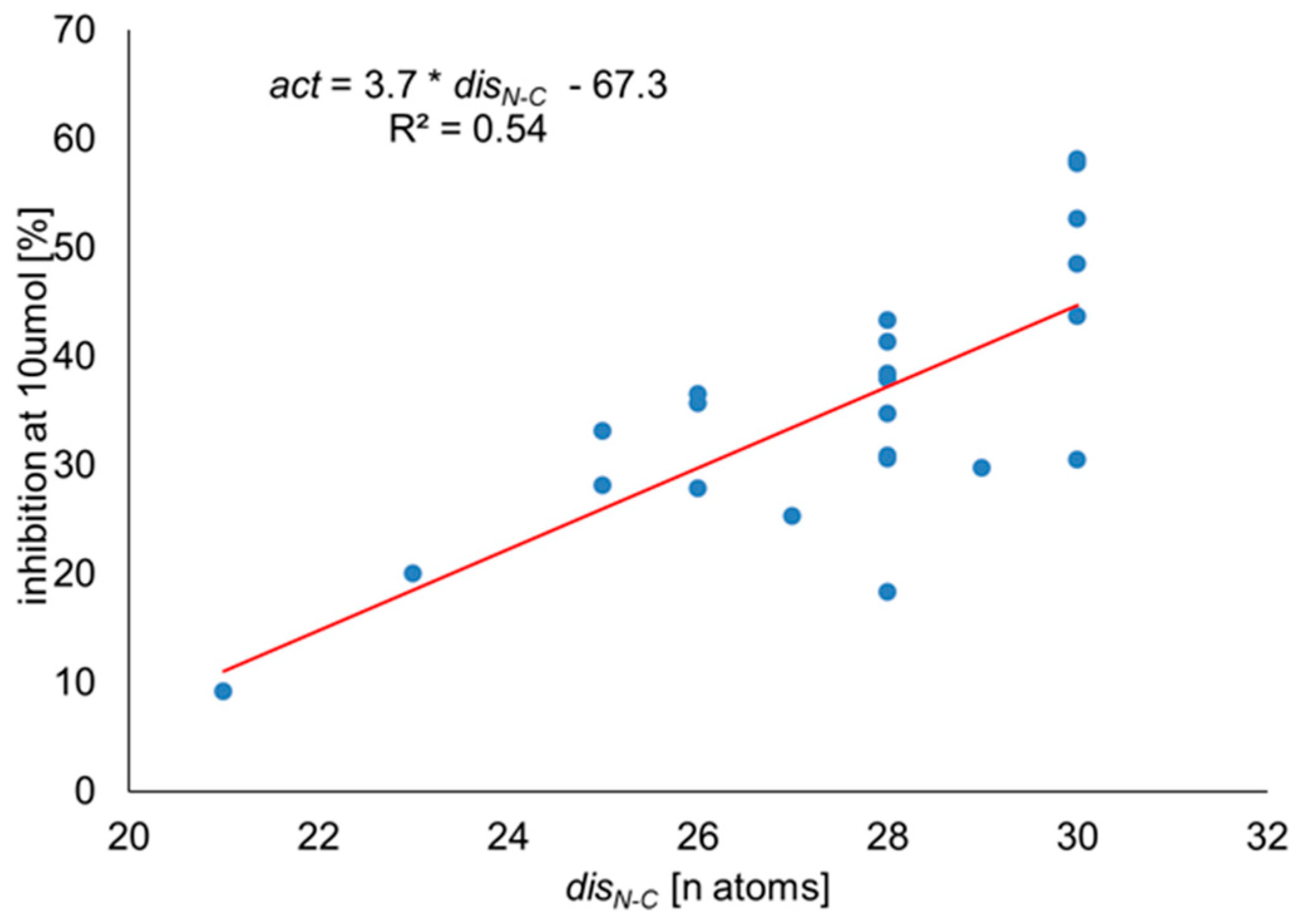
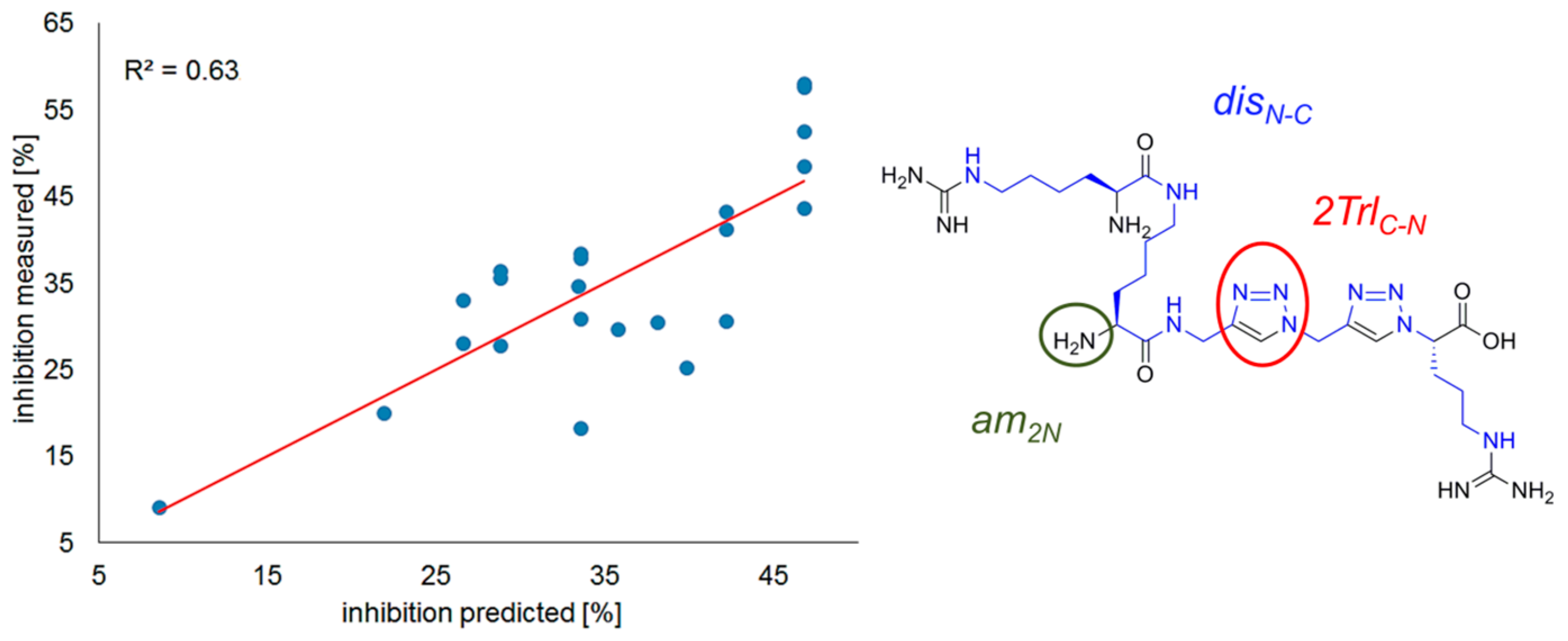
