
Effects of Silymarin on the In Vivo Pharmacokinetics of Simvastatin and Its Active Metabolite in Rats


Abstract
1. Introduction
2. Results
2.1. Method Optimization
2.1.1. Optimization of Chromatography and Mass Spectrometry Conditions
2.1.2. Optimization of Plasma Sample Preparation
2.2. Method Validation
2.2.1. Selectivity
2.2.2. Linearity and Lower Limit of Quantitation (LLOQ)
2.2.3. Accuracy and Precision
2.2.4. Extraction Recovery and Matrix Effect
2.2.5. Dilution Integrity
2.2.6. Stability Studies
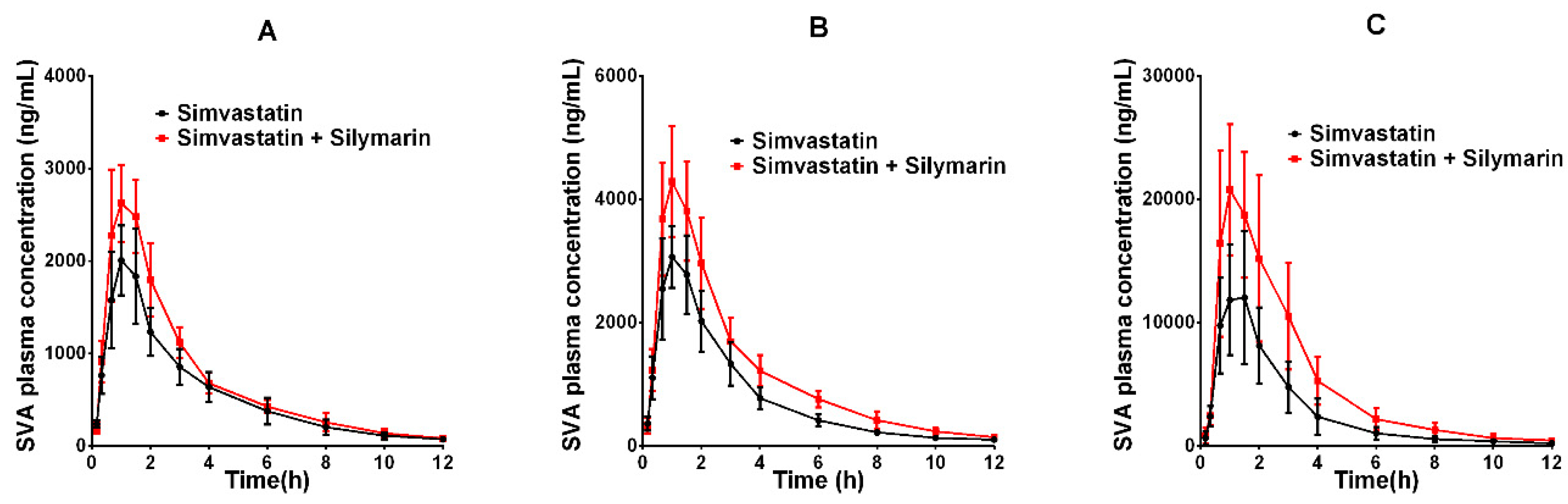
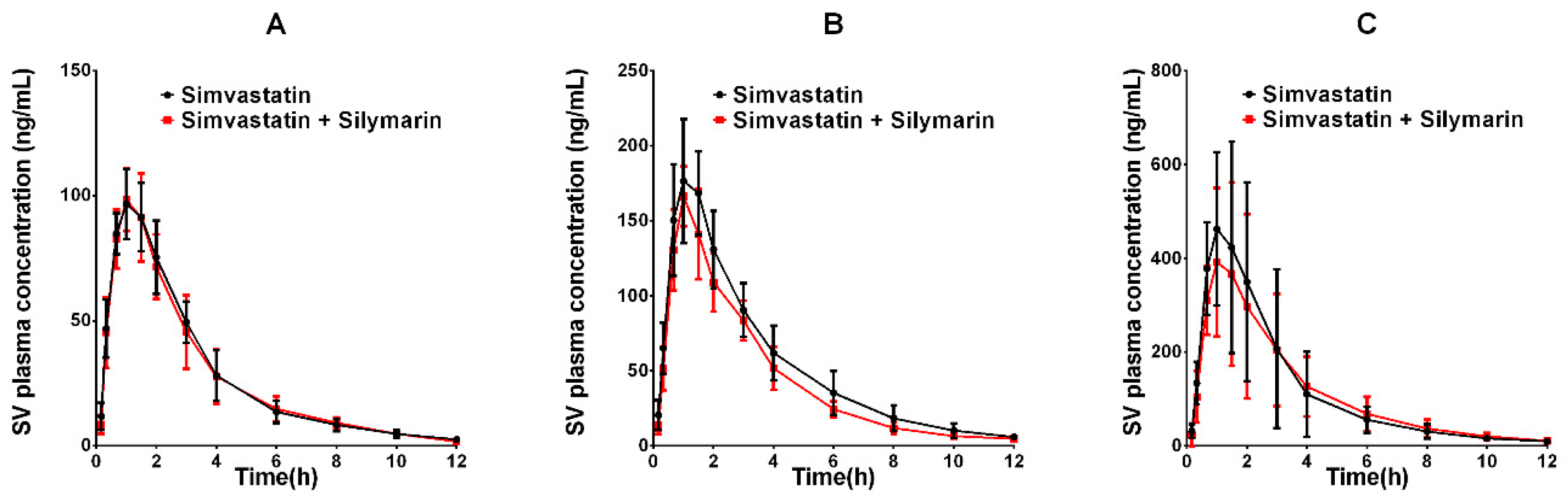
2.3. Effects of Silymarin on Simvastatin Pharmacokinetics
3. Discussion
4. Materials and Methods
4.1. Reagents and Chemicals
4.2. UPLC–MS/MS Conditions
4.3. Preparation of Calibration Standards and Quality Control Samples
4.4. Sample Preparation
4.5. Bioanalytical Method Validation
4.5.1. Selectivity
4.5.2. Linearity and Lower Limit of Quantitation (LLOQ)
4.5.3. Accuracy and Precision
4.5.4. Extraction Recovery and Matrix Effect
4.5.5. Dilution Integrity
4.5.6. Stability Studies
4.6. Herb–Drug Interaction Studies
4.6.1. Animals
4.6.2. Drug Administration
4.6.3. Blood Sampling
4.6.4. Analysis of Pharmacokinetic Data
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Statistics 2017: Monitoring Health for the SDGs. Available online: https://www.who.int/gho/publications/world_health_statistics/2017/en/ (accessed on 27 March 2019).
- Catapano, A.L.; Graham, I.; De Backer, G.; Wiklund, O.; Chapman, M.J.; Drexel, H.; Hoes, A.W.; Jennings, C.S.; Landmesser, U.; Pedersen, T.R.; et al. 2016 ESC/EAS Guidelines for the Management of Dyslipidaemias. Eur. Heart J. 2016, 37, 2999–3058. [Google Scholar] [CrossRef]
- Heart Protection Study Collaborative Group. Effects on 11-year mortality and morbidity of lowering LDL cholesterol with simvastatin for about 5 years in 20,536 high-risk individuals: A randomised controlled trial. Lancet 2011, 378, 2013–2020. [Google Scholar] [CrossRef]
- Strandberg, T.E.; Pyorala, K.; Cook, T.J.; Wilhelmsen, L.; Faergeman, O.; Thorgeirsson, G.; Pedersen, T.R.; Kjekshus, J. Mortality and incidence of cancer during 10-year follow-up of the Scandinavian Simvastatin Survival Study (4S). Lancet 2004, 364, 771–777. [Google Scholar] [CrossRef]
- Brunham, L.R.; Lansberg, P.J.; Zhang, L.; Miao, F.; Carter, C.; Hovingh, G.K.; Visscher, H.; Jukema, J.W.; Stalenhoef, A.F.; Ross, C.J.; et al. Differential effect of the rs4149056 variant in SLCO1B1 on myopathy associated with simvastatin and atorvastatin. Pharm. J. 2012, 12, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Canestaro, W.J.; Austin, M.A.; Thummel, K.E. Genetic factors affecting statin concentrations and subsequent myopathy: A HuGENet systematic review. Genet. Med. 2014, 16, 810–819. [Google Scholar] [CrossRef]
