
Development of Oxygen-Bridged Pyrazole-Based Structures as Cannabinoid Receptor 1 Ligands

 , , ,
, , ,
Abstract
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Biology
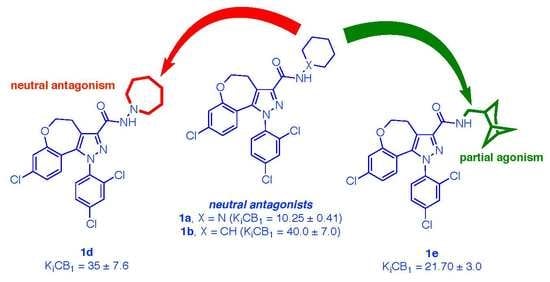
2.2.1. Radioligand Binding Assays
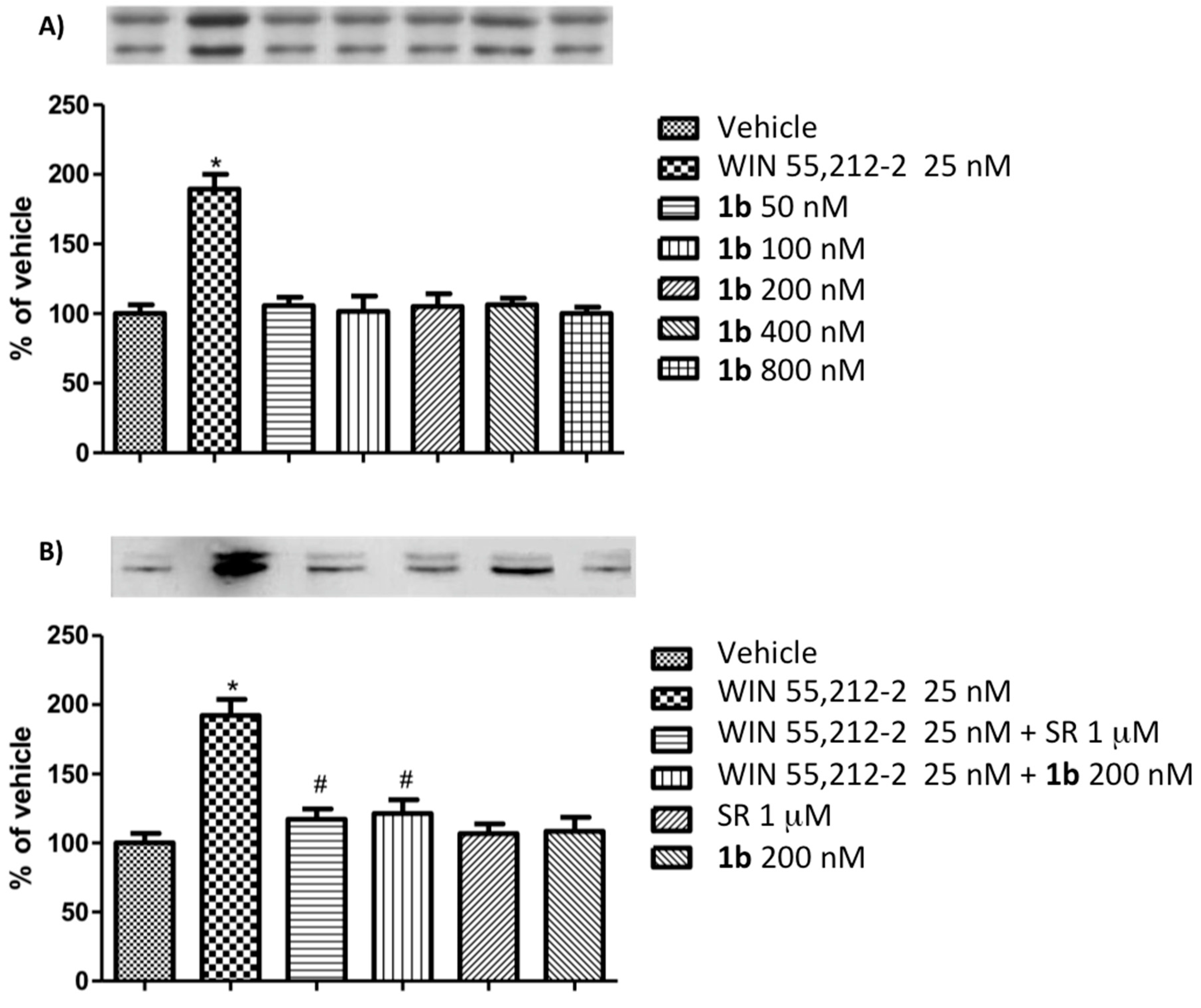
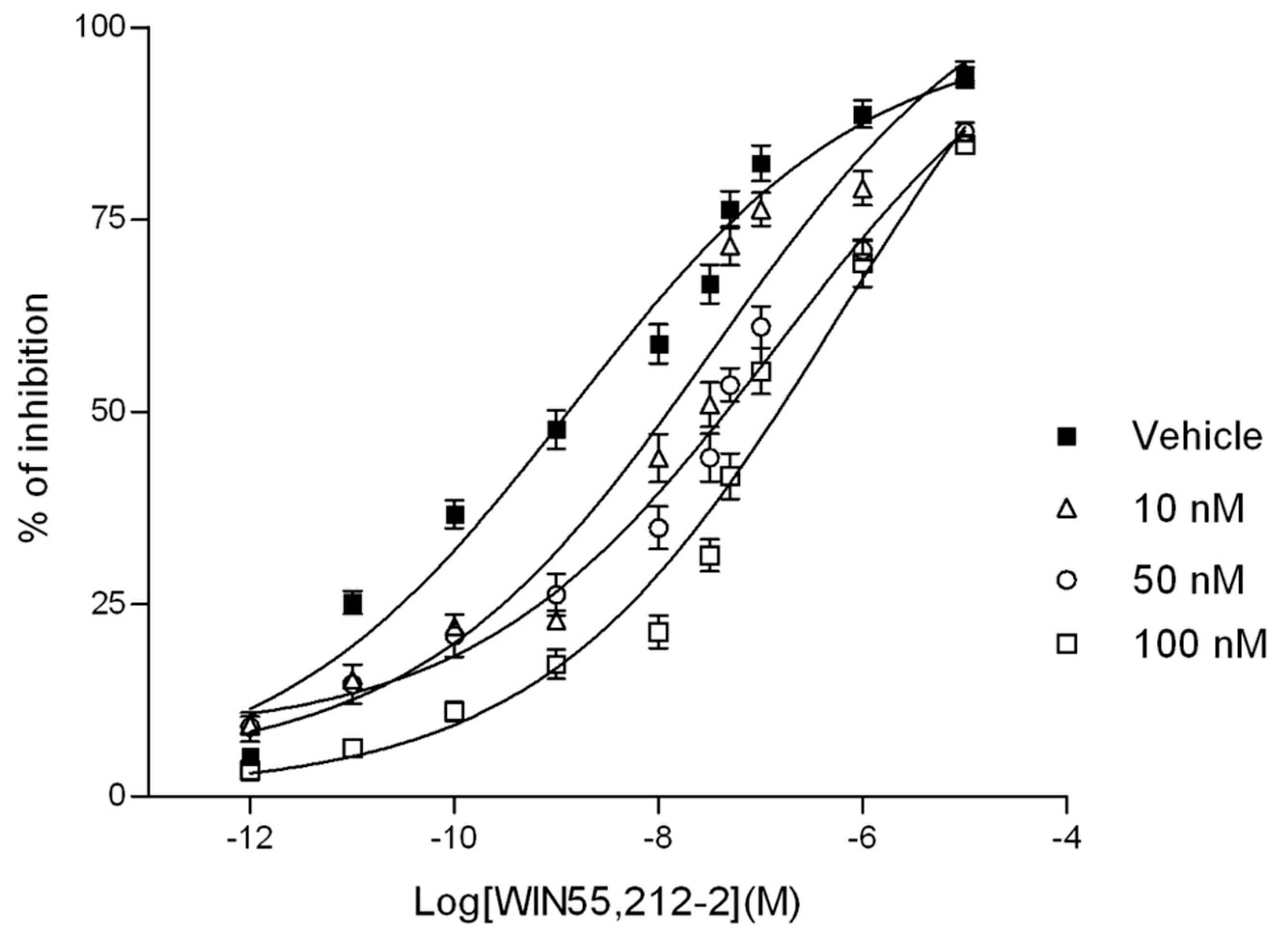
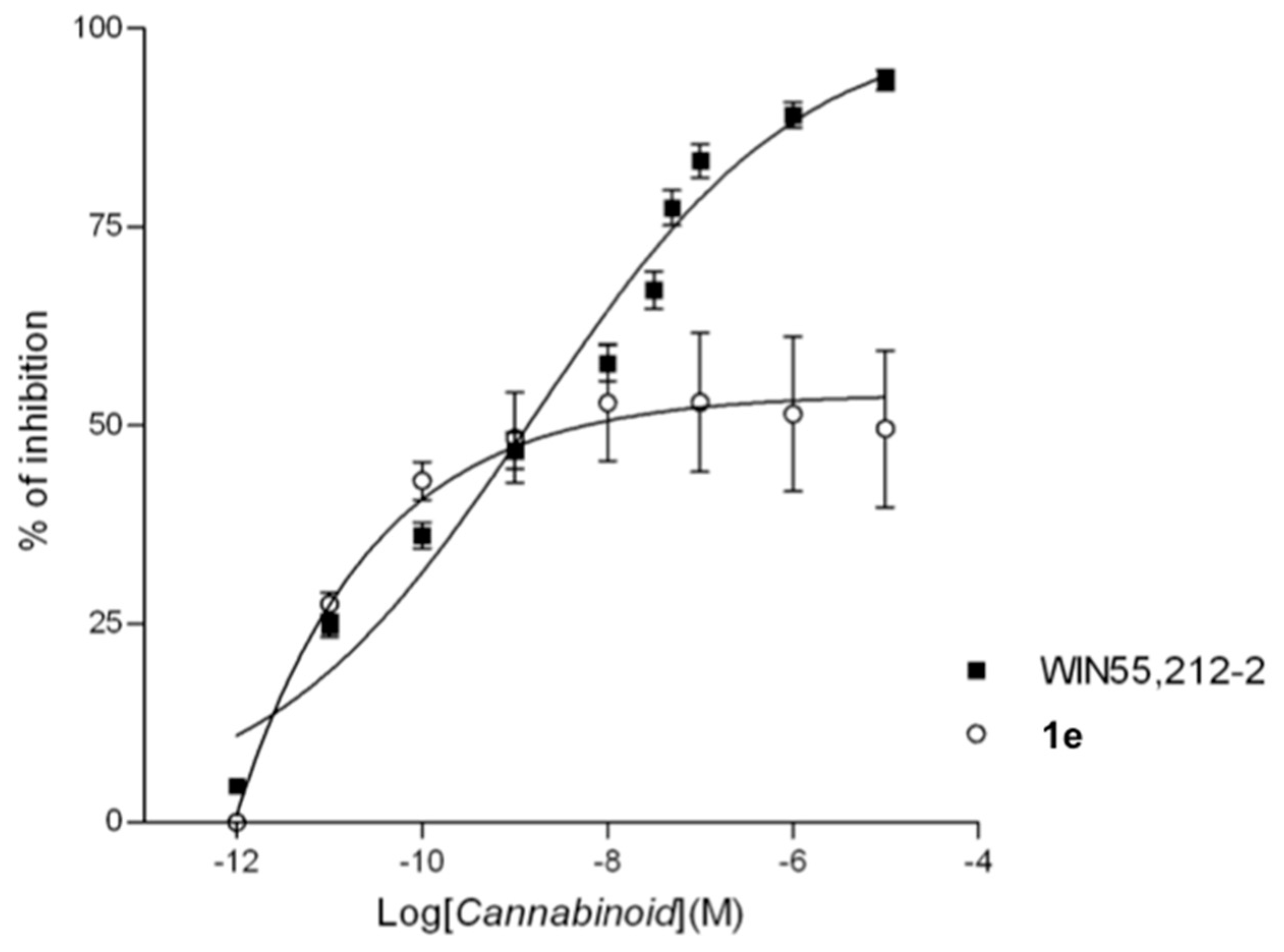
2.2.2. In Vitro Assays
2.2.3. Isolated Organs (Mouse Vas Deferens)
2.2.4. In Vivo Assays: Gastrointestinal Transit (GIT)
2.2.5. In Vivo Assays: Food Intake
2.3. Modelling
3. Experimental Section
3.1. General Procedures
3.1.1. Synthesis of 4-(3-Chlorophenoxy)butyrric Acid (3)
3.1.2. Synthesis of 8-Chloro-1-oxo-2,3,4,5-tetrahydrobenzocycloeptan-5-one (4)
3.1.3. Synthesis of Ethyl 2-(8-Chloro-5-hydroxy-2,3-dihydrobenzo[b]oxepin-4-yl)-2-oxoacetate (5)
3.1.4. Synthesis of Ethyl 8-chloro-1-(2,4-dichlorophenyl)-4,5-dihydrobenzo-1H-6-oxa-cyclohepta[1,2-c]pyrazole-3-carboxylate (6)
3.1.5. Synthesis of 8-Chloro-1-(2,4-dichlorophenyl)-4,5-dihydrobenzo-1H-6-oxa-cyclohepta[1,2-c]pyrazole-3-carboxylic acid (7)
3.1.6. General Procedure for the Synthesis of Carbohydrazides (1c,d) and Carboxamides (1e–j)
3.1.7. 8-Chloro-1-(2,4-dichlorophenyl)-N-pyrrolidin-1-yl-4,5-dihydrobenzo-1H-6-oxa-cyclohepta[1,2-c]pyrazole-3-carbohydrazide (1c) [20]
3.1.8. 8-Chloro-1-(2,4-dichlorophenyl)-N-homopiperidin-1-yl-4,5-dihydrobenzo-1H-6-oxa-cyclohepta[1,2-c]pyrazole-3-carbohydrazide (1d)
3.1.9. 8-Chloro-1-(2,4-dichlorophenyl)-N-(6,6-dimethyl-bicyclo[3.1.1]hept-2-yl-methyl)-4,5-dihydrobenzo-1H-6-oxa-cyclohepta[1,2-c]pyrazole-3-carboxamide (1e)
3.1.10. 8-Chloro-1-(2,4-dichlorophenyl)-N-phenyl-4,5-dihydrobenzo-1H-6-oxa-cyclohepta[1,2-c]pyrazole-3-carboxamide (1f)
3.1.11. 8-Chloro-1-(2,4-dichlorophenyl)-N-(p-chlorophenyl)-4,5-dihydrobenzo-1H-6-oxa-cyclohepta[1,2-c]pyrazole-3-carboxamide (1g)
