
Soluble Heparin and Heparan Sulfate Glycosaminoglycans Interfere with Sonic Hedgehog Solubilization and Receptor Binding


Abstract
:1. Introduction
2. Results
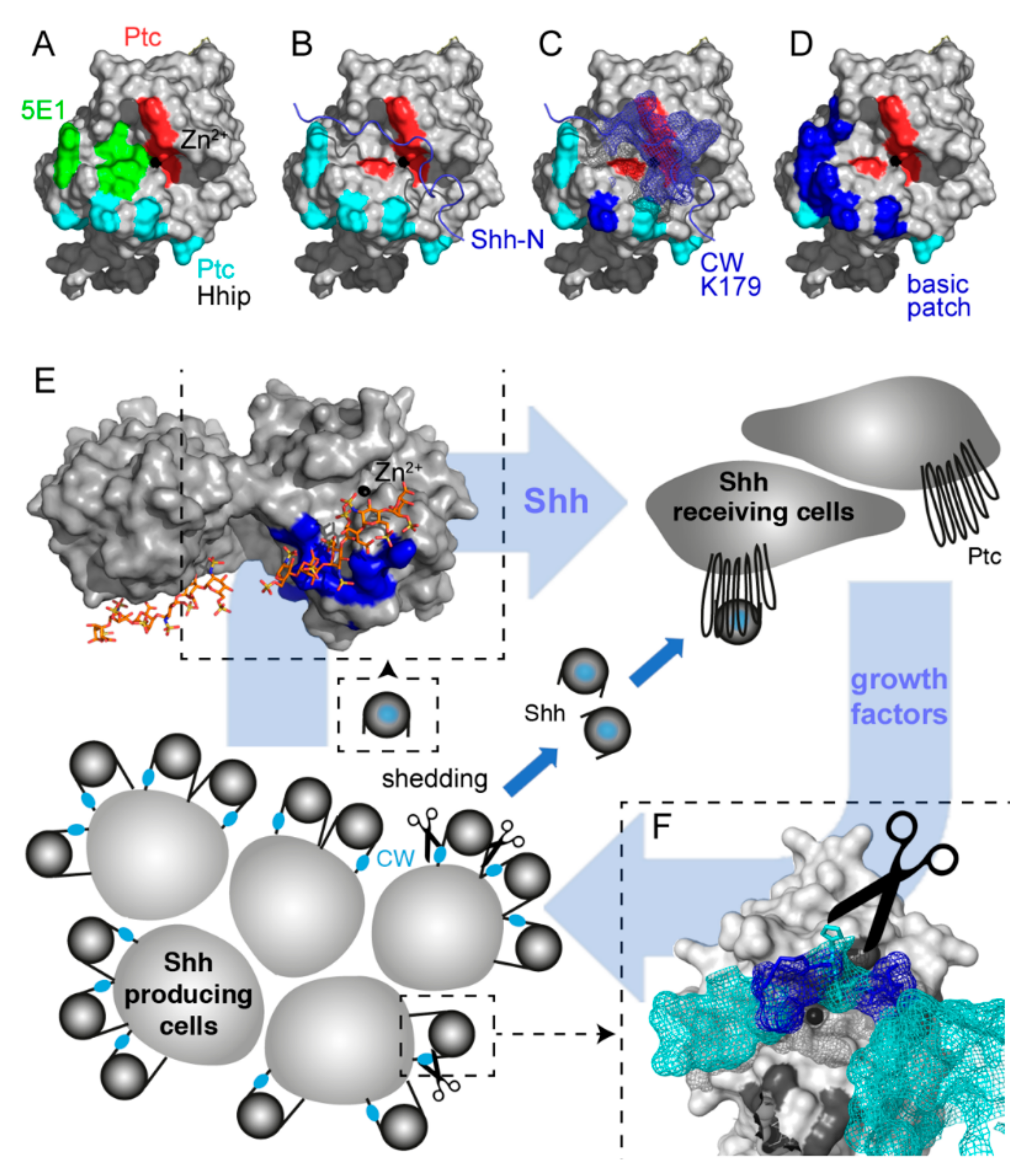
2.1. A “Hotspot” for Hh Binding to Ptc and Hh Activity Regulators
2.2. Proteolytic CW Processing Releases Shh In Vitro
2.3. Proteolytic CW Cleavage is Essential for Hh Biofunction In Vivo
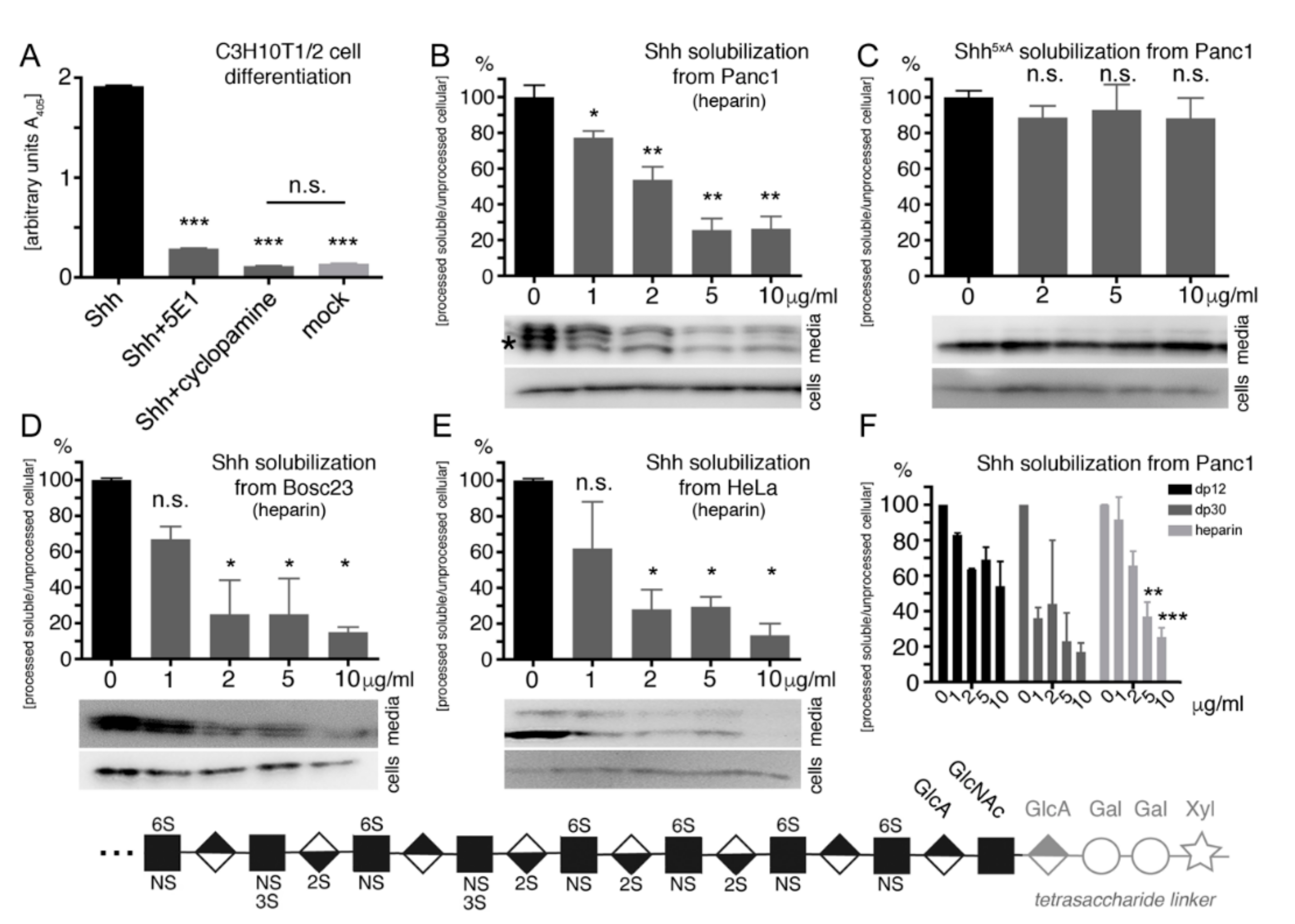
2.4. Heparin Competes with Proteolytic Shh Processing and Release from Cancer Cells
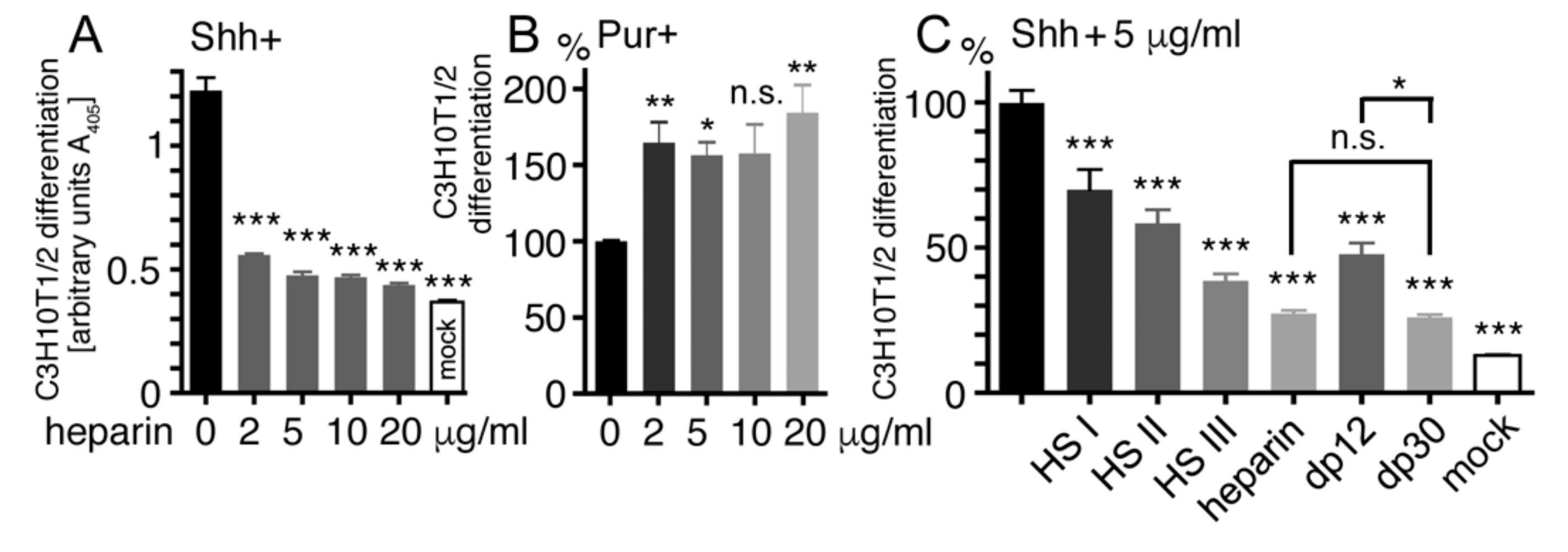
2.5. Heparin Competes with Ptc for Shh Binding
2.6. Oligosaccharide Structure Determination for Shh Binding
3. Discussion
4. Materials and Methods
4.1. Heparin and HS
4.2. Cloning and Expression of Recombinant Proteins
4.3. Cell Culture and Protein Analysis
4.4. Shh Reporter Assays
4.5. Reverse Transcription Polymerase Chain Reaction (RT-PCR)
4.6. Fly Lines
4.7. Statistical Analysis
4.8. Bioinformatics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ahn, S.; Joyner, A.L. Dynamic changes in the response of cells to positive hedgehog signaling during mouse limb patterning. Cell 2004, 118, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Briscoe, J.; Chen, Y.; Jessell, T.M.; Struhl, G. A hedgehog-insensitive form of patched provides evidence for direct long-range morphogen activity of sonic hedgehog in the neural tube. Mol. Cell 2001, 7, 1279–1291. [Google Scholar] [CrossRef]
- Charron, F.; Stein, E.; Jeong, J.; McMahon, A.P.; Tessier-Lavigne, M. The morphogen sonic hedgehog is an axonal chemoattractant that collaborates with netrin-1 in midline axon guidance. Cell 2003, 113, 11–23. [Google Scholar] [CrossRef]