- Ramsey, L.B.; Johnson, S.G.; Caudle, K.E.; Haidar, C.E.; Voora, D.; Wilke, R.A.; Maxwell, W.D.; McLeod, H.L.; Krauss, R.M.; Roden, D.M.; et al. The clinical pharmacogenetics implementation consortium guideline for SLCO1B1 and simvastatin-induced myopathy: 2014 update. Clin. Pharmacol. Ther. 2014, 96, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Wilke, R.A.; Ramsey, L.B.; Johnson, S.G.; Maxwell, W.D.; McLeod, H.L.; Voora, D.; Krauss, R.M.; Roden, D.M.; Feng, Q.; Cooper-Dehoff, R.M.; et al. The clinical pharmacogenomics implementation consortium: CPIC guideline for SLCO1B1 and simvastatin-induced myopathy. Clin. Pharmacol. Ther. 2012, 92, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Snowden, S.G.; Grapov, D.; Settergren, M.; D’Alexandri, F.L.; Haeggstrom, J.Z.; Fiehn, O.; Hyotylainen, T.; Pedersen, T.L.; Newman, J.W.; Oresic, M.; et al. High-dose simvastatin exhibits enhanced lipid-lowering effects relative to simvastatin/ezetimibe combination therapy. Circ. Cardiovasc. Genet. 2014, 7, 955–964. [Google Scholar] [CrossRef]
- Choi, H.Y.; Bae, K.S.; Cho, S.H.; Ghim, J.L.; Choe, S.; Jung, J.A.; Jin, S.J.; Kim, H.S.; Lim, H.S. Impact of CYP2D6, CYP3A5, CYP2C19, CYP2A6, SLCO1B1, ABCB1, and ABCG2 gene polymorphisms on the pharmacokinetics of simvastatin and simvastatin acid. Pharm. Genom. 2015, 25, 595–608. [Google Scholar] [CrossRef]
- Goosen, T.C.; Bauman, J.N.; Davis, J.A.; Yu, C.; Hurst, S.I.; Williams, J.A.; Loi, C.M. Atorvastatin glucuronidation is minimally and nonselectively inhibited by the fibrates gemfibrozil, fenofibrate, and fenofibric acid. Drug Metab. Dispos. 2007, 35, 1315–1324. [Google Scholar] [CrossRef] [PubMed]
- Hoch, M.; Hoever, P.; Alessi, F.; Theodor, R.; Dingemanse, J. Pharmacokinetic interactions of almorexant with midazolam and simvastatin, two CYP3A4 model substrates, in healthy male subjects. Eur. J. Clin. Pharmacol. 2013, 69, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Itkonen, M.K.; Tornio, A.; Neuvonen, M.; Neuvonen, P.J.; Niemi, M.; Backman, J.T. Clopidogrel Has No Clinically Meaningful Effect on the Pharmacokinetics of the Organic Anion Transporting Polypeptide 1B1 and Cytochrome P450 3A4 Substrate Simvastatin. Drug Metab. Dispos. 2015, 43, 1655–1660. [Google Scholar] [CrossRef]
- Moron, B.; Verma, A.K.; Das, P.; Taavela, J.; Dafik, L.; Diraimondo, T.R.; Albertelli, M.A.; Kraemer, T.; Maki, M.; Khosla, C.; et al. CYP3A4-catalyzed simvastatin metabolism as a non-invasive marker of small intestinal health in celiac disease. Am. J. Gastroenterol. 2013, 108, 1344–1351. [Google Scholar] [CrossRef] [PubMed]