3.1.12. 8-Chloro-1-(2,4-dichlorophenyl)-N-tolyl-4,5-dihydrobenzo-1H-6-oxa-cyclohepta[1,2-c] pyrazole-3-carboxamide (1h)
3.1.13. 8-Chloro-1-(2,4-dichlorophenyl)-N-(p-methoxyphenyl)-4,5-dihydrobenzo-1H-6-oxa-cyclohepta[1,2-c]pyrazole-3-carboxamide (1i)
3.1.14. 8-Chloro-1-(2,4-dichlorophenyl)-N-(p-trifluoromethylphenyl)-4,5-dihydrobenzo-1H-6-oxa-cyclohepta[1,2-c]pyrazole-3-carbohydrazide (1j)
3.2. Evaluation of Biological Activity
3.2.1. Chemicals and Drugs for in Vitro and in Vivo Assays
3.2.2. Radioligand Binding Methods
3.2.3. In Vitro Assays
3.2.4. Isolated Organs
3.2.5. Gastrointestinal Transit
3.2.6. Food Intake
3.3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Howlett, A.C.; Barth, F.; Bonner, T.I.; Cabral, G.; Casellas, P.; Devane, W.A.; Felder, C.C.; Herkenham, M.; Mackie, K.; Martin, B.R.; et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol. Rev. 2002, 54, 161–202. [Google Scholar] [CrossRef]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Mackie, K. Cannabinoid receptors as therapeutic targets. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 101–122. [Google Scholar] [CrossRef]
- Sloan, M.E.; Gowin, J.L.; Ramchandani, V.A.; Hurd, Y.L.; Le Foll, B. The endocannabinoid system as a target for addiction treatment: Trials and tribulations. Neuropharmacology 2017, 124, 73–83. [Google Scholar] [CrossRef]
- Giannone, F.A.; Baldassarre, M.; Domenicali, M.; Zaccherini, G.; Trevisani, F.; Bernardi, M.; Caraceni, P. Reversal of liver fibrosis by the antagonism of endocannabinoid CB1 receptor in a rat model of CCl(4)-induced advanced cirrhosis. Lab. Investig. 