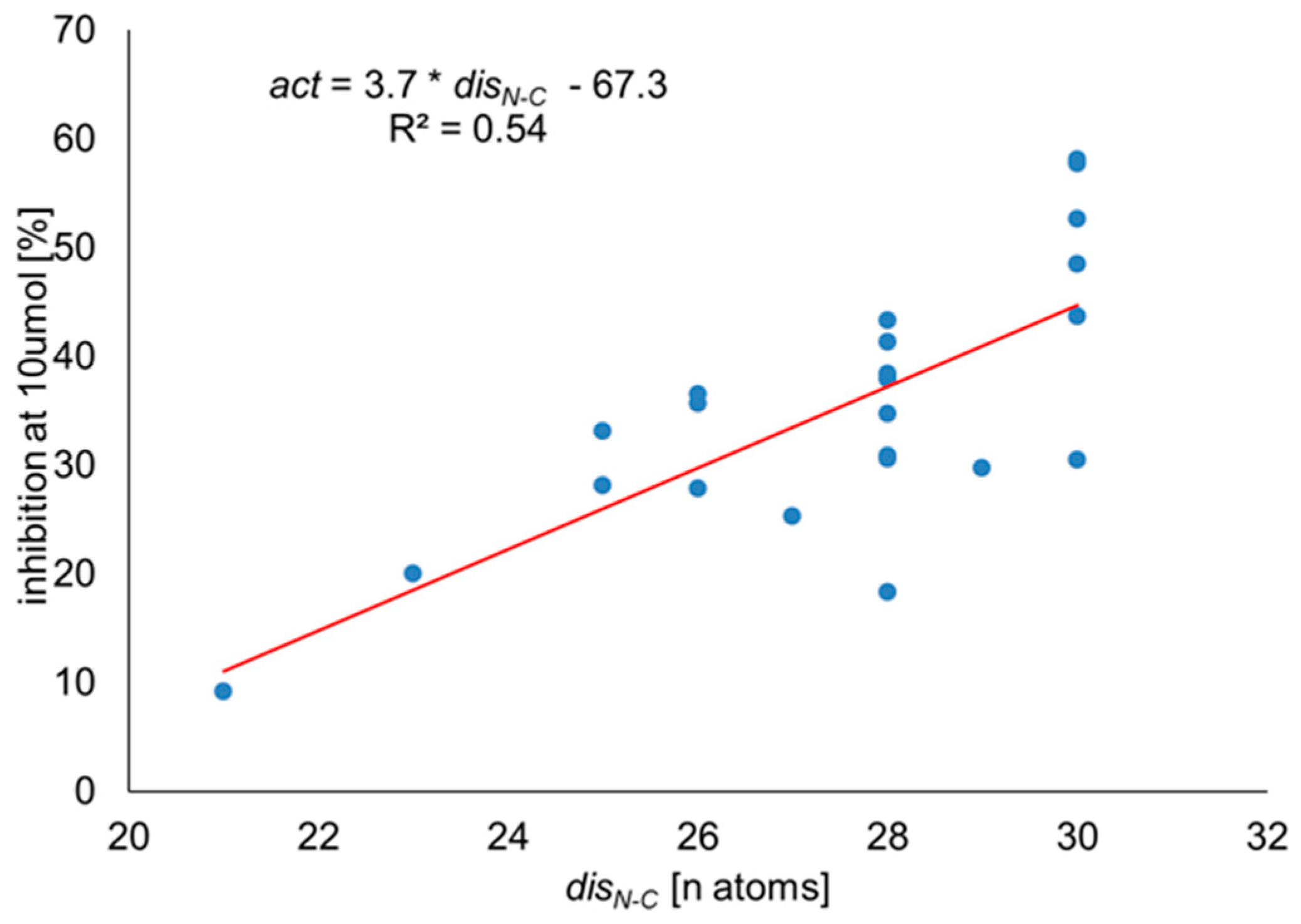
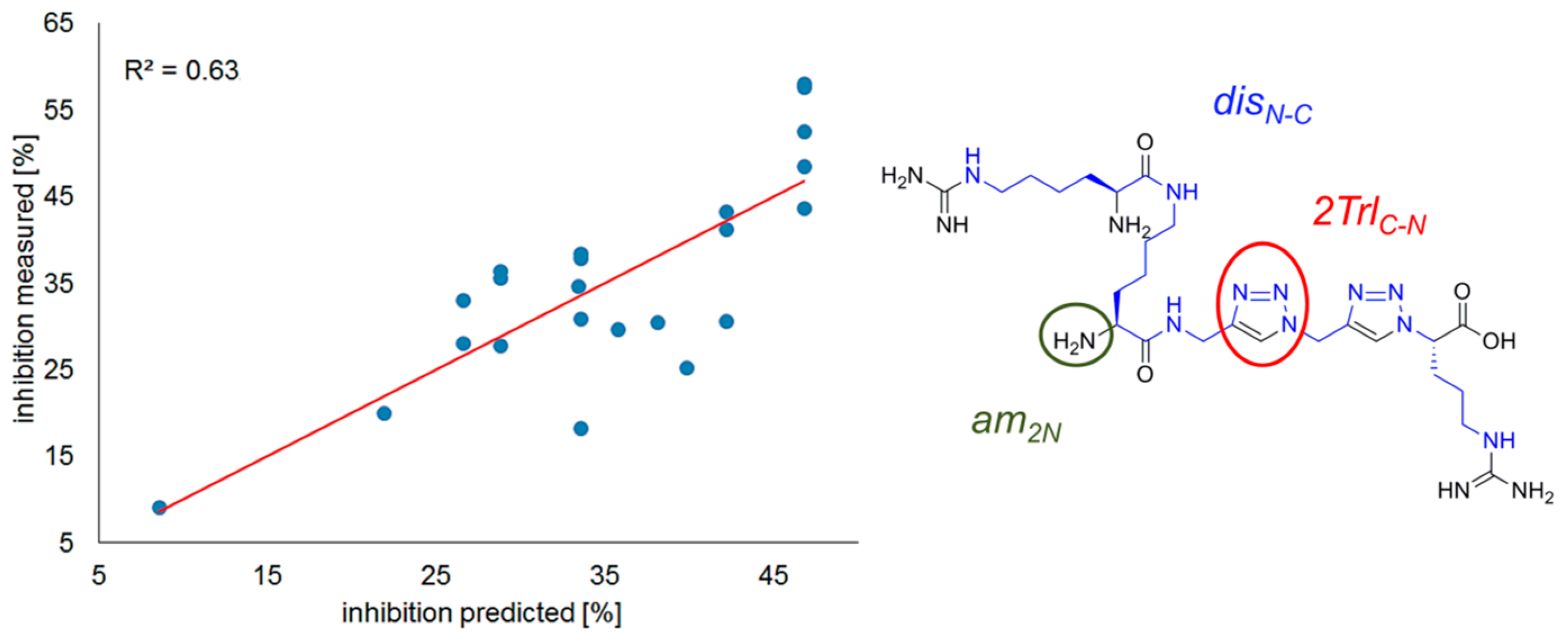
Correlation Analysis
R2 = 0.63, n = 23.
