HPLC-DAD Determination of Nitrite and Nitrate in Human Saliva Utilizing a Phosphatidylcholine Column

 , ,
, ,
Abstract
1. Introduction
2. Results and Discussion
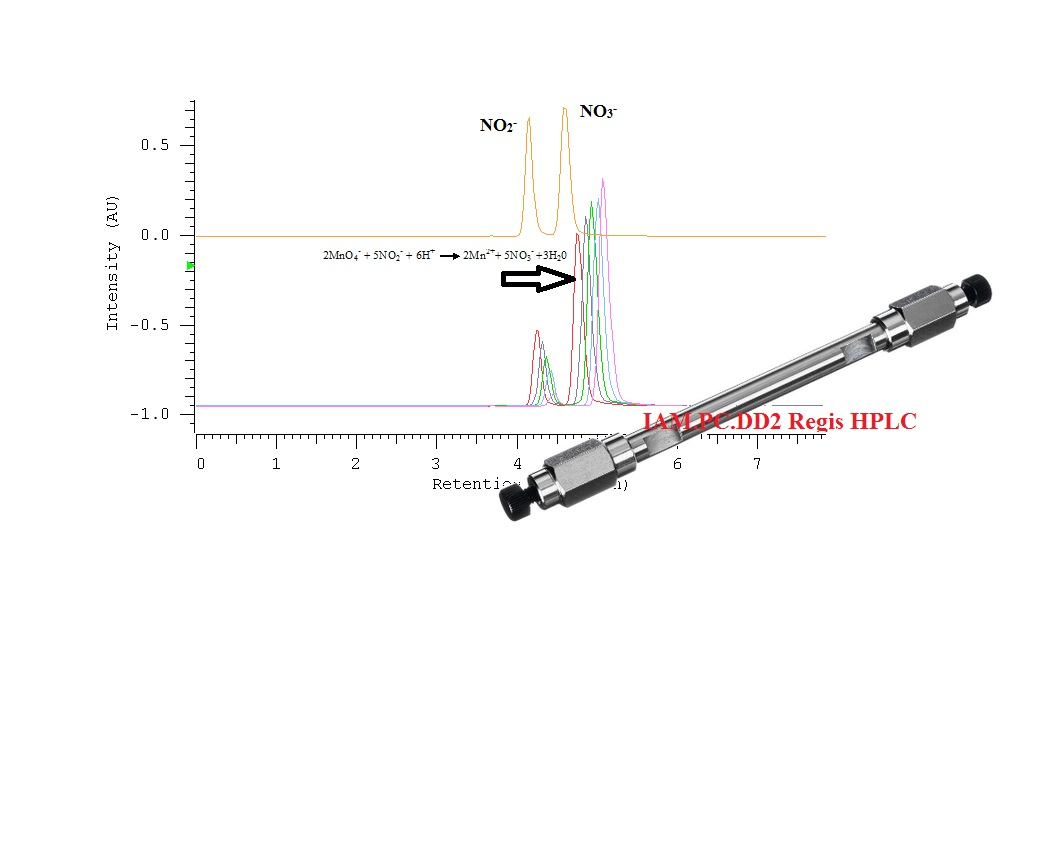
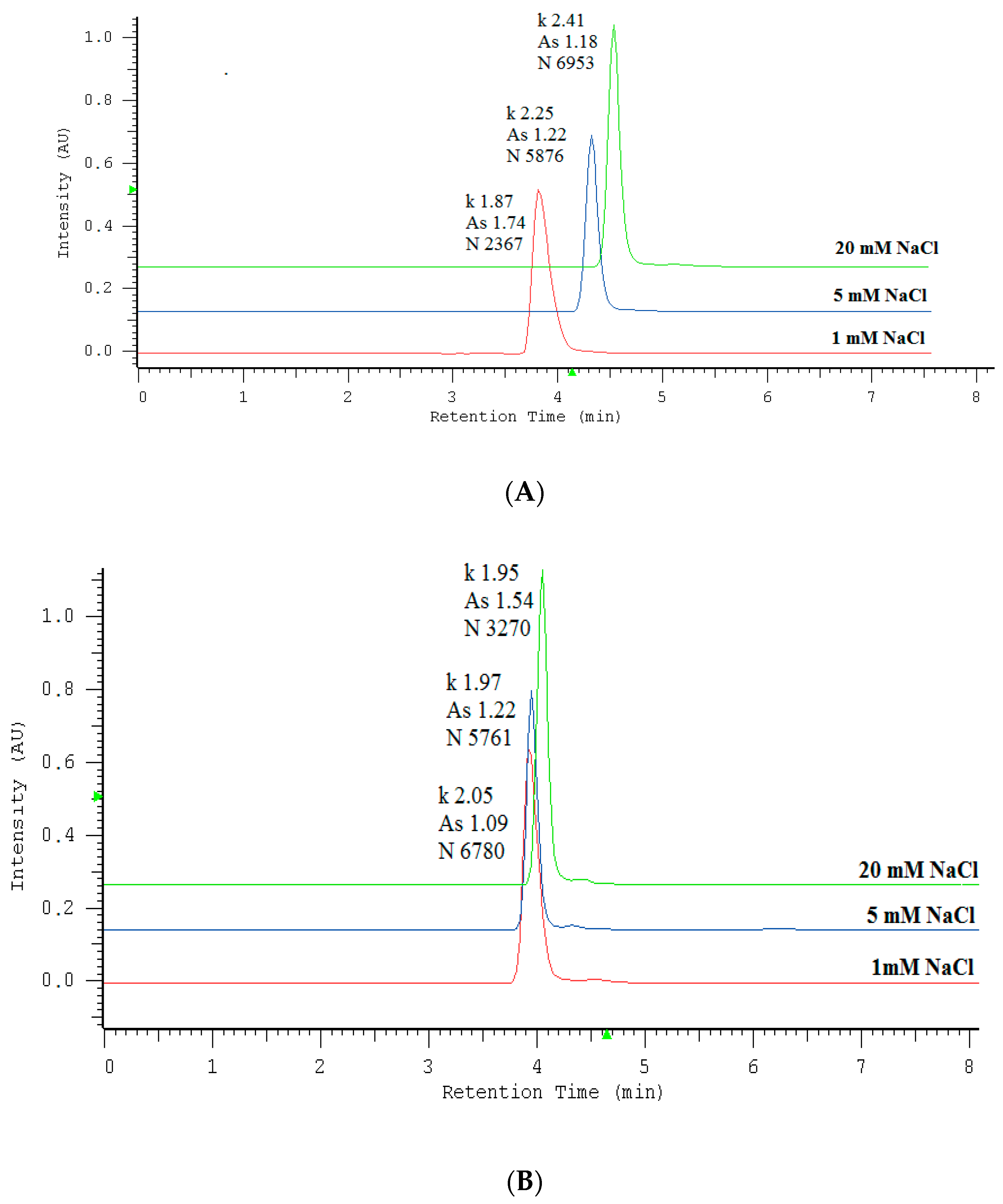
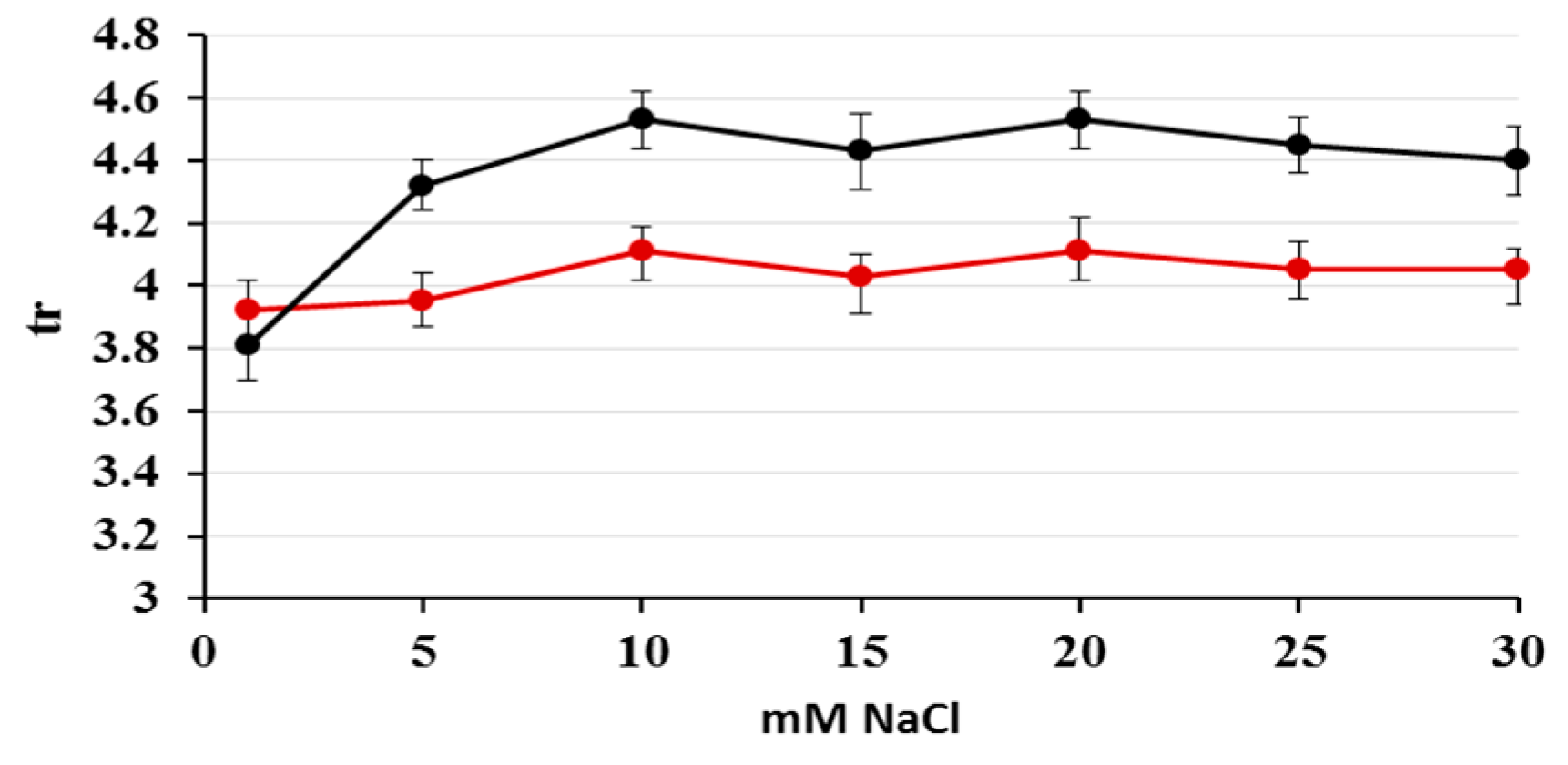
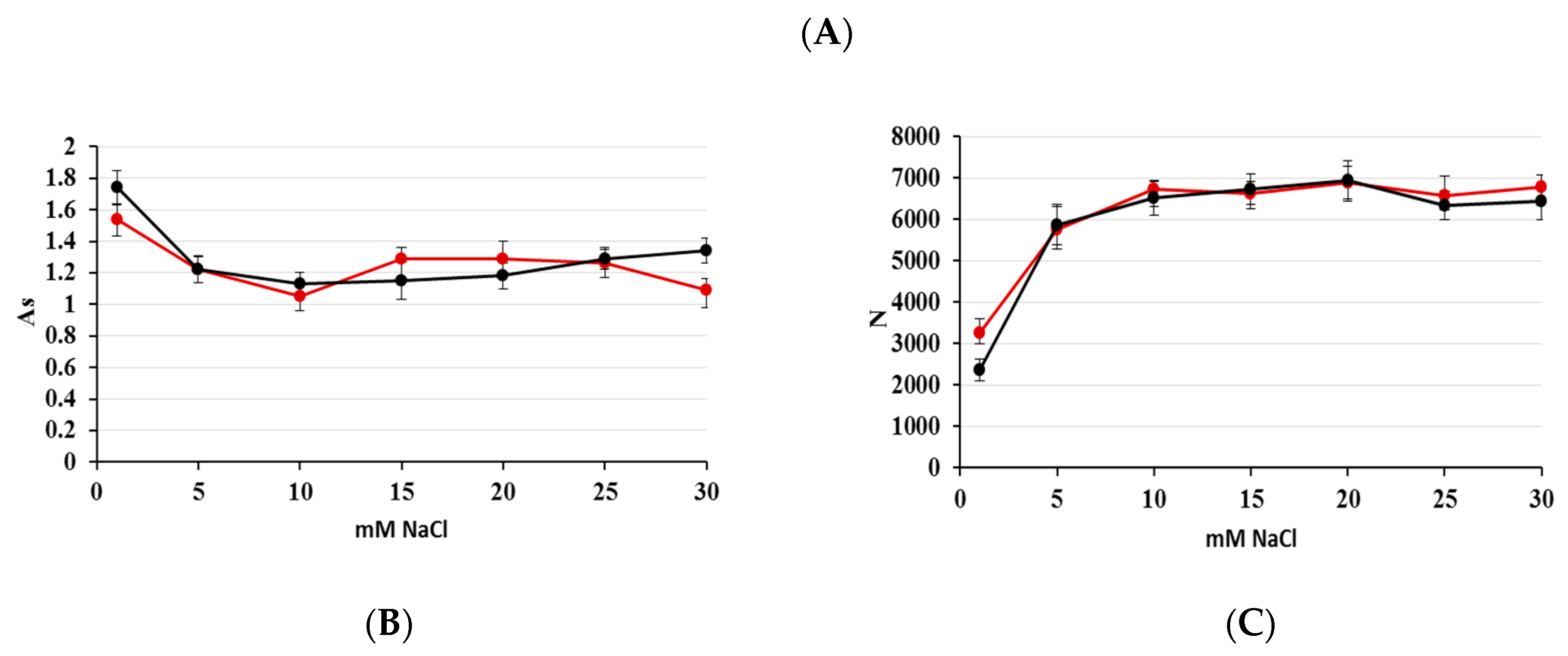
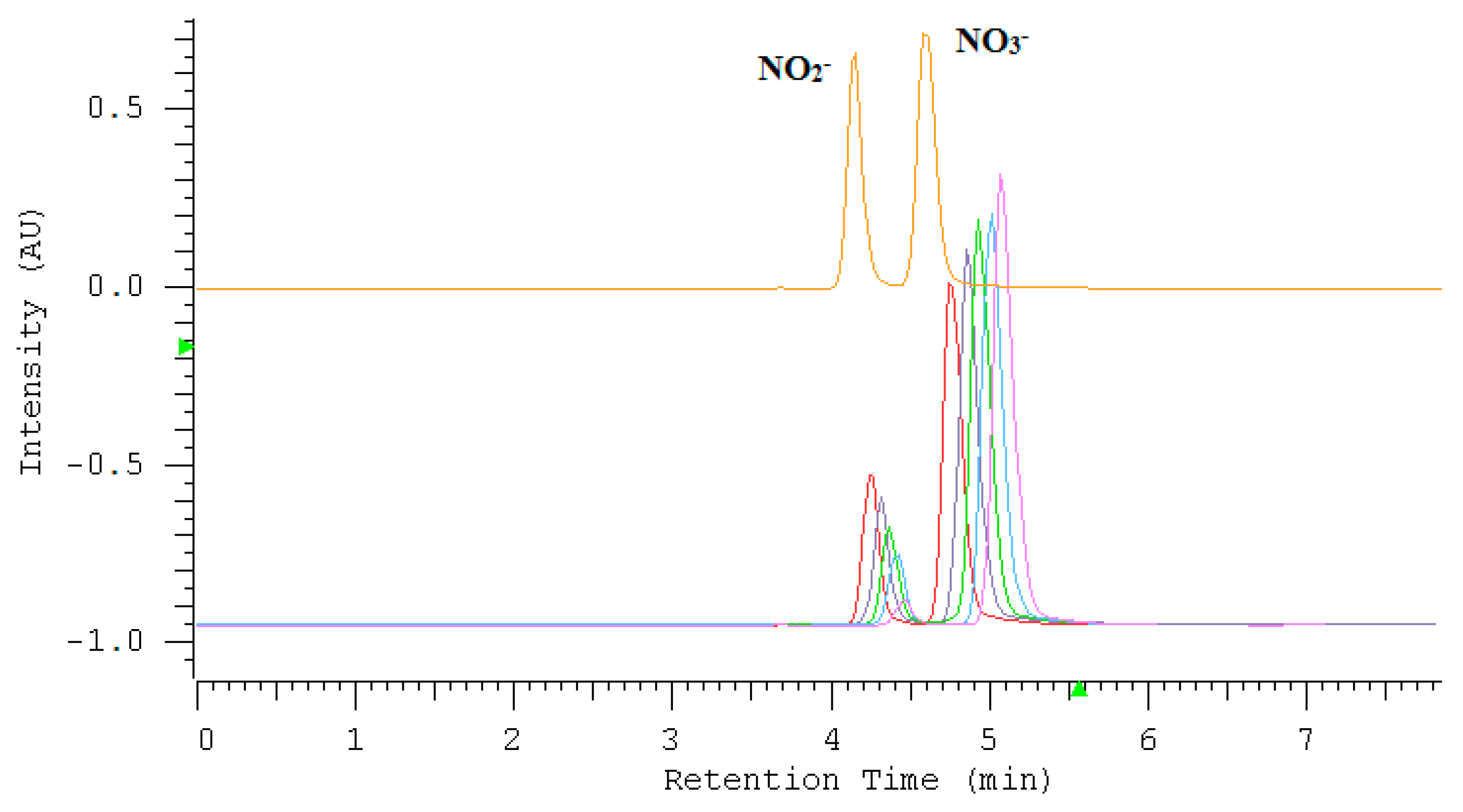
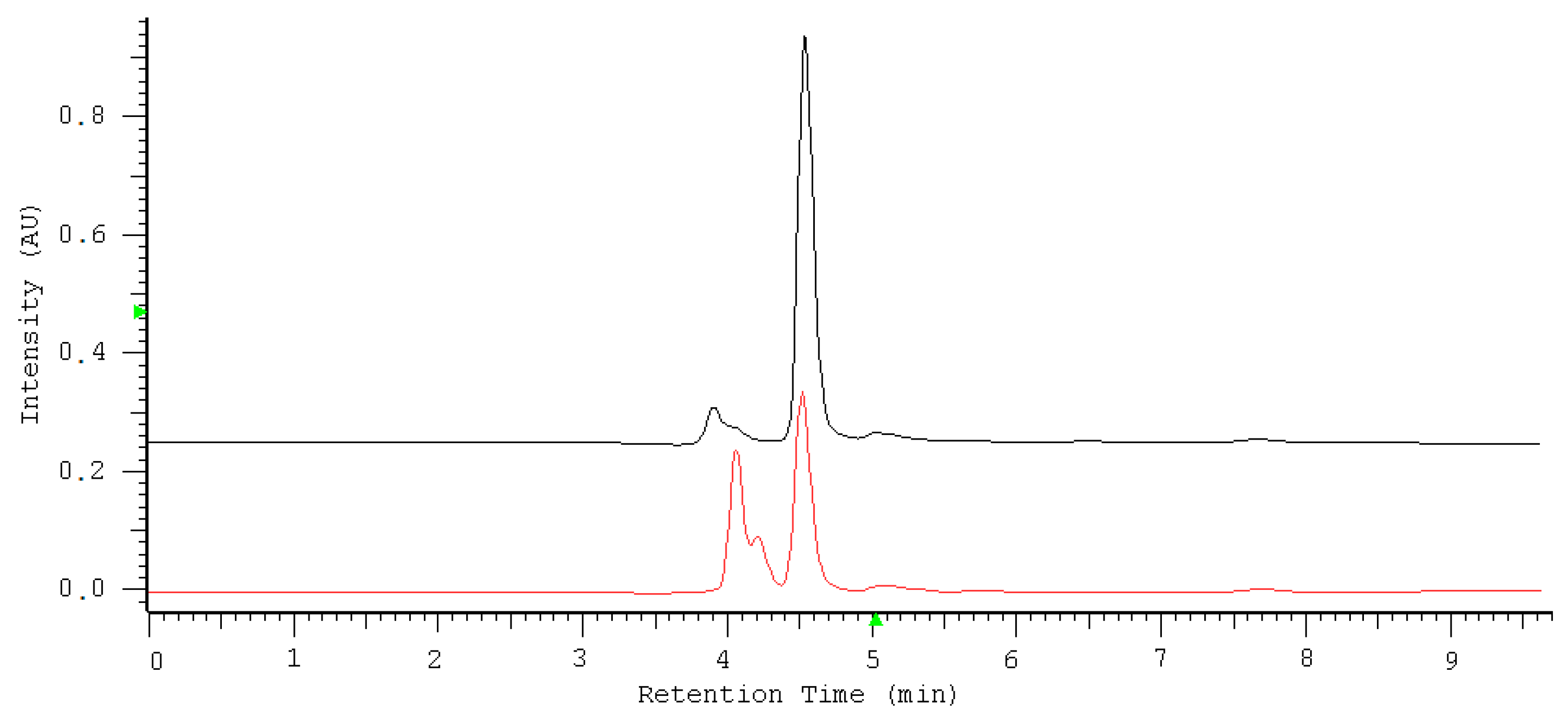
2.1. Optimization of Nitrate/Nitrite Separation
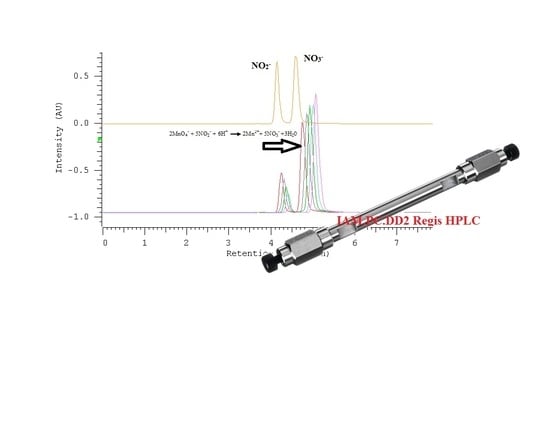
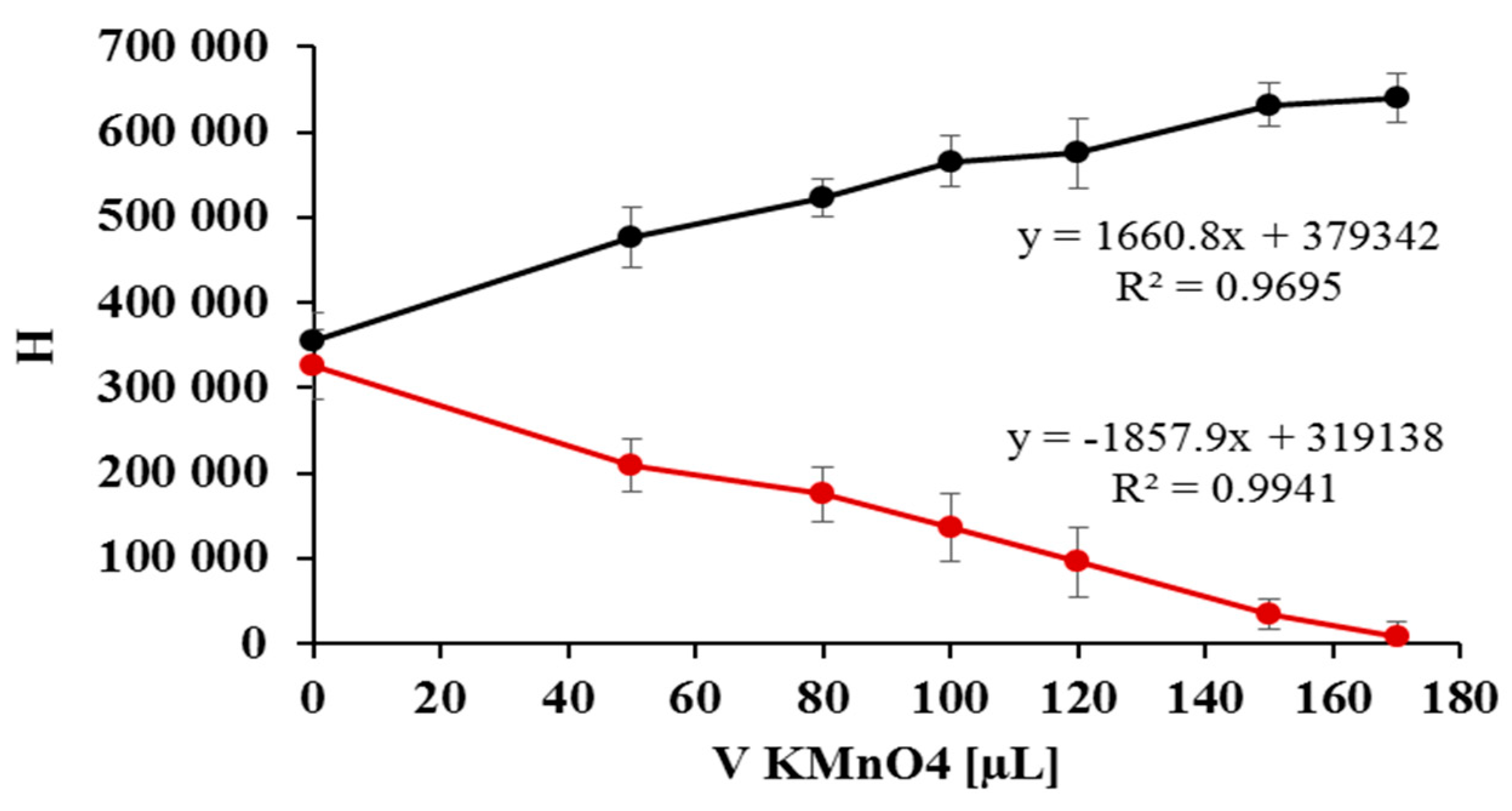
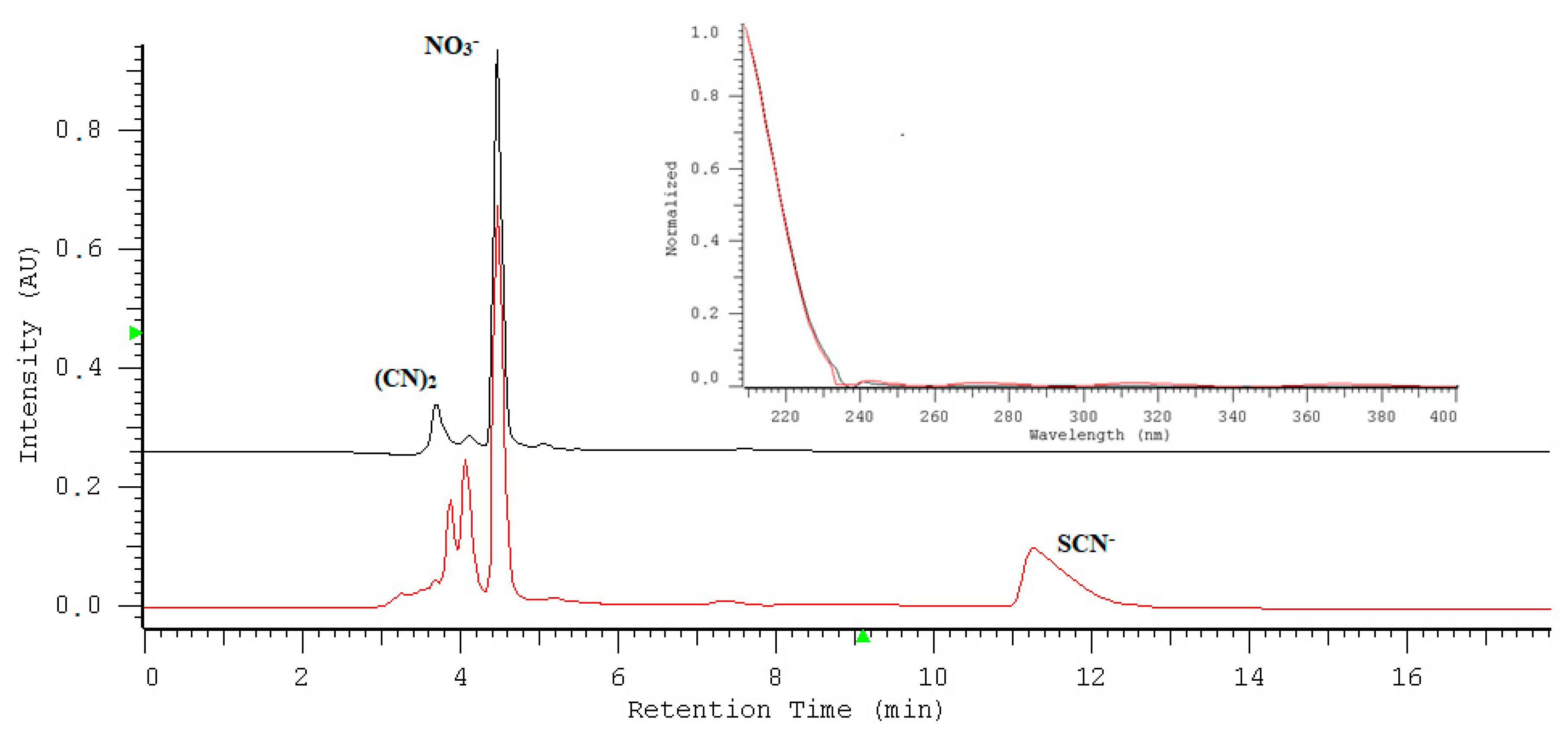
2.2. Quantification of Nitrate/Nitrite by Oxidation Using Potassium Permanganate
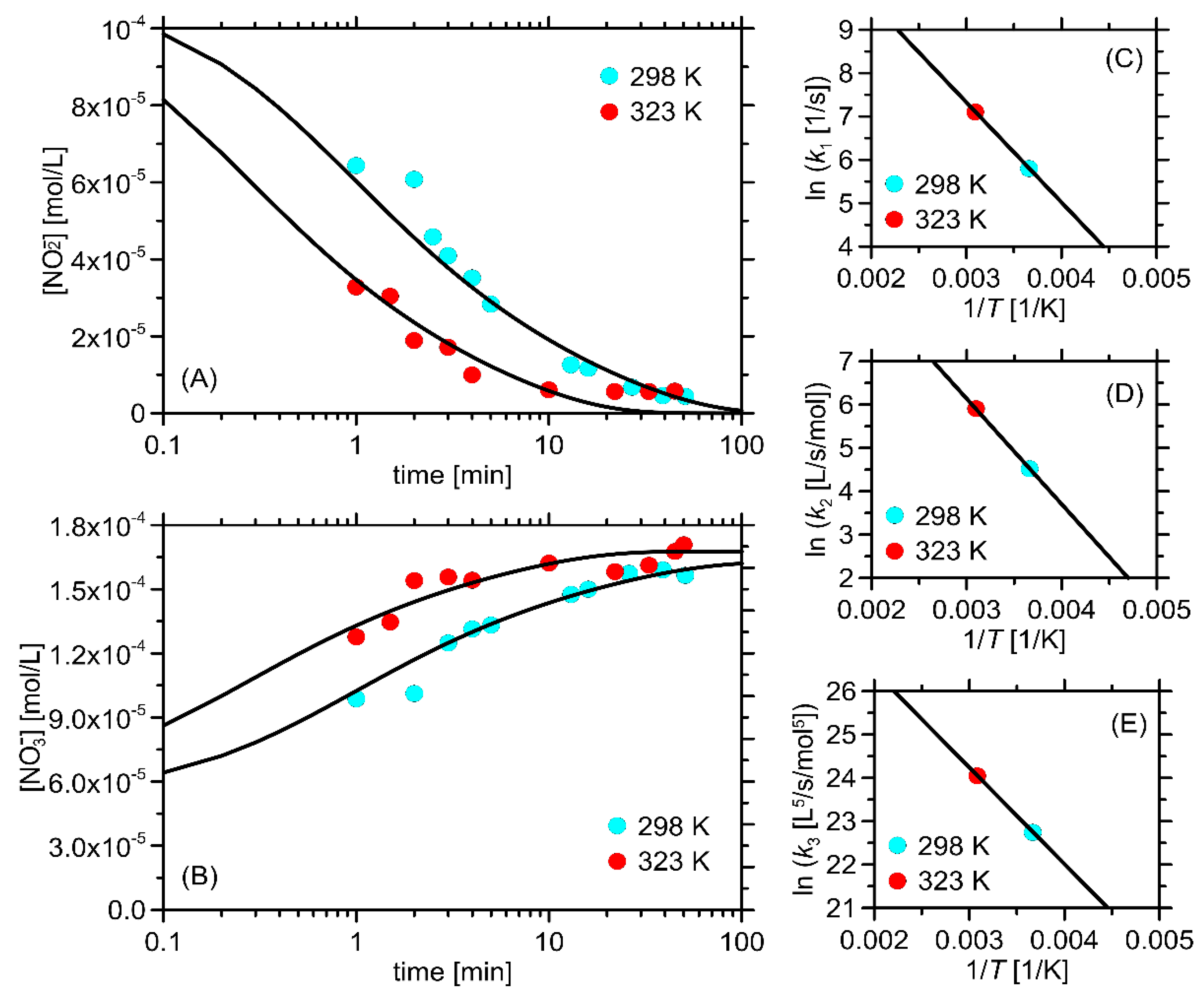
2.3. The Kinetic Study of the Permanganate–Nitrite Reaction in Acidic Media
2.4. Linearity and Sensitivity
2.5. Recovery Study
2.6. Stability Tests
2.7. Application of the Assay
3. Materials and Methods
3.1. Materials and Sample
3.2. HPLC Conditions
3.3. Method Validation
3.3.1. Linearity, Recovery, and Precision
3.3.2. Recovery and Precision
3.4. Saliva Sample Preparation
3.5. Data Analysis
4. Conclusion
Author Contributions
Funding
Conflicts of Interest
References