2012, 92, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Succu, S.; Mascia, M.S.; Sanna, F.; Melis, T.; Argiolas, A.; Melis, M.R. The cannabinoid CB1 receptor antagonist SR 141716A induces penile erection by increasing extra-cellular glutamic acid in the paraventricular nucleus of male rats. Behav. Brain Res. 2006, 169, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Jones, D. End of the line for cannabinoid receptor 1 as an anti-obesity target? Nat. Rev. Drug Discov. 2008, 7, 961–962. [Google Scholar] [CrossRef] [PubMed]
- Mastinu, A.; Pira, M.; Pinna, G.A.; Pisu, C.; Casu, M.A.; Reali, R.; Marcello, S.; Murineddu, G.; Lazzari, P. NESS06M reduces body weight with an improved profile relative to SR141716A. Pharmacol. Res. 2013, 74, 94–108. [Google Scholar] [CrossRef]
- Fois, G.R.; Fattore, L.; Murineddu, G.; Salis, A.; Pintore, G.; Asproni, B.; Pinna, G.A.; Diana, M. The novel cannabinoid antagonist SM-11 reduces hedonic aspect of food intake through a dopamine-dependent mechanism. Pharmacol. Res. 2016, 113, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Protiva, M.; Seidlová, V.; Svátek, E.; Hradil, F. Benzocycloheptanes and heterocyclic analogues as potential drugs. II. Amines of 8-chloro-2,3,4,5-tetrahydro-1-benzoxepin series. Collect. Czech. Chem. Commun. 1972, 37, 868–886. [Google Scholar] [CrossRef]
- Murineddu, G.; Ruiu, S.; Mussinu, J.M.; Loriga, G.; Grella, G.E.; Carai, M.A.M.; Lazzari, P.; Pani, L.; Pinna, G.A. Tricyclic pyrazoles. Part 2: Synthesis and biological evaluation of novel 4,5-dihydro-1H-benzo[g]indazole-based ligands for cannabinoid receptors. Bioorg. Med. Chem. 2005, 13, 3309–3320. [Google Scholar] [CrossRef]
- Davis, M.I.; Ronesi, J.; Lovinger, D.M. A Predominant Role for Inhibition of the Adenylate Cyclase/Protein Kinase A Pathway in ERK Activation by Cannabinoid Receptor 1 in N1E-115 Neuroblastoma Cells. J. Biol. Chem. 2003, 278, 48973–48980. [Google Scholar] [CrossRef]