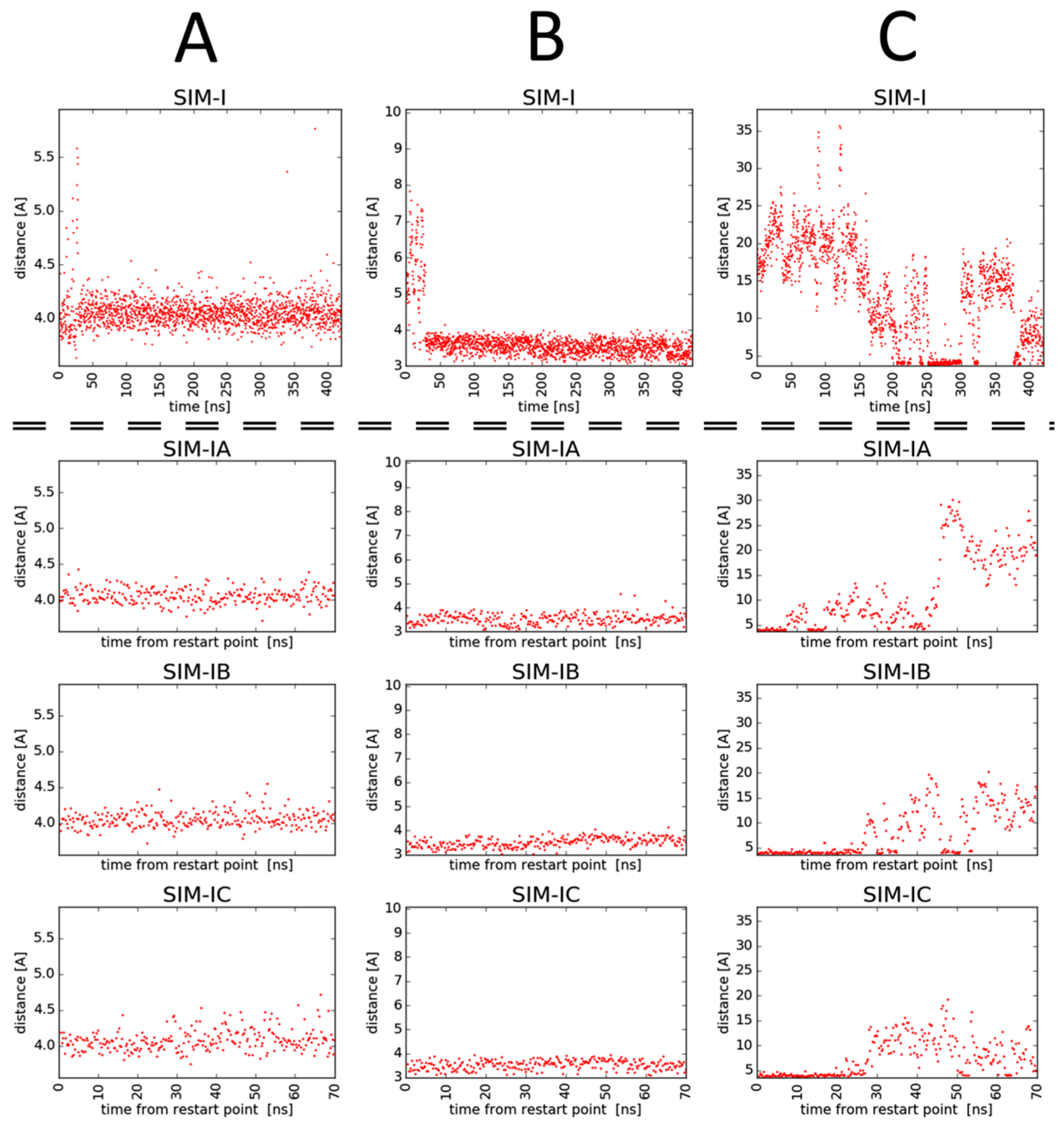
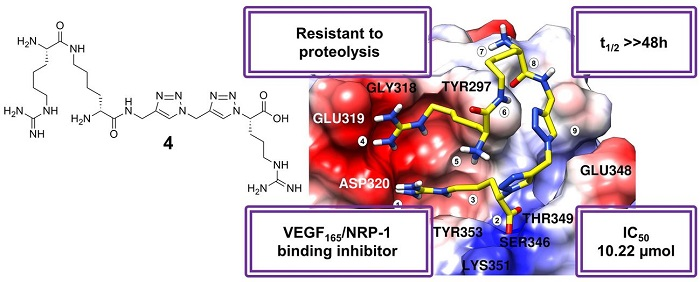
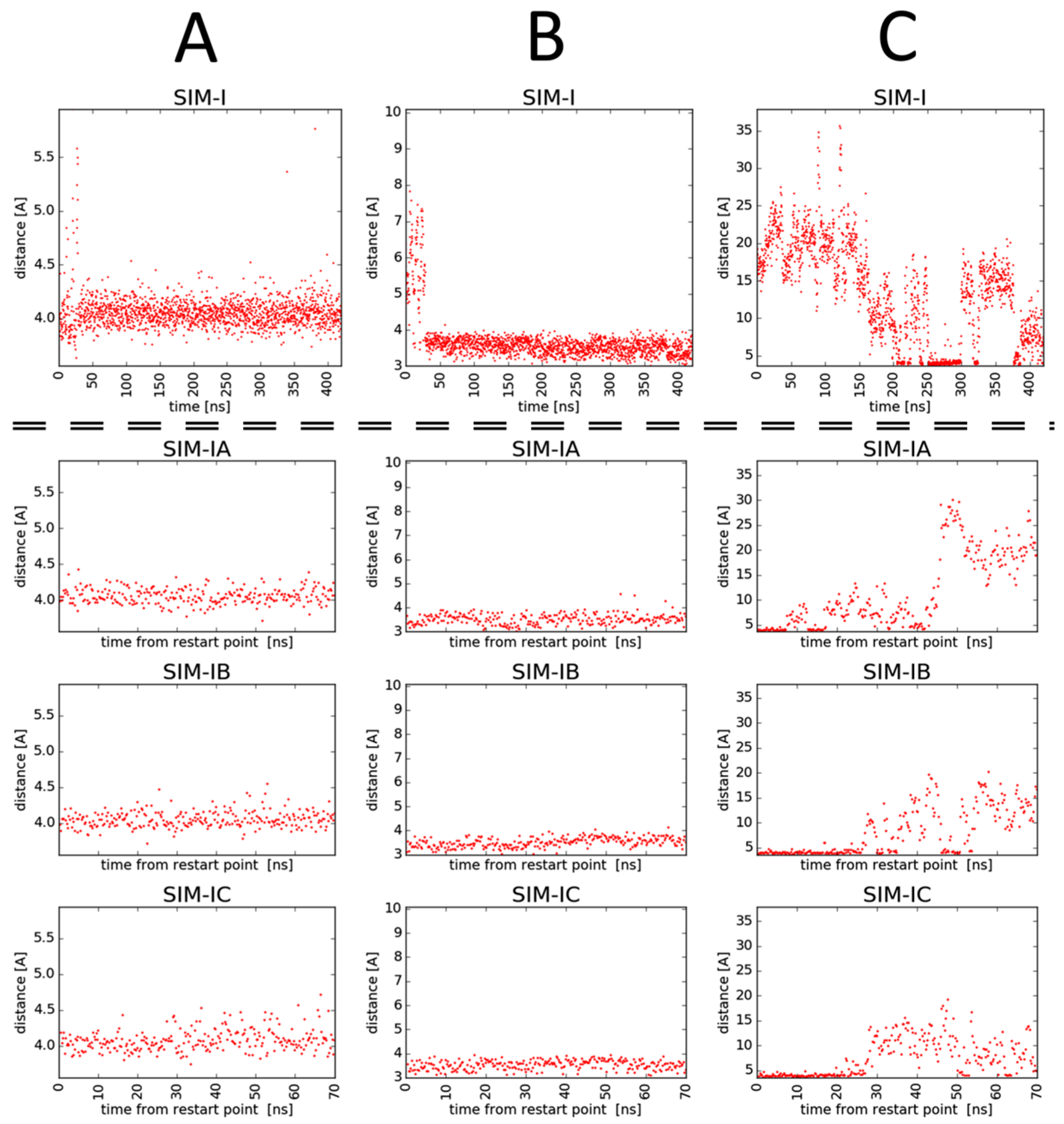
2.3. Molecular Modelling
- -
- Ionic interaction of C-terminal Arg guanidine with Asp320;
- -
- Hydrogen bonds of C-terminal Arg carboxylate with Ser346 and Thr349;
- -
- Dispersive contacts of C-terminal Arg aliphatic portion with the residue lining the binding cleft Tyr353, Tyr 297, Trp301, Thr316, Gly414, Ile415, and Ser416;
- -
- Ionic interaction of Har guanidine with Glu319 or an H-bond of this moiety with carbonyl group of Gly318.
2.4. In Vitro Proteolysis of Triazolopeptides
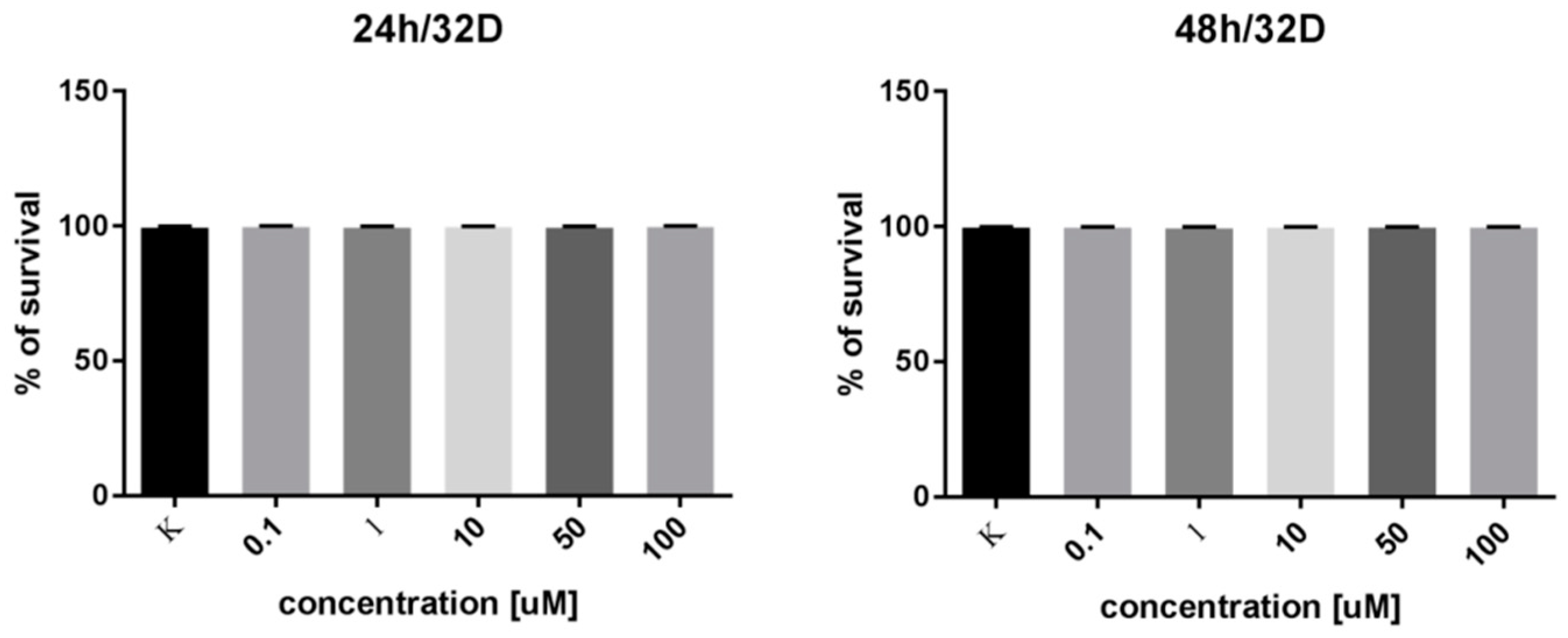
2.5. In Vitro Cell Survival Test
3. Materials and Methods
3.1. Synthesis
3.1.1. Wong Diazotransfer
3.1.2. Fmoc-Propargylamine (24) Preparation
3.1.3. Click Reaction on Solid Support
3.1.4. Common Procedures for Peptide Chain Elongation on Solid Support
3.1.5. Guanylation Reaction of Lys to Gain Har
3.1.6. Peptidotriazole Cleavage from Solid Support
3.1.7. HPLC Analysis, Purification, and HRMS Characterization
3.2. ELISA Assays of Inhibitory Activity
3.3. Correlation Analysis
3.4. Molecular Dynamics
3.5. Stability
3.6. In Vitro Cell Survival Test
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fong, G.-H.; Rossant, J.; Gertsenstein, M.; Breitman, M.L. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 1995, 376, 66–70. [Google Scholar] [CrossRef]
- Shay, S.; Hua-Quan, M.; Masashi, N.; Seiji, T.; Michael, K. VEGF165 mediates formation of complexes containing VEGFR-2 and neuropilin-1 that enhance VEGF165-receptor binding. J. Cell. Biochem. 2002, 85, 357–368. [Google Scholar]
- Fuh, G. The interaction of Neuropilin-1 with Vascular Endothelial Growth Factor and its receptor Flt-1. J. Biol. Chem. 2000, 275, 26690–26695. [Google Scholar] [CrossRef]
- Tordjman, R.; Lepelletier, Y.; Lemarchandel, V.; Cambot, M.; Gaulard, P.; Hermine, O.; Roméo, P.-H. A neuronal receptor, neuropilin-1, is essential for the initiation of the primary immune response. Nat. Immunol. 2002, 3, 477. [Google Scholar] [CrossRef]
- Bruder, D.; Probst-Kepper, M.; Westendorf, A.M.; Geffers, R.; Beissert, S.; Loser, K.; von Boehmer, H.; Buer, J.; Hansen, W. Frontline: Neuropilin-1: A surface marker of regulatory T cells. Eur. J. Immunol. 2004, 34, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Mizui, M.; Kikutani, H. Neuropilin-1: The Glue between Regulatory T Cells and Dendritic Cells? Immunity 2008, 28, 302–303. [Google Scholar] [CrossRef] [PubMed]
- Jubb, A.M.; Strickland, L.A.; Liu, S.D.; Mak, J.; Schmidt, M.; Koeppen, H.; Jubb M., A.; Strickland, A.L.; Liu, S.D.; Mak, J.; et al. Neuropilin-1 expression in cancer and development. J. Pathol. 2012, 226, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Soker, S.; Takashima, S.; Miao, H.Q.; Neufeld, G.; Klagsbrun, M. Neuropilin-1 Is Expressed by Endothelial and Tumor Cells as an Isoform-Specific Receptor for Vascular Endothelial Growth Factor. Cell 1998, 92, 735–745. [Google Scholar] [CrossRef]