- Harfe, B.D.; Scherz, P.J.; Nissim, S.; Tian, H.; McMahon, A.P.; Tabin, C.J. Evidence for an expansion-based temporal Shh gradient in specifying vertebrate digit identities. Cell 2004, 118, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.; Wolf, A.; Wagner, M.; Kuhlmann, J.; Waldmann, H. The cholesterol membrane anchor of the Hedgehog protein confers stable membrane association to lipid-modified proteins. Proc. Natl. Acad. Sci. USA 2004, 101, 8531–8536. [Google Scholar] [CrossRef]
- Porter, J.A.; Young, K.E.; Beachy, P.A. Cholesterol modification of hedgehog signaling proteins in animal development. Science 1996, 274, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Pepinsky, R.B.; Zeng, C.; Wen, D.; Rayhorn, P.; Baker, D.P.; Williams, K.P.; Bixler, S.A.; Ambrose, C.M.; Garber, E.A.; Miatkowski, K.; et al. Identification of a palmitic acid-modified form of human Sonic hedgehog. J. Biol. Chem. 1998, 273, 14037–14045. [Google Scholar] [CrossRef]
- Amanai, K.; Jiang, J. Distinct roles of Central missing and Dispatched in sending the Hedgehog signal. Development 2001, 128, 5119–5127. [Google Scholar]
- Chamoun, Z.; Mann, R.K.; Nellen, D.; von Kessler, D.P.; Bellotto, M.; Beachy, P.A.; Basler, K. Skinny hedgehog, an acyltransferase required for palmitoylation and activity of the hedgehog signal. Science 2001, 293, 2080–2084. [Google Scholar] [CrossRef]
- Lee, J.D.; Treisman, J.E. Sightless has homology to transmembrane acyltransferases and is required to generate active Hedgehog protein. Curr. Biol. 2001, 11, 1147–1152. [Google Scholar] [CrossRef]