- Pertwee, R.G.; Stevenson, L.A.; Elrick, D.B.; Mechoulam, R.; Corbett, A.D. Inhibitory effects of certain enantiomeric cannabinoids in the mouse vas deferens and the myenteric plexus preparation of guinea-pig small intestine. Br. J. Pharmacol. 1992, 105, 980–984. [Google Scholar] [CrossRef]
- Schlicker, E.; Kathmann, M. Modulation of transmitter release via presynaptic cannabinoid receptors. Trends Pharmacol. Sci. 2001, 22, 565–572. [Google Scholar] [CrossRef]
- Ruiu, S.; Pinna, G.A.; Marchese, G.; Mussinu, J.M.; Saba, P.; Tambaro, S.; Casti, P.; Vargiu, R.; Pani, L. Synthesis and characterization of NESS 0327: A novel putative antagonist of the CB1 cannabinoid receptor. J. Pharmacol. Exp. Ther. 2003, 306, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Murineddu, G.; Ruiu, S.; Loriga, G.; Manca, I.; Lazzari, P.; Reali, R.; Pani, L.; Toma, L.; Pinna, G.A. Tricyclic pyrazoles. 3. Synthesis, biological evaluation, and molecular modeling of analogues of the cannabinoid antagonist 8-chloro-1-(2′,4′-dichlorophenyl)-N-piperidin-1-yl-1,4,5,6-tetrahydrobenzo[6,7]cyclohepta[1,2-c]pyrazole-3-carboxamide. J. Med. Chem. 2005, 48, 7351–7362. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. Cannabinoids and the gastrointestinal tract. Gut 2001, 48, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V.; Goparaju, S.K.; Wang, L.; Liu, J.; Batkai, S.; Jarai, Z.; Fezza, F.; Miura, G.I.; Palmiter, R.D.; Sugiura, T.; et al. Leptin-regulated endocannabinoids are involved in maintaining food intake. Nature 2001, 410, 822–825. [Google Scholar] [CrossRef] [PubMed]
- Lohray, B.B.; Lohray, V.B.; Srivastava, B. Tricyclic Pyrazoles Derivatives as Cannabinoid Receptor Modulators. US Patent 8,030,345 B2, 4 October 2011. [Google Scholar]