- Dennis, M.J.; Wilson, L.A. Nitrates and nitrites. In Encyclopedia of Food Sciences and Nutrition, 2nd ed.; Trugo, L., Finglas, P.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Fan, A.M. Biomarkers in toxicology, risk assessment, and environmental chemical regulations. In Biomarkers in Toxicology; Academic Press: Cambridge, MA, USA, 2014; pp. 1057–1080. [Google Scholar]
- Lundberg, J.O.; Weitzberg, E. Nitric oxide formation from inorganic nitrate and nitrite. In Nitric Oxide, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2010; pp. 539–553. [Google Scholar]
- Reddy, D.; Lancaster, J.R.; Cornforth, D.P. Nitrite inhibition of Clostridium botulinum: electron spin resonance detection of iron-nitric oxide complexes. Science 1983, 221, 769–770. [Google Scholar] [CrossRef] [PubMed]
- Macdougall, D.B.; Mottram, D.S.; Rhodes, D.N. Contribution of nitrite and nitrate to the colour and flavor of cured meats. J. Sci. Food Agric. 1975, 26, 1743–1754. [Google Scholar] [CrossRef]
- Froehlich, D.A.; Gullett, E.; Usborne, W. Effect of Nitrite and Salt on the Color, Flavor and Overall Acceptability of Ham. J. Food Sci. 1983, 48, 152–154. [Google Scholar] [CrossRef]
- Bryan, N.S.; Petrosino, J.F. Nitrate-Reducing oral bacteria: Linking oral and systemic health. In Nitrite and Nitrate in Human Health and Disease; Humana Press: Cham, Switzerland, 2017; pp. 21–31. [Google Scholar]