- Micchelli, C.A.; The, I.; Selva, E.; Mogila, V.; Perrimon, N. Rasp, a putative transmembrane acyltransferase, is required for Hedgehog signaling. Development 2002, 129, 843–851. [Google Scholar] [PubMed]
- Jakobs, P.; Exner, S.; Schurmann, S.; Pickhinke, U.; Bandari, S.; Ortmann, C.; Kupich, S.; Schulz, P.; Hansen, U.; Seidler, D.G.; et al. Scube2 enhances proteolytic Shh processing from the surface of Shh-producing cells. J. Cell Sci. 2014, 127, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Damhofer, H.; Veenstra, V.L.; Tol, J.A.; van Laarhoven, H.W.; Medema, J.P.; Bijlsma, M.F. Blocking Hedgehog release from pancreatic cancer cells increases paracrine signaling potency. J. Cell Sci. 2015, 128, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Dierker, T.; Dreier, R.; Petersen, A.; Bordych, C.; Grobe, K. Heparan Sulfate-modulated, Metalloprotease-mediated Sonic Hedgehog Release from Producing Cells. J. Biol. Chem. 2009, 284, 8013–8022. [Google Scholar] [CrossRef] [PubMed]
- Ohlig, S.; Farshi, P.; Pickhinke, U.; van den Boom, J.; Hoing, S.; Jakuschev, S.; Hoffmann, D.; Dreier, R.; Scholer, H.R.; Dierker, T.; et al. Grobe, K. Sonic hedgehog shedding results in functional activation of the solubilized protein. Dev. Cell 2011, 20, 764–774. [Google Scholar] [CrossRef]
- Edwards, D.R.; Handsley, M.M.; Pennington, C.J. The ADAM metalloproteinases. Mol. Asp. Med. 2008, 29, 258–289. [Google Scholar] [CrossRef]