- Pertwee, R.G.; Ross, R.A.; Craib, S.J.; Thomas, A. (−)-Cannabidiol antagonizes cannabinoid receptor agonists and noradrenaline in the mouse vas deferens. Eur. J. Pharmacol. 2002, 456, 99–106. [Google Scholar] [CrossRef]
- Greig, I.R.; Ross, R.A.; Pertwee, R.G. 1,5-Diaryl-pyrazoles as Cannabinoid Receptor Neutral Antagonists Useful as Therapeutic Agents. US Patent 0,022,611, 28, January, 2010. [Google Scholar]
- Coutts, A.A.; Brewster, N.; Ingram, T.; Razdan, R.K.; Pertwee, R.G. Comparison of novel cannabinoid partial agonists and SR14176A in the guinea-pig small intestine. Br. J. Pharmacol. 2000, 129, 645–652. [Google Scholar] [CrossRef]
- Lazzari, P.; Sanna, A.; Mastinu, A.; Cabasino, S.; Manca, I.; Pani, L. Weight loss induced by rimonabant is associated with altered leptin expression and hypothalamic leptin signalling in diet induced obese mice. Behav. Brain Res. 2011, 217, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Tallett, A.J.; Blundell, J.E.; Rodgers, R.J. Grooming, scratching and feeding: Role of response competition in acute anorectic response to rimonabant in male rats. Psychopharmacology 2007, 195, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Tallett, A.J.; Blundell, J.E.; Rodgers, R.J. Effects of acute low-dose combined treatment with rimonabant and sibutramine on appetite and weight gain in rats. Pharmacol. Biochem. Behav. 2010, 97, 92–100. [Google Scholar] [CrossRef]
- Gary-Bobo, M.; Elachouri, G.; Gallas, J.F.; Janiak, P.; Marini, P.; Ravinet-Trillou, C.; Chabbert, M.; Cruccioli, N.; Pfersdorff, C.; Roque, C.; et al. Rimonabant reduces obesity-associated hepatic steatosis and features of metabolic syndrome in obese zucker fa/fa rats. Hepatology 2007, 46, 122–129. [Google Scholar] [CrossRef]