- Tiso, M.; Schechter, A.N. Nitrate Reduction to Nitrite, Nitric Oxide and Ammonia by Gut Bacteria under Physiological Conditions. PLoS One 2015, 10, e0119712. [Google Scholar]
- González, P.J.; Correia, C.; Moura, I.; Brondino, C.D.; Moura, J.J. Bacterial nitrate reductases: Molecular and biological aspects of nitrate reduction. J. Inorg. Biochem. 2006, 100, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Iijima, K.; Moriya, A.; McElroy, K.; Scobie, G.; Fyfe, V.; McColl, K. Conditions for acid catalysed luminal nitrosation are maximal at the gastric cardia. Gut 2003, 52, 1095–1101. [Google Scholar] [CrossRef]
- Lawley, P. N-nitroso compounds. In Chemical Carcinogenesis and Mutagenesis I; Springer: Berlin Heidelberg, Germany, 1990; pp. 409–469. [Google Scholar]
- Olajos, E.; Coulston, F. Comparative toxicology of n-nitroso compounds and their carcinogenic potential to man. Ecotoxicol. Environ. Saf. 1978, 2, 317–367. [Google Scholar] [CrossRef]
- Honikel, K.O. The use and control of nitrate and nitrite for the processing of meat products. Meat Sci. 2008, 78, 68–76. [Google Scholar] [CrossRef]
- Kleinbongard, K.; Dejam, A.; Lauer, T.; Jax, T.; Kerber, S.; Gharini, P.; Balzer, J.; Zotz, R.B.; Scharf, R.E.; Willers, R.; et al. Plasma nitrite concentrations reflect the degree of endothelial dysfunction in humans. Free Radic. Biol. Med. 2006, 40, 295–302. [Google Scholar] [CrossRef]