- Soker, S.; Fidder, H.; Neufeld, G.; Klagsbrun, M. Characterization of novel vascular endothelial growth factor (VEGF) receptors on tumor cells that bind VEGF165 via its exon 7-encoded domain. J. Biol. Chem. 1996, 271, 5761–5767. [Google Scholar] [CrossRef] [PubMed]
- Goel, H.L.; Mercurio, A.M. VEGF targets the tumour cell. Nat. Rev. Cancer 2013, 13, 871. [Google Scholar] [CrossRef]
- Bachelder, R.E.; Crago, A.; Chung, J.; Wendt, M.A.; Shaw, L.M.; Robinson, G.; Mercurio, A.M. Vascular Endothelial Growth Factor Is an Autocrine Survival Factor for Neuropilin-expressing Breast Carcinoma Cells. Cancer Res. 2001, 61, 5736–5740. [Google Scholar] [PubMed]
- Hansen, W. Neuropilin 1 guides regulatory T cells into vegf-producing melanoma. Oncoimmunology 2013, 2. [Google Scholar] [CrossRef]
- Djordjevic, S.; Driscoll, P.C. Targeting VEGF signalling via the neuropilin co-receptor. Drug Discov. Today 2013, 18, 447–455. [Google Scholar] [CrossRef]
- Peng, K.; Bai, Y.; Zhu, Q.; Hu, B.; Xu, Y. Targeting VEGF–neuropilin interactions: A promising antitumor strategy. Drug Discov. Today 2019, 24, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Niland, S.; Eble, J.A. Neuropilins in the Context of Tumor Vasculature. Int. J. Mol. Sci. 2019, 20, 639. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.; Mota, F.; Steadman, D.; Soudy, C.; Miyauchi, J.T.; Crosby, S.; Jarvis, A.; Reisinger, T.; Winfield, N.; Evans, G.; et al. Small Molecule Neuropilin-1 Antagonists Combine Antiangiogenic and Antitumor Activity with Immune Modulation through Reduction of Transforming Growth Factor Beta (TGFβ) Production in Regulatory T-Cells. J. Med. Chem. 2018, 61, 4135–4154. [Google Scholar] [CrossRef] [PubMed]
- Novoa, A.; Pellegrini-Moïse, N.; Bechet, D.; Barberi-Heyob, M.; Chapleur, Y. Sugar-based peptidomimetics as potential inhibitors of the vascular endothelium growth factor binding to neuropilin-1. Bioorg. Med. Chem. 2010, 18, 3285–3298. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.-Q.; Lepelletier, Y.; Montès, M.; Borriello, L.; Jarray, R.; Grépin, R.; Leforban, B.; Loukaci, A.; Benhida, R.; Hermine, O.; et al. NRPa-308, a new neuropilin-1 antagonist, exerts in vitro anti-angiogenic and anti-proliferative effects and in vivo anti-cancer effects in a mouse xenograft model. Cancer Lett. 2018, 414, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Borriello, L.; Montès, M.; Lepelletier, Y.; Leforban, B.; Liu, W.-Q.Q.; Demange, L.; Delhomme, B.; Pavoni, S.; Jarray, R.; Boucher, J.L.; et al. Structure-based discovery of a small non-peptidic Neuropilins antagonist exerting in vitro and in vivo anti-tumor activity on breast cancer model. Cancer Lett. 2014, 349, 120–127. [Google Scholar] [CrossRef]
- Starzec, A.; Miteva, M.A.; Ladam, P.; Villoutreix, B.O.; Perret, G.Y. Discovery of novel inhibitors of vascular endothelial growth factor-A-Neuropilin-1 interaction by structure-based virtual screening. Bioorg. Med. Chem. 2014, 22, 4042–4048. [Google Scholar] [CrossRef]