- Prueksaritanont, T.; Subramanian, R.; Fang, X.; Ma, B.; Qiu, Y.; Lin, J.H.; Pearson, P.G.; Baillie, T.A. Glucuronidation of statins in animals and humans: A novel mechanism of statin lactonization. Drug Metab. Dispos. 2002, 30, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Aller, R.; Izaola, O.; Gomez, S.; Tafur, C.; Gonzalez, G.; Berroa, E.; Mora, N.; Gonzalez, J.M.; de Luis, D.A. Effect of silymarin plus vitamin E in patients with non-alcoholic fatty liver disease. A randomized clinical pilot study. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 3118–3124. [Google Scholar] [PubMed]
- Fried, M.W.; Navarro, V.J.; Afdhal, N.; Belle, S.H.; Wahed, A.S.; Hawke, R.L.; Doo, E.; Meyers, C.M.; Reddy, K.R. Effect of silymarin (milk thistle) on liver disease in patients with chronic hepatitis C unsuccessfully treated with interferon therapy: A randomized controlled trial. JAMA 2012, 308, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Gillessen, A.; Herrmann, W.A.; Kemper, M.; Morath, H.; Mann, K. Effect of silymarin on liver health and quality of life. Results of a non-interventional study. MMW Fortschr. Med. 2014, 156, 120–126. [Google Scholar] [CrossRef]
- Moayedi Esfahani, B.A.; Reisi, N.; Mirmoghtadaei, M. Evaluating the safety and efficacy of silymarin in beta-thalassemia patients: A review. Hemoglobin 2015, 39, 75–80. [Google Scholar] [CrossRef]
- Perez, H.J.; Carrillo, S.C.; Garcia, E.; Ruiz-Mar, G.; Perez-Tamayo, R.; Chavarria, A. Neuroprotective effect of silymarin in a MPTP mouse model of Parkinson’s disease. Toxicology 2014, 319, 38–43. [Google Scholar] [CrossRef]
- Poruba, M.; Kazdova, L.; Oliyarnyk, O.; Malinska, H.; Matuskova, Z.; Tozzi di Angelo, I.; Skop, V.; Vecera, R. Improvement bioavailability of silymarin ameliorates severe dyslipidemia associated with metabolic syndrome. Xenobiotica 2015, 45, 751–756. [Google Scholar] [CrossRef]
- Doehmer, J.; Weiss, G.; McGregor, G.P.; Appel, K. Assessment of a dry extract from milk thistle (Silybum marianum) for interference with human liver cytochrome-P450 activities. Toxicol. In Vitro 2011, 25, 21–27. [Google Scholar] [CrossRef]
- Malekinejad, H.; Rezabakhsh, A.; Rahmani, F.; Hobbenaghi, R. Silymarin regulates the cytochrome P450 3A2 and glutathione peroxides in the liver of streptozotocin-induced diabetic rats. Phytomedicine 2012, 19, 583–590. [Google Scholar] [CrossRef]