- Li, H.; Meininger, C.J.; Wu, G. Rapid determination of nitrite by reversed-phase high-performance liquid chromatography with fluorescence detection. J. Chromatogr. B: Biomed. Sci. Appl. 2000, 746, 199–207. [Google Scholar] [CrossRef]
- Helaleh, M.I.; Korenaga, T. Ion chromatographic method for simultaneous determination of nitrate and nitrite in human saliva. J. Chromatogr. B: Biomed. Sci. Appl. 2000, 744, 433–437. [Google Scholar] [CrossRef]
- Schaller, D.; Hilder, E.F.; Haddad, P.H. Monolithic stationary phases for fast ion chromatography and capillary electrochromatography of inorganic ions. J. Sep. Sci. 2006, 29, 1705–1719. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, E.; Sillanpää, M. Magnetic hydroxyapatite nanoparticles: An efficient adsorbent for the separation and removal of nitrate and nitrite ions from environmental samples. J. Sep. Sci. 2015, 38, 164–169. [Google Scholar] [CrossRef]
- Wang, X.F.; Fan, J.C.; Quan, R.R.; Wang, J.J. Rapid determination of nitrite in foods in acidic conditions by high-performance liquid chromatography with fluorescence detection. J. Sep. Sci. 2016, 29, 2263–2269. [Google Scholar] [CrossRef]