- Jarvis, A.; Allerston, C.K.; Jia, H.; Herzog, B.; Garza-Garcia, A.; Winfield, N.; Ellard, K.; Aqil, R.; Lynch, R.; Chapman, C.; et al. Small Molecule Inhibitors of the Neuropilin-1 Vascular Endothelial Growth Factor A (VEGF-A) Interaction. J. Med. Chem. 2010, 53, 2215–2226. [Google Scholar] [CrossRef] [PubMed]
- Soker, S.; Gollamudi-Payne, S.; Fidder, H.; Charmahelli, H.; Klagsbrun, M. Inhibition of Vascular Endothelial Growth Factor (VEGF)-induced Endothelial Cell Proliferation by a Peptide Corresponding to the Exon 7-Encoded Domain of VEGF165. J. Biol. Chem. 1997, 272, 31582–31588. [Google Scholar] [CrossRef]
- Jia, H.; Bagherzadeh, A.; Hartzoulakis, B.; Jarvis, A.; Löhr, M.; Shaikh, S.; Aqil, R.; Cheng, L.; Tickner, M.; Esposito, D.; et al. Characterization of a bicyclic peptide neuropilin-1 (NP-1) antagonist (EG3287) reveals importance of vascular endothelial growth factor exon 8 for NP-1 binding and role of NP-1 in KDR signaling. J. Biol. Chem. 2006, 281, 13493–13502. [Google Scholar] [CrossRef] [PubMed]
- Grabowska, K.; Puszko, A.K.; Lipiński, P.F.J.; Laskowska, A.K.; Wileńska, B.; Witkowska, E.; Misicka, A. Design, synthesis and in vitro biological evaluation of a small cyclic peptide as inhibitor of vascular endothelial growth factor binding to neuropilin-1. Bioorg. Med. Chem. Lett. 2016, 26, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Getz, J.A.; Cheneval, O.; Craik, D.J.; Daugherty, P.S. Design of a Cyclotide Antagonist of Neuropilin-1 and -2 That Potently Inhibits Endothelial Cell Migration. ACS Chem. Biol. 2013, 8, 1147–1154. [Google Scholar] [CrossRef]
- Grabowska, K.; Puszko, A.K.; Lipiński, P.F.J.; Laskowska, A.K.; Wileńska, B.; Witkowska, E.; Perret, G.Y.; Misicka, A. Structure-activity relationship study of a small cyclic peptide H-c[Lys-Pro-Glu]-Arg-OH: A potent inhibitor of Vascular Endothelial Growth Factor interaction with Neuropilin-1. Bioorg. Med. Chem. 2016, 25, 5–8. [Google Scholar] [CrossRef]
- Binétruy-Tournaire, R.; Demangel, C.; Malavaud, B.; Vassy, R.; Rouyre, S.; Kraemer, M.; Plouët, J.; Derbin, C.; Perret, G.; Mazié, J.C. Identification of a peptide blocking vascular endothelial growth factor (VEGF)-mediated angiogenesis. EMBO J. 2000, 19, 1525–1533. [Google Scholar] [CrossRef] [PubMed]
- Fedorczyk, B.; Lipiński, P.F.J.; Tymecka, D.; Puszko, A.K.; Wilenska, B.; Perret, G.Y.; Misicka, A. Conformational latitude—Activity relationship of KPPR tetrapeptide analogues toward their ability to inhibit binding of Vascular Endothelial Growth Factor 165 to Neuropilin-1. J. Pept. Sci. 2017, 23, 445–454. [Google Scholar] [CrossRef]
- Tymecka, D.; Puszko, A.K.; Lipiński, P.F.J.; Fedorczyk, B.; Wilenska, B.; Sura, K.; Perret, G.Y.; Misicka, A. Branched pentapeptides as potent inhibitors of the vascular endothelial growth factor 165 binding to Neuropilin-1: Design, synthesis and biological activity. Eur. J. Med. Chem. 2018, 158, 453–462. [Google Scholar] [CrossRef]
- Tymecka, D.; Lipiński, P.F.J.P.F.J.; Fedorczyk, B.; Puszko, A.; Wileńska, B.; Perret, G.Y.G.Y.; Misicka, A.; Lipi, P.F.J.; Puszko, A.; Wile, B.; et al. Structure-activity relationship study of tetrapeptide inhibitors of the Vascular Endothelial Growth Factor A binding to Neuropilin-1. Peptides 2017, 94, 25–32. [Google Scholar] [CrossRef]