- Schurmann, S.; Steffes, G.; Manikowski, D.; Kastl, P.; Malkus, U.; Bandari, S.; Ohlig, S.; Ortmann, C.; Rebollido-Rios, R.; Otto, M.; et al. Proteolytic processing of palmitoylated Hedgehog peptides specifies the 3-4 intervein region of the Drosophila wing. ELife 2018, 7, e33033. [Google Scholar] [CrossRef] [PubMed]
- Kastl, P.; Manikowski, D.; Steffes, G.; Schurmann, S.; Bandari, S.; Klambt, C.; Grobe, K. Disrupting Hedgehog Cardin-Weintraub sequence and positioning changes cellular differentiation and compartmentalization in vivo. Development 2018, 145. [Google Scholar] [CrossRef]
- Cardin, A.D.; Weintraub, H.J. Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis 1989, 9, 21–32. [Google Scholar] [CrossRef]
- Ohlig, S.; Pickhinke, U.; Sirko, S.; Bandari, S.; Hoffmann, D.; Dreier, R.; Farshi, P.; Gotz, M.; Grobe, K. An emerging role of sonic hedgehog shedding as a modulator of heparan sulfate interactions. J. Biol. Chem. 2012, 287, 43708–43719. [Google Scholar] [CrossRef]
- Ortmann, C.; Pickhinke, U.; Exner, S.; Ohlig, S.; Lawrence, R.; Jboor, H.; Dreier, R.; Grobe, K. Sonic hedgehog processing and release are regulated by glypican heparan sulfate proteoglycans. J. Cell Sci. 2015, 128, 2374–2385. [Google Scholar] [CrossRef]
- Rubin, J.B.; Choi, Y.; Segal, R.A. Cerebellar proteoglycans regulate sonic hedgehog responses during development. Development 2002, 129, 2223–2232. [Google Scholar]
- Chang, S.C.; Mulloy, B.; Magee, A.I.; Couchman, J.R. Two distinct sites in sonic hedgehog combine for heparan sulfate interactions and cell signaling functions. J. Biol. Chem. 2011, 286, 44391–44402. [Google Scholar] [CrossRef]