- Kurzyca, I.; Niedzielski, P.; Frankowski, M. Simultaneous speciation analysis of inorganic nitrogen with the use of ion chromatography in highly salinated environmental samples. J. Sep. Sci. 2016, 39, 3482–3487. [Google Scholar] [CrossRef] [PubMed]
- Evenhuis, C.J.; Buchberger, W.; Hilder, E.F.; Flook, K.J.; Pohl, C.A.; Nesterenko, P.N.; Haddad, P.R. Separation of inorganic anions on a high capacity porous polymeric monolithic column and application to direct determination of anions in seawater. J. Sep. Sci. 2008, 31, 2598–2604. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, I.; Silva, S. Quantification of residual nitrite and nitrate in ham by reverse-phase high performance liquid chromatography/diode array detector. Talanta 2008, 74, 1598–1602. [Google Scholar] [CrossRef] [PubMed]
- Kodamatani, H.; Yamazaki, S.; Saito, K.; Tomiyasu, T.; Komatsu, Y. Selective determination method for measurement of nitrate in water samples using high-performance liquid chromatography with post-column photochemical reaction and chemiluminescence detection. J. Chromatogr. A 2009, 1216, 3163–3167. [Google Scholar] [CrossRef]
- Niedzielski, P.; Kurzyca, I.; Siepak, J. A new tool for inorganic nitrogen speciation study: Simultaneous determination of ammonium ion, nitrite and nitrate by ion chromatography with post-column ammonium derivatization by Nessler reagent and diode-array detection in rain water samples. Anal. Chim. Acta 2006, 577, 220–224. [Google Scholar] [CrossRef]