- Starzec, A.; Vassy, R.; Martin, A.; Lecouvey, M.; Di Benedetto, M.; Crépin, M.; Perret, G.Y. Antiangiogenic and antitumor activities of peptide inhibiting the vascular endothelial growth factor binding to neuropilin-1. Life Sci. 2006, 79, 2370–2381. [Google Scholar] [CrossRef]
- Puszko, A.K.; Sosnowski, P.; Tymecka, D.; Raynaud, F.; Hermine, O.; Lepelletier, Y.; Misicka, A. Neuropilin-1 peptide-like ligands with proline mimetics, tested using the improved chemiluminescence affinity detection method. Medchemcomm 2019, 10, 332–340. [Google Scholar] [CrossRef]
- Tron, G.C.; Pirali, T.; Billington, R.A.; Canonico, P.L.; Sorba, G.; Genazzani, A.A. Click chemistry reactions in medicinal chemistry: Applications of the 1,3-dipolar cycloaddition between azides and alkynes. Med. Res. Rev. 2008, 28, 278–308. [Google Scholar] [CrossRef]
- Diness, F.; Schoffelen, S.; Meldal, M. Advances in Merging Triazoles with Peptides and Proteins. In Peptidomimetics I.; Lubell, W.D., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 267–304. ISBN 978-3-319-49119-6. [Google Scholar]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-Triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chemie Int. Ed. 2002, 41, 2596–2599. [Google Scholar]
- Ben Haj Salah, K.; Das, S.; Ruiz, N.; Andreu, V.; Martinez, J.; Wenger, E.; Amblard, M.; Didierjean, C.; Legrand, B.; Inguimbert, N. How are 1,2,3-triazoles accommodated in helical secondary structures? Org. Biomol. Chem. 2018, 16, 3576–3583. [Google Scholar] [CrossRef]
- Nyffeler, P.T.; Liang, C.H.; Koeller, K.M.; Wong, C.H. The chemistry of amine-azide interconversion: Catalytic diazotransfer and regioselective azide reduction. J. Am. Chem. Soc. 2002, 124, 10773–10778. [Google Scholar] [CrossRef]
- Lundquist IV, J.T.; Pelletier, J.C. Improved solid-phase peptide synthesis method utilizing α-azide-protected amino acids. Org. Lett. 2001, 3, 781–783. [Google Scholar] [CrossRef]
- Punna, S.; Finn, M.G. A Convenient Colorimetric Test for Aliphatic Azides. Synlett 2004, 2004, 99–100. [Google Scholar]
- Bernatowicz, M.S.; Youling, W.; Matsueda, G.R. 1H-Pyrazole-1-carboxamidine Hydrochloride: An Attractive Reagent for Guanylation of Amines and Its Application to Peptide Synthesis. J. Org. Chem. 1992, 57, 2497–2502. [Google Scholar] [CrossRef]
- Starzec, A.; Ladam, P.; Vassy, R.; Badache, S.; Bouchemal, N.; Navaza, A.; du Penhoat, C.H.; Perret, G.Y. Structure-function analysis of the antiangiogenic ATWLPPR peptide inhibiting VEGF165 binding to neuropilin-1 and molecular dynamics simulations of the ATWLPPR/neuropilin-1 complex. Peptides 2007, 28, 2397–2402. [Google Scholar] [CrossRef] [PubMed]
- Vander Kooi, C.W.; Jusino, M.A.; Perman, B.; Neau, D.B.; Bellamy, H.D.; Leahy, D.J. Structural basis for ligand and heparin binding to neuropilin B domains. Proc. Natl. Acad. Sci. USA 2007, 104, 6152–6157. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.W.; Xu, P.; Li, X.; Vander Kooi, C.W. Structural Basis for Selective Vascular Endothelial Growth Factor-A (VEGF-A) Binding to Neuropilin-1. J. Biol. Chem. 2012, 287, 11082–11089. [Google Scholar] [CrossRef]