- Sridar, C.; Goosen, T.C.; Kent, U.M.; Williams, J.A.; Hollenberg, P.F. Silybin inactivates cytochromes P450 3A4 and 2C9 and inhibits major hepatic glucuronosyltransferases. Drug Metab. Dispos. 2004, 32, 587–594. [Google Scholar] [CrossRef]
- D’Andrea, V.; Perez, L.M.; Sanchez Pozzi, E.J. Inhibition of rat liver UDP-glucuronosyltransferase by silymarin and the metabolite silibinin-glucuronide. Life Sci. 2005, 77, 683–692. [Google Scholar] [CrossRef]
- Gao, C.; Shi, R.; Wang, T.; Tan, H.; Xu, H.; Ma, Y. Interaction between oblongifolin C and UDP-glucuronosyltransferase isoforms in human liver and intestine microsomes. Xenobiotica 2015, 45, 578–585. [Google Scholar] [CrossRef]
- Jancova, P.; Siller, M.; Anzenbacherova, E.; Kren, V.; Anzenbacher, P.; Simanek, V. Evidence for differences in regioselective and stereoselective glucuronidation of silybin diastereomers from milk thistle (Silybum marianum) by human UDP-glucuronosyltransferases. Xenobiotica 2011, 41, 743–751. [Google Scholar] [CrossRef]
- Wu, J.W.; Lin, L.C.; Tsai, T.H. Drug-drug interactions of silymarin on the perspective of pharmacokinetics. J. Ethnopharmacol. 2009, 121, 185–193. [Google Scholar] [CrossRef]
- Lee, K.S.; Chae, S.W.; Park, J.H.; Park, J.H.; Choi, J.M.; Rhie, S.J.; Lee, H.J. Effects of single or repeated silymarin administration on pharmacokinetics of risperidone and its major metabolite, 9-hydroxyrisperidone in rats. Xenobiotica 2013, 43, 303–310. [Google Scholar] [CrossRef]
- Liao, S.; Jin, X.; Li, J.; Zhang, T.; Zhang, W.; Shi, W.; Fan, S.; Wang, X.; Wang, J.; Zhong, B.; et al. Effects of Silymarin, Glycyrrhizin, and Oxymatrine on the Pharmacokinetics of Ribavirin and Its Major Metabolite in Rats. Phytother. Res. 2016, 30, 618–626. [Google Scholar] [CrossRef]
- Malekinejad, H.; Rokhsartalab-Azar, S.; Hassani-Dizaj, S.; Alizadeh-Fanalou, S.; Rezabakhsh, A.; Amniattalab, A. Effects of silymarin on the pharmacokinetics of atorvastatin in diabetic rats. Eur. J. Drug Metab. Pharmacokinet. 2014, 39, 311–320. [Google Scholar] [CrossRef]
- Voruganti, S.; Yamsani, S.K.; Yamsani, M.R. Effect of silibinin on the pharmacokinetics of nitrendipine in rabbits. Eur. J. Drug Metab. Pharmacokinet. 2014, 39, 277–281. [Google Scholar] [CrossRef]
- El-Shitany, N.A.; El-Haggar, S.; El-desoky, K. Silymarin prevents adriamycin-induced cardiotoxicity and nephrotoxicity in rats. Food Chem. Toxicol. 2008, 46, 2422–2428. [Google Scholar] [CrossRef]
- Skottova, N.; Kazdova, L.; Oliyarnyk, O.; Vecera, R.; Sobolova, L.; Ulrichova, J. Phenolics-rich extracts from Silybum marianum and Prunella vulgaris reduce a high-sucrose diet induced oxidative stress in hereditary hypertriglyceridemic rats. Pharmacol. Res. 2004, 50, 123–130. [Google Scholar] [CrossRef]