- Maynadier, M.; Basile, I.; Gary-Bobo, M. Adiponectin normalization: A clue to the anti-metabolic syndrome action of Rimonabant. Drug Discov. Today 2009, 14, 192–197. [Google Scholar] [CrossRef]
- Després, J.P.; Ross, R.; Boka, G.; Alméras, N.; Lemieux, I. ADAGIO-lipids investigators. Effect of rimonabant on the high-triglyceride/low-HDL-cholesterol dyslipidemia, intraabdominal adiposity, and liver fat: The ADAGIO-Lipids trial. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Hua, T.; Vemuri, K.; Pu, M.; Qu, L.; Won Han, G.; Wu, Y.; Zhao, S.; Shui, W.; Li, S.; Korde, A.; et al. Crystal Structure of the Human Cannabinoid Receptor CB1. Cell 2016, 167, 750–762.e14. [Google Scholar] [CrossRef]
- Shao, Z.; Yin, J.; Chapman, K.; Grzemska, M.; Clark, L.; Wang, J.; Rosenbaum, D.M. High-resolution crystal structure of the human CB1 cannabinoid receptor. Nature 2016, 540, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitaion of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50% inhibition (IC50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar]
- Murineddu, G.; Lazzari, P.; Ruiu, S.; Sanna, A.; Loriga, G.; Manca, I.; Falzoi, M.; Dessì, C.; Curzu, M.M.; Chelucci, G.; et al. Tricyclic Pyrazoles. 4. Synthesis and biological evaluation of analogues of the robust and selective CB2 cannabinoid ligand 1-(2′,4′-dichlorophenyl)-6-methyl-N-piperidin-1-yl-1,4-dihydroindeno[1,2-c]pyrazole-3-carboxamide. J. Med. Chem. 2006, 49, 7502–7512. [Google Scholar] [CrossRef] [PubMed]
- Arunlakshana, O.; Schild, H.O. Some quantitative uses of drug antagonists. Br. J. Pharmacol. 1959, 14, 48–58. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision, B.01; Gaussian Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