- Mota, F.; Fotinou, C.; Rhana, R.; Chan, A.W.E.; Yelland, T.; Arooz, M.T.; O’Leary, A.P.; Hutton, J.; Frankel, P.; Zachary, I.; et al. Architecture and hydration of the arginine-binding site of neuropilin-1. FEBS J. 2018, 285, 1290–1304. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E.I.; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. Amber14; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Marion, A.; Góra, J.; Kracker, O.; Fröhr, T.; Latajka, R.; Sewald, N.; Antes, I. Amber-Compatible Parametrization Procedure for Peptide-like Compounds: Application to 1,4- and 1,5-Substituted Triazole-Based Peptidomimetics. J. Chem. Inf. Model. 2018, 58, 90–110. [Google Scholar] [CrossRef]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 3 and 4 are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Sequence | RT [min.] | General Formula | Calc. MS [m/z] | Meas. MS [m/z] |
|---|---|---|---|---|---|
| “linker” subseries | |||||
| 1 | Lys(Har)-GlyΨ[Trl]Arg | 11.98 1 | C22H43N13O4 | 554.3634 4 | 554.3640 4 |
| 2 | D-Lys(Har)-GlyΨ[Trl]Arg | 11.96 1 | C22H43N13O4 | 554.3634 4 | 554.3656 4 |
| 3 | Lys(Har)-GlyΨ[Trl]GlyΨ[Trl]Arg | 13.22 1 | C25H46N16O4 | 635.3961 4 | 635.3979 4 |
| 4 | D-Lys(Har)-GlyΨ[Trl]GlyΨ[Trl]Arg | 13.30 1 | C25H46N16O4 | 635.3961 4 | 635.3980 4 |
| 5 | D-Lys(D-Har)-GlyΨ[Trl]GlyΨ[Trl]Arg | 13.33 1 | C25H46N16O4 | 635.3961 4 | 635.3982 4 |
| 6 | Lys(Har)-Pro-GlyΨ[Trl]Arg | 14.65 1 | C27H50N14O5 | 651.4161 4 | 651.4187 4 |
| 7 | D-Lys(Har)-Pro-GlyΨ[Trl]Arg | 14.65 1 | C27H50N14O5 | 651.4161 4 | 651.4190 4 |
| 8 | Lys(Har)-Phe-GlyΨ[Trl]Arg | 14.89 2 | C31H52N14O5 | 701.4318 4 | 701.4339 4 |
| 9 | D-Lys(Har)-Phe-GlyΨ[Trl]Arg | 15.04 2 | C31H52N14O5 | 701.4318 4 | 701.4338 4 |
| 10 | Lys(Har)-GlyΨ[Trl]Ala-Arg | 13.17 1 | C25H48N14O5 | 625.4005 4 | 625.3994 4 |
| 11 | Lys(Har)-GlyΨ[Trl]Gly-Arg | 12.19 1 | C24H46N14O5 | 611.3848 4 | 611.3878 4 |
| 12 | Har-Pro-GlyΨ[Trl]Arg | 13.78 1 | C21H38N12O4 | 523.3212 4 | 523.3221 4 |
| 13 | Har-GlyΨ[Trl]GlyΨ[Trl]GlyΨ[Trl]Arg | 14.52 1 | C22H37N17O3 | 588.3338 4 | 588.3362 4 |
| “arm” subseries | |||||
| 14 | Lys(Fmoc-Har)-GlyΨ[Trl]GlyΨ[Trl]Arg | 16.50 3 | C40H56N16O6 | 429.2357 5 | 429.2371 5 |
| 15 | D-Lys(Fmoc-Har)-GlyΨ[Trl]GlyΨ[Trl]Arg | 16.53 3 | C40H56N16O6 | 429.2357 5 | 429.2373 5 |
| 16 | Har-Lys-GlyΨ[Trl]GlyΨ[Trl]Arg | 13.05 1 | C25H46N16O4 | 635.3961 4 | 635.3991 4 |
| 17 | Dab(Har)-GlyΨ[Trl]GlyΨ[Trl]Arg | 12.46 1 | C23H42N16O4 | 607.3648 4 | 607.3675 4 |
| 18 | Dap(Har)-GlyΨ[Trl]GlyΨ[Trl]Arg | 11.92 1 | C22H40N16O4 | 593.3491 4 | 593.3517 4 |
| 19 | Har-6Ahx-GlyΨ[Trl]GlyΨ[Trl]]Arg | 16.83 1 | C25H45N15O4 | 620.3852 4 | 620.3878 4 |
| 20 | Har-5Ava-GlyΨ[Trl]GlyΨ[Trl]Arg | 15.23 1 | C24H43N15O4 | 606.3695 4 | 606.3721 4 |
| 21 | Har-Ala-GlyΨ[Trl]GlyΨ[Trl]]Arg | 13.46 1 | C22H39N15O4 | 578.3382 4 | 578.3406 4 |