- Gapper, L.; Fong, B.; Otter, D.; Indyk, H.; Woollard, D. Determination of nitrite and nitrate in dairy products by ion exchange LC with spectrophotometric detection. Int. Dairy J. 2004, 14, 881–887. [Google Scholar] [CrossRef]
- Wang, Q.-H.; Yu, L.-J.; Liu, Y.; Lin, L.; Lu, R.; Zhu, J.; He, L.; Lu, Z.-L. Methods for the detection and determination of nitrite and nitrate: A review. Talanta 2017, 165, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Haddad, P.R.; Tanaka, K.; Mori, M.; Tekura, K.; Hase, K.; Ohno, M.; Kamo, N. Creation and characteristics of phosphatidylcholine stationary phases for the chromatographic separation of inorganic anions. J. Chromatogr. A 2003, 997, 237–242. [Google Scholar] [CrossRef]
- Yang, C.Y.; Cai, J.; Liu, H.; Pidgeon, C. Immobilized Artificial Membranes — screens for drug membrane interactions. Adv. Drug Delivery Rev. 1997, 23, 229–256. [Google Scholar] [CrossRef]
- Wiedmer, S.K.; Jussila, M.S.; Riekkola, M.L. Phospholipids and liposomes in liquid chromatographic and capillary electromigration techniques. Trends Anal. Chem. 2004, 23, 562–582. [Google Scholar] [CrossRef]