Sample Availability: Samples of the compounds are available from the authors. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Compound 1 | R | Receptor Affinity | Selectivity | |
| KiCB1 (nM) a | KiCB2(nM) b | KiCB2/KiCB1 | ||
| a [9] |  | 10.25 ± 0.41 | >5000 | - |
| b [10] |  | 40.0 ± 7.0 | 3725 ± 25 | 93:1 |
| c |  | 55.10 ± 5.0 | 4500 ± 500 | 82:1 |
| d |  | 35.0 ± 7.6 | 2000 ± 50 | 57:1 |
| e |  | 21.70 ± 3.0 | 325 ± 38 | 15:1 |
| f |  | 233.0 ± 33.0 | 4125 ± 620 | 18:1 |
| g |  | 318.0 ± 36.0 | >5000 | - |
| h |  | 429.36 ± 49.0 | >5000 | - |
| i |  | 330.0 ± 45.0 | >5000 | - |
| j |  | 459.0 ± 57.0 | >5000 | - |
| SR141716A | 1.80 ± 0.075 | 514 ± 30 | 285:1 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murineddu, G.; Asproni, B.; Corona, P.; Piras, S.; Lazzari, P.; Ruiu, S.; Legnani, L.; Toma, L.; Pinna, G.A. Development of Oxygen-Bridged Pyrazole-Based Structures as Cannabinoid Receptor 1 Ligands. Molecules 2019, 24, 1656. https://doi.org/10.3390/molecules24091656
Murineddu G, Asproni B, Corona P, Piras S, Lazzari P, Ruiu S, Legnani L, Toma L, Pinna GA. Development of Oxygen-Bridged Pyrazole-Based Structures as Cannabinoid Receptor 1 Ligands. Molecules. 2019; 24(9):1656. https://doi.org/10.3390/molecules24091656
Chicago/Turabian StyleMurineddu, Gabriele, Battistina Asproni, Paola Corona, Sandra Piras, Paolo Lazzari, Stefania Ruiu, Laura Legnani, Lucio Toma, and Gérard A. Pinna. 2019. "Development of Oxygen-Bridged Pyrazole-Based Structures as Cannabinoid Receptor 1 Ligands" Molecules 24, no. 9: 1656. https://doi.org/10.3390/molecules24091656
APA StyleMurineddu, G., Asproni, B., Corona, P., Piras, S., Lazzari, P., Ruiu, S., Legnani, L., Toma, L., & Pinna, G. A. (2019). Development of Oxygen-Bridged Pyrazole-Based Structures as Cannabinoid Receptor 1 Ligands. Molecules, 24(9), 1656. https://doi.org/10.3390/molecules24091656