- Poruba, M.; Matuskova, Z.; Kazdova, L.; Oliyarnyk, O.; Malinska, H.; Tozzi di Angelo, I.; Vecera, R. Positive effects of different drug forms of silybin in the treatment of metabolic syndrome. Physiol. Res. 2015, 64, 507–512. [Google Scholar]
- El Karbane, M.; Azougagh, M.; Amood, A.L.K.M.; Bouchafra, H.; Cherrah, Y.; Bouklouze, A. Development and validation of a reversed-phase HPLC method for simultaneous analysis of butylhydroxyanisol, simvastatin and its impurities in tablet dosage forms. Ann. Pharm. Fr. 2014, 72, 244–255. [Google Scholar] [CrossRef]
- Magdy, N.; Ayad, M.F. Two smart spectrophotometric methods for the simultaneous estimation of Simvastatin and Ezetimibe in combined dosage form. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2015, 137, 685–691. [Google Scholar] [CrossRef]
- Liu, M.; Su, X.; Li, G.; Zhao, G.; Zhao, L. Validated UPLC-MS/MS method for simultaneous determination of simvastatin, simvastatin hydroxy acid and berberine in rat plasma: Application to the drug-drug pharmacokinetic interaction study of simvastatin combined with berberine after oral administration in rats. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2015, 1006, 8–15. [Google Scholar]
- Alakhali, K.M. Method Validation for Analysis of Simvastatin in Human Plasma Using Liquid Chromatography Tandem Mass Spectrometry (LC-MS-MS). J. Clin. Diagn. Res. 2013, 7, 2739–2743. [Google Scholar] [CrossRef]
- Senthamil Selvan, P.; Pal, T.K. Chromatography-tandem mass spectrometry method for the simultaneous quantitation of metoprolol succinate and simvastatin in human plasma. J. Pharm. Biomed. Anal. 2009, 49, 780–785. [Google Scholar] [CrossRef]
- Fujino, H.; Saito, T.; Tsunenari, Y.; Kojima, J.; Sakaeda, T. Metabolic properties of the acid and lactone forms of HMG-CoA reductase inhibitors. Xenobiotica 2004, 34, 961–971. [Google Scholar] [CrossRef]
- Prueksaritanont, T.; Ma, B.; Yu, N. The human hepatic metabolism of simvastatin hydroxy acid is mediated primarily by CYP3A, and not CYP2D6. Br. J. Clin. Pharmacol. 2003, 56, 120–124. [Google Scholar] [CrossRef]
- Macan, M.; Vuksic, A.; Zunec, S.; Konjevoda, P.; Lovric, J.; Kelava, M.; Stambuk, N.; Vrkic, N.; Bradamante, V. Effects of simvastatin on malondialdehyde level and esterase activity in plasma and tissue of normolipidemic rats. Pharmacol. Rep. 2015, 67, 907–913. [Google Scholar] [CrossRef]
- Wang, X.; Zhu, H.J.; Markowitz, J.S. Carboxylesterase 1-mediated drug-drug interactions between clopidogrel and simvastatin. Biol. Pharm. Bull. 2015, 38, 292–297. [Google Scholar] [CrossRef]