- Marcus, Y. Thermodynamics of solvation of ions. Part 5.—Gibbs free energy of hydration at 298.15 K. J. Chem. Soc. Faraday Trans. 1991, 87, 2995–2999. [Google Scholar] [CrossRef]
- Nostro, P.L.; Ninham, B.W. Hofmeister phenomena: an update on ion specificity in biology. Chem. Rev. 2012, 112, 2286–2322. [Google Scholar] [CrossRef]
- Flieger, J. The effect of chaotropic mobile phase additives on the separation of selected alkaloids in reversed-phase high-performance liquid chromatography. J. Chromatogr. A 2006, 1113, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.; LoBrutto, R.; Kazakevich, Y. Effect of the counter-anion type and concentration on the liquid chromatography retention of β-blockers. J. Chromatogr. A 2002, 964, 179–187. [Google Scholar] [CrossRef]
- Dózsa, L.; Beck, M.T. The mechanism of the permanganate-nitrite reaction. Inorg. Chim. Acta 1970, 4, 219–225. [Google Scholar] [CrossRef]
- International Conference on Harmonization (ICH) of Technical Requirements for the Registration of Pharmaceuticals for Human Use. Validation of analytical procedures: Text and Methodology; ICH-Q2B: Geneva, Switzerland, 1996; Available online: https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1__Guideline.pdf (accessed on 6 May 2019).
- The R Project for Statistical Computing. Available online: https://www.R-project.org/ (accessed on 5 May 2019).