| 22 | Har-Gly-GlyΨ[Trl]GlyΨ[Trl]Arg | 13.06 1 | C21H37N15O4 | 564.3226 4 | 564.3244 4 |
| 23 | Har-GlyΨ[Trl]GlyΨ[Trl]Arg | 13.06 1 | C19H34N14O3 | 507.3020 4 | 507.3011 4 |
| No | Sequence | Inhibition of bt-VEGF165 Binding to NRP-1 [10 µM] 1 | IC50 [µM] |
|---|---|---|---|
| A7R | Ala-Thr-Trp-Lys-Pro-Pro-Arg | 61.0 ± 0.4 3 | 5.86 3 |
| KPPR | Lys-Pro-Pro-Arg | 64.5 | 4.60 4 |
| Lys(Har)-Dap/Dab-Pro-Arg | 96.8/98.5 | 0.2/0.2 4 | |
| “linker” subseries | |||
| 1 | Lys(Har)-GlyΨ[Trl]Arg | 28.1 ± 1.2 | - |
| 2 | D-Lys(Har)-GlyΨ[Trl]Arg | 33.1 ± 1.9 | - |
| 3 | Lys(Har)-GlyΨ[Trl]GlyΨ[Trl]Arg | 58.1 ± 2.1 | 8.39 2 |
| 4 | D-Lys(Har)-GlyΨ[Trl]GlyΨ[Trl]Arg | 52.6 ± 1.3 | 10.22 2 |
| 5 | D-Lys(D-Har)-GlyΨ[Trl]GlyΨ[Trl]Arg | 48.5 ± 2.9 | 9.11 2 |
| 6 | Lys(Har)-Pro-GlyΨ[Trl]Arg | 18.3 ± 0.9 | - |
| 7 | D-Lys(Har)-Pro-GlyΨ[Trl]Arg | 38.4 ± 1.5 | - |
| 8 | Lys(Har)-Phe-GlyΨ[Trl]Arg | 30.9 ± 2.1 | - |
| 9 | D-Lys(Har)-Phe-GlyΨ[Trl]Arg | 37.9 ± 1.1 | - |
| 10 | Lys(Har)-GlyΨ[Trl]Ala-Arg | 43.2 ± 1.5 | - |
| 11 | Lys(Har)-GlyΨ[Trl]Gly-Arg | 30.6 ± 1.1 | - |
| 12 | Har-Pro-GlyΨ[Trl]Arg | 9.2 ± 0.8 | - |
| 13 | Har-GlyΨ[Trl]GlyΨ[Trl]GlyΨ[Trl]Arg | 34.8 ± 1.5 | - |
| “arm” subseries | |||
| 14 | Lys(Fmoc-Har)-GlyΨ[Trl]GlyΨ[Trl]Arg | 57.7 ± 1.8 | - |
| 15 | D-Lys(Fmoc-Har)-GlyΨ[Trl]GlyΨ[Trl]Arg | 43.7 ± 0.5 | - |
| 16 | Har-Lys-GlyΨ[Trl]GlyΨ[Trl]Arg | 36.5 ± 1.4 | - |
| 17 | Dab(Har)-GlyΨ[Trl]GlyΨ[Trl]Arg | 41.3 ± 0.5 | - |
| 18 | Dap(Har)-GlyΨ[Trl]GlyΨ[Trl]Arg | 25.3 ± 3.5 | - |
| 19 | Har-6Ahx-GlyΨ[Trl]GlyΨ[Trl]Arg | 30.5 ± 0.6 | - |
| 20 | Har-5Ava-GlyΨ[Trl]GlyΨ[Trl]Arg | 29.7 ± 1.8 | - |
| 21 | Har-Ala-GlyΨ[Trl]GlyΨ[Trl]Arg | 27.8 ± 2.1 | - |
| 22 | Har-Gly-GlyΨ[Trl]GlyΨ[Trl]Arg | 35.7 ± 1.6 | - |
| 23 | Har-GlyΨ[Trl]GlyΨ[Trl]Arg | 20.0 ± 2.1 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fedorczyk, B.; Lipiński, P.F.J.; Puszko, A.K.; Tymecka, D.; Wilenska, B.; Dudka, W.; Perret, G.Y.; Wieczorek, R.; Misicka, A. Triazolopeptides Inhibiting the Interaction between Neuropilin-1 and Vascular Endothelial Growth Factor-165. Molecules 2019, 24, 1756. https://doi.org/10.3390/molecules24091756
Fedorczyk B, Lipiński PFJ, Puszko AK, Tymecka D, Wilenska B, Dudka W, Perret GY, Wieczorek R, Misicka A. Triazolopeptides Inhibiting the Interaction between Neuropilin-1 and Vascular Endothelial Growth Factor-165. Molecules. 2019; 24(9):1756. https://doi.org/10.3390/molecules24091756
Chicago/Turabian StyleFedorczyk, Bartlomiej, Piotr F. J. Lipiński, Anna K. Puszko, Dagmara Tymecka, Beata Wilenska, Wioleta Dudka, Gerard Y. Perret, Rafal Wieczorek, and Aleksandra Misicka. 2019. "Triazolopeptides Inhibiting the Interaction between Neuropilin-1 and Vascular Endothelial Growth Factor-165" Molecules 24, no. 9: 1756. https://doi.org/10.3390/molecules24091756
APA StyleFedorczyk, B., Lipiński, P. F. J., Puszko, A. K., Tymecka, D., Wilenska, B., Dudka, W., Perret, G. Y., Wieczorek, R., & Misicka, A. (2019). Triazolopeptides Inhibiting the Interaction between Neuropilin-1 and Vascular Endothelial Growth Factor-165. Molecules, 24(9), 1756. https://doi.org/10.3390/molecules24091756