- Iwuchukwu, O.F.; Feng, Q.; Wei, W.Q.; Jiang, L.; Jiang, M.; Xu, H.; Denny, J.C.; Wilke, R.A.; Krauss, R.M.; Roden, D.M.; et al. Genetic variation in the UGT1A locus is associated with simvastatin efficacy in a clinical practice setting. Pharmacogenomics 2014, 15, 1739–1747. [Google Scholar] [CrossRef]
- Gufford, B.T.; Chen, G.; Vergara, A.G.; Lazarus, P.; Oberlies, N.H.; Paine, M.F. Milk Thistle Constituents Inhibit Raloxifene Intestinal Glucuronidation: A Potential Clinically Relevant Natural Product-Drug Interaction. Drug Metab. Dispos. 2015, 43, 1353–1359. [Google Scholar] [CrossRef]
- Lee, C.K.; Choi, J.S. Effects of silibinin, inhibitor of CYP3A4 and P-glycoprotein in vitro, on the pharmacokinetics of paclitaxel after oral and intravenous administration in rats. Pharmacology 2010, 85, 350–356. [Google Scholar] [CrossRef]
- Sgorlon, S.; Stefanon, B.; Sandri, M.; Colitti, M. Nutrigenomic activity of plant derived compounds in health and disease: Results of a dietary intervention study in dog. Res. Vet. Sci. 2016, 109, 142–148. [Google Scholar] [CrossRef]
- Holtzman, C.W.; Wiggins, B.S.; Spinler, S.A. Role of P-glycoprotein in statin drug interactions. Pharmacotherapy 2006, 26, 1601–1607. [Google Scholar] [CrossRef]
- Ishigami, M.; Kawabata, K.; Takasaki, W.; Ikeda, T.; Komai, T.; Ito, K.; Sugiyama, Y. Drug interaction between simvastatin and itraconazole in male and female rats. Drug Metab. Dispos. 2001, 29, 1068–1072. [Google Scholar]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisited. FASEB J. 2008, 22, 659–661. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds used in this study are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Setting | SV | LV | SVA | LVA |
|---|---|---|---|---|
| Quantifier transition | 441.4→325.2 | 427.4→325.2 | 435.4→319.2 | 421.3→319.2 |
| Qualifier transition | 441.4→295.3 | 427.4→295.3 | 435.4→115.2 | 421.3→101.1 |
| Declustering potential (DP), V | 130 | 130 | −100 | −100 |
| Collision energy (CE), V | 32 | 30 | −25 | −25 |
| Collision cell exit potential (CXP), V | 10 | 10 | −10 | −10 |
| Entrance potential (EP), V | 14 | 14 | −17 | −17 |
| Analytes | Spiked Con. (ng/mL) | Intra-Day (n = 5) | Inter-Day (n = 15) | ||||
|---|---|---|---|---|---|---|---|
| Measured con. (ng/mL) | Precision (RSD %) | Accuracy (RE %) | Measured con. (ng/mL) | Precision (RSD %) | Accuracy (RE %) | ||
| SVA | 5.0 | 5.23 ± 0.36 | 6.9 | 4.6 | 5.11 ± 0.32 | 6.3 | 2.2 |
| 10.0 | 9.72 ± 0.38 | 3.9 | −2.9 | 9.71 ± 0.38 | 4.0 | −2.9 | |
| 100.0 | 99.8 ± 4.13 | 4.1 | −0.2 | 98.79 ± 3.42 | 3.5 | −1.2 | |
| 1600.0 | 1644 ± 70.77 | 4.3 | 2.8 | 1642 ± 75.42 | 4.6 | 2.6 | |