- Najdenkoska, A. Development of HPLC method for analysis of nitrite and nitrate in vegetable. J. Agric. Food Environ. Sci. 2016, 67, 33–39. [Google Scholar]
Sample Availability: Not available. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Linear Regression Parameters | Nitrates NO3− | Nitrites NO2− | |
|---|---|---|---|
| LOD * [ng mL−1] | 4.56 | 4.21 | |
| LOQ * [ng mL−1] | 15.21 | 14.03 | |
| Linear range ** [µg mL−1] | 0.50–35.00 | 0.50–15.00 | |
| Regression equation ***(y = ax + b) **** | a ± sa | 7809.31 ± 97.92 | 8966.61 ± 194.40 |
| b ± sb | 1739.12 ± 5114.79 | −1743.60 ± 9500.16 | |
| R2seF | 0.9992 6360.71 8703.09 | 0.9972 2127.55 18575.24 | |
| Parameters | Nitrate | ||
| Fortification level (µg/mL) | 3.00 | 7.50 | 15.00 |
| Average recovery (n = 6) (%) | 98.12 | 98.65 | 93.33 |
| Intra-day precision, (RSDr,%) | 3.21 | 2.65 | 3.41 |
| Inter-day precision, (RSDwR,%) | 4.73 | 4.71 | 4.20 |
| Parameters | Nitrite | ||
| Fortification level (ng/mL) | 3.00 | 7.50 | 15.00 |
| Average recovery (n = 6) (%) | 86.90 | 87.74 | 104.23 |
| Intra-day precision, (RSDr,%) | 3.51 | 3.83 | 3.34 |
| Inter-day precision, (RSDwR,%) | 5.90 | 4.19 | 4.76 |
| Water System | 3 µg mL−1 | 7.5 µg mL−1 | 15 µg mL−1 | ||
| Without oxidation | tr | 4.54 (±0.03) | 4.58 (±0.05) | 4.52 (±0.01) | |
| H | 23604 (±342) | 58198 (±989) | 115198 (±1727) | ||
| As | 1.15 (±0.02) | 1.29 (±0.03) | 1.28 (±0.02) | ||
| N | 5591 (±331) | 5771 (±295) | 5859 (±307) | ||
| P | 0.9996 (±0.0003) | 0.9996 (±0.0001) | 1.0000 (±0.0000) | ||
| R2 | 0.9907 (±0.0014) | 0.9907 (±0.0009) | 0.9910 (±0.0003) | ||
| After oxidation | tr | 4.57 (±0.05) | 4.53 (±0.03) | 4.53 (±0.02) | |
| H | 59305 (±771) | 136869 (±1916) | 262669 (±3415) | ||
| As | 1.26 (±0.03) | 1.34 (±0.04) | 1.33 (±0.04) | ||
| N | 6122 (±262) | 5960 (±311) | 5944 (±270) | ||
| P | 1.0000 (±0.0000) | 0.9996 (±0.0002) | 0.9999 (±0.0001) | ||
| R2 | 0.9908 (±0.0011) | 0.9910 (±0.0007) | 0.9908 (±0.0009) | ||
| Samples of Saliva | Without oxidation | tr | 4.53 (±0.03) | 4.55 (±0.04) | 4.58 (±0.06) |
| H | 72097 (±901) | 108175 (±1293) | 167554 (±1508) | ||
| As | 1.36 (±0.03) | 1.28 (±0.02) | 1.25 (±0.04) | ||
| N | 5446 (±369) | 5365 (±291) | 5850 (±283) | ||
| P | 0.9614 (±0.0097) | 0.9731 (±0.0054) | 0.9982 (±0.0016) | ||
| R2 | 0.9909 (±0.0012) | 0.9908 (±0.0008) | 0.9907 (±0.0008) | ||
| After oxidation | tr | 4.51 (±0.02) | 4.53 (±0.01) | 4.55 (±0.04) | |
| H | 123968 (±1759) | 197560 (±2374) | 340325 (±3544) | ||
| As | 1.37 (±0.05) | 1.22 (±0.02) | 1.35 (±0.03) | ||
| N | 6499 (±321) | 6036 (± 279) | 6450 (±332) | ||
| P | 0.9997 (±0.0002) | 0.9998 (±0.0002) | 1.0000 (±0.0000) | ||
| R2 | 0.9906 (±0.0005) | 0.9904 (±0.0012) | 0.9904 (±0.0007) | ||
| Nitrate | Nitrite | ||
|---|---|---|---|
| Concentration Range [µg mL−1] | Mean ± SD [µg mL−1] | Concentration Range [µg mL−1] | Mean ± SD [µg mL−1] |
| 8.98–18.52 | 13.76 ± 3.20 | 3.50–5.34 | 4.24 ± 0.65 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tatarczak-Michalewska, M.; Flieger, J.; Kawka, J.; Płaziński, W.; Flieger, W.; Blicharska, E.; Majerek, D. HPLC-DAD Determination of Nitrite and Nitrate in Human Saliva Utilizing a Phosphatidylcholine Column. Molecules 2019, 24, 1754. https://doi.org/10.3390/molecules24091754
Tatarczak-Michalewska M, Flieger J, Kawka J, Płaziński W, Flieger W, Blicharska E, Majerek D. HPLC-DAD Determination of Nitrite and Nitrate in Human Saliva Utilizing a Phosphatidylcholine Column. Molecules. 2019; 24(9):1754. https://doi.org/10.3390/molecules24091754
Chicago/Turabian StyleTatarczak-Michalewska, Małgorzata, Jolanta Flieger, Justyna Kawka, Wojciech Płaziński, Wojciech Flieger, Eliza Blicharska, and Dariusz Majerek. 2019. "HPLC-DAD Determination of Nitrite and Nitrate in Human Saliva Utilizing a Phosphatidylcholine Column" Molecules 24, no. 9: 1754. https://doi.org/10.3390/molecules24091754
APA StyleTatarczak-Michalewska, M., Flieger, J., Kawka, J., Płaziński, W., Flieger, W., Blicharska, E., & Majerek, D. (2019). HPLC-DAD Determination of Nitrite and Nitrate in Human Saliva Utilizing a Phosphatidylcholine Column. Molecules, 24(9), 1754. https://doi.org/10.3390/molecules24091754