| 0.5 | 0.52 ± 0.05 | 9.3 | 3.2 | 0.508 ± 0.04 | 8.2 | 1.7 | |
| SV | 1.0 | 1.00 ± 0.04 | 3.5 | −0.9 | 0.992 ± 0.03 | 3.1 | −0.8 |
| 10.0 | 9.84 ± 0.55 | 5.6 | −1.6 | 9.83 ± 0.41 | 4.1 | −1.7 | |
| 160 | 157.2 ± 6.83 | 4.4 | −1.7 | 155.2 ± 6.62 | 4.3 | −3.0 | |
| Analytes | Spiked con. (ng/mL) | Extraction Recovery (%) | Matrix Effect (%) |
|---|---|---|---|
| SVA | 10 | 78.4 ± 3.9 | 92.9 ± 3.0 |
| 100 | 81.6 ± 3.9 | 93.9 ± 5.9 | |
| 1600 | 80.1 ± 2.7 | 97.7 ± 4.8 | |
| SV | 1.0 | 76.7 ± 4.4 | 92.6 ± 4.8 |
| 10 | 82.3 ± 3.2 | 97.8 ± 1.6 | |
| 160 | 81.5 ± 2.1 | 93.3 ± 3.0 | |
| LVA | 100 | 81.3 ± 3.7 | 95.5 ± 1.8 |
| LV | 10 | 79.5 ± 4.2 | 97.9 ± 2.2 |
| Analytes | Spiked con. (ng/mL) | Dilution Factor | Measured con. (ng/mL) | Precision (RSD %) | Accuracy (RE %) |
|---|---|---|---|---|---|
| SVA | 10,000 | 10 | 9491.8 ± 451.1 | 4.8 | −5.1 |
| 10,000 | 20 | 9276.2 ± 484.5 | 5.2 | −7.2 | |
| SV | 1000 | 10 | 947.0 ± 44.4 | 4.7 | −5.3 |
| 1000 | 20 | 956.5 ± 57.4 | 6.0 | −4.3 |
| Analytes | Storage Conditions | Spiked con. (ng/mL) | Measured con. (ng/mL) | Precision (RSD %) | Accuracy (RE %) |
|---|---|---|---|---|---|
| SVA | Short-term stability (25 °C, 2 h) | 10 | 9.83 ± 0.64 | 6.5 | −1.7 |
| 1600 | 1629 ± 60.22 | 3.7 | 1.9 | ||
| Long-term stability (−80 °C, 7 days) | 10 | 9.91 ± 0.63 | 6.3 | −0.9 | |
| 1600 | 1608 ± 33.31 | 2.1 | 0.6 | ||
| Freeze–thaws stability (−80 to 25 °C) | 10 | 9.95 ± 0.40 | 4.0 | −0.5 | |
| 160 | 1623 ± 79.81 | 4.9 | 1.5 | ||
| Post-preparation stability (4 °C, 24 h) | 10 | 9.91 ± 0.50 | 5.1 | −0.9 | |
| 160 | 1663 ± 68.78 | 4.1 | 3.9 | ||
| SV | Short-term stability (25 °C, 2 h) | 1.0 | 0.956 ± 0.05 | 5.4 | −4.4 |
| 160 | 155.9 ± 8.15 | 5.2 | −2.6 | ||
| Long-term stability (−80 °C, 7 days) | 1.0 | 1.008 ± 0.05 | 4.8 | 0.8 | |
| 160 | 159.0 ± 11.10 | 7.0 | −0.6 | ||
| Freeze–thaws stability (−80 °C to 25 °C) | 1.0 | 0.977 ± 0.03 | 3.3 | −2.3 | |
| 160 | 155.5 ± 10.96 | 7.0 | −2.8 | ||
| Post-preparation stability (4 °C, 24 h) | 1.0 | 0.972 ± 0.05 | 5.6 | −2.8 | |
| 160 | 156.2 ± 6.63 | 4.2 | −2.3 |
| Low Dose (20 mg/kg) | Middle Dose (40 mg/kg) | High Dose (80 mg/kg) | ||||
|---|---|---|---|---|---|---|
| PK Parameter (Unit) | with Silymarin | without Silymarin | with Silymarin | without Silymarin | with Silymarin | without Silymarin |
| SV | ||||||
| AUC0–12h (ng*h/mL) | 327.0 ± 66.7 | 332.2 ± 48.5 | 530.3 ± 70.6 | 641.5 ± 117.2 | 1357.4 ± 635.2 | 1409.0 ± 736.6 |
| AUC0-∞ (ng*h/mL) | 332.7 ± 67.0 | 340.8 ± 50.7 | 541.4 ± 72.2 | 658.3 ± 124.2 | 1399.3 ± 632.0 | 1442.6 ± 740.5 |
| AUMC (h2*ng/mL) | 982.5 ± 240.8 | 984.6 ± 224.6 | 1571.2 ± 280.8 | 2061.9 ± 600.9 | 4277.2 ± 1884.4 | 3929.8 ± 2214.5 |
| Cmax (ng/mL) | 101.4 ± 11.8 | 104.4 ± 9.3 | 179.1 ± 10.5 | 203.1 ± 24.9 | 435.7 ± 171.8 | 482.8 ± 168.8 |
| Tmax (h) a | 1.0(0.67,1.12) | 1.0(0.67,1.5) | 1.0(0.92,1.12) | 1.0(0.67,1.5) | 1.0(0.92,1.5) | 1.0(0.92,1.12) |
| t1/2 (h) | 2.0 ± 0.2 | 2.3 ± 0.4 | 2.1 ± 0.2 | 2.1 ± 0.2 | 2.4 ± 0.7 | 2.4 ± 0.4 |
| MRT (h) | 3.0 ± 0.2 | 2.9 ± 0.3 | 3.0 ± 0.2 | 3.2 ± 0.4 | 3.2 ± 0.3 | 2.7 ± 0.4 |
| CL (L/h/kg) | 62.1 ± 11.0 | 59.7 ± 8.1 | 74.6 ± 9.2 | 62.8 ± 14.1 | 66.1 ± 25.5 | 69.8 ± 37.4 |
| λz (1/h) | 0.35 ± 0.03 | 0.31 ± 0.05 | 0.32 ± 0.02 | 0.32 ± 0.02 | 0.30 ± 0.09 | 0.30 ± 0.05 |
| SVA | ||||||
| AUC0–12h (ng*h/mL) | 8559.1 ± 831.1 * | 6701.5 ± 1275.3 | 13,977.7 ± 2522.7 * | 9477.1 ± 1786.7 | 62,658.6 ± 15,666.7 * | 33,376.9 ± 13,560.7 |
| AUC0-∞ (ng*h/mL) | 8797.4 ± 872.2 * | 6933.3 ± 1315.8 | 14,445.3 ± 2600.0 * | 9776.4 ± 1785.3 | 63,887.5 ± 16,153.3 * | 34,015.5 ± 13,875.1 |
| AUMC (h2*ng/mL) | 26,604.5 ± 3472.7 | 21,744.3 ± 5392.1 | 44,543.6 ± 8982.8 * | 27,939.8 ± 5737.6 | 177,142.2 ± 54,541.3 * | 87,850.4 ± 41,089.1 |
| Cmax (ng/mL) | 2795.5 ± 495.0 * | 2255.0 ± 266.0 | 4623.3 ± 690.4 * | 3276.7 ± 573.0 | 21,833.3 ± 5689.9 * | 12,936.7 ± 5313.4 |
| Tmax (h) a | 1.25(0.92,1.5) | 1.0(0.67,1.5) | 1.0(0.67,1.1) | 1.0(0.67,1.5) | 1.0(0.92,1.63) | 1.25(0.92,1.5) |
| t1/2 (h) | 2.2 ± 0.2 | 2.4 ± 0.4 | 2.4 ± 0.3 | 2.5 ± 0.4 | 2.1 ± 0.4 | 2.3 ± 0.3 |
| MRT (h) | 3.1 ± 0.3 | 3.2 ± 0.2 | 3.2 ± 0.2 | 3.0 ± 0.2 | 2.8 ± 0.4 | 2.6 ± 0.2 |
| λz (1/h) | 0.32 ± 0.02 | 0.29 ± 0.05 | 0.29 ± 0.03 | 0.28 ± 0.04 | 0.33 ± 0.07 | 0.3 ± 0.04 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Wu, Y.; Li, Y.-J.; Meng, L.; Ding, C.-Y.; Dong, Z.-J. Effects of Silymarin on the In Vivo Pharmacokinetics of Simvastatin and Its Active Metabolite in Rats. Molecules 2019, 24, 1666. https://doi.org/10.3390/molecules24091666
Li Y, Wu Y, Li Y-J, Meng L, Ding C-Y, Dong Z-J. Effects of Silymarin on the In Vivo Pharmacokinetics of Simvastatin and Its Active Metabolite in Rats. Molecules. 2019; 24(9):1666. https://doi.org/10.3390/molecules24091666
Chicago/Turabian StyleLi, Ying, Yin Wu, Ya-Jing Li, Lu Meng, Cong-Yang Ding, and Zhan-Jun Dong. 2019. "Effects of Silymarin on the In Vivo Pharmacokinetics of Simvastatin and Its Active Metabolite in Rats" Molecules 24, no. 9: 1666. https://doi.org/10.3390/molecules24091666
APA StyleLi, Y., Wu, Y., Li, Y.-J., Meng, L., Ding, C.-Y., & Dong, Z.-J. (2019). Effects of Silymarin on the In Vivo Pharmacokinetics of Simvastatin and Its Active Metabolite in Rats. Molecules, 24(9), 1666. https://doi.org/10.3390/molecules24091666