- Whalen, D.M.; Malinauskas, T.; Gilbert, R.J.; Siebold, C. Structural insights into proteoglycan-shaped Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 16420–16425. [Google Scholar] [CrossRef]
- Sarrazin, S.; Lamanna, W.C.; Esko, J.D. Heparan sulfate proteoglycans. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef]
- Gallagher, J.T. Heparan sulfate: Growth control with a restricted sequence menu. J. Clin. Investig. 2001, 108, 357–361. [Google Scholar] [CrossRef]
- Park, P.W.; Reizes, O.; Bernfield, M. Cell surface heparan sulfate proteoglycans: Selective regulators of ligand-receptor encounters. J. Biol. Chem. 2000, 275, 29923–29926. [Google Scholar] [CrossRef] [PubMed]
- Tumova, S.; Woods, A.; Couchman, J.R. Heparan sulfate proteoglycans on the cell surface: Versatile coordinators of cellular functions. Int. J. Biochem. Cell Biol. 2000, 32, 269–288. [Google Scholar] [CrossRef]
- Toida, T.; Yoshida, H.; Toyoda, H.; Koshishi, I.; Imanari, T.; Hileman, R.E.; Fromm, J.R.; Linhardt, R.J. Structural differences and the presence of unsubstituted amino groups in heparan sulphates from different tissues and species. Biochem. J. 1997, 322, 499–506. [Google Scholar] [CrossRef]
- Lindahl, U.; Kusche-Gullberg, M.; Kjellén, L. Regulated diversity of heparan sulfate. J. Biol. Chem. 1998, 273, 24979–24982. [Google Scholar] [CrossRef]
- Shriver, Z.; Capila, I.; Venkataraman, G.; Sasisekharan, R. Heparin and heparan sulfate: Analyzing structure and microheterogeneity. Handb. Exp. Pharmacol. 2012, 207, 159–176. [Google Scholar]
- Timar, J.; Lapis, K.; Dudas, J.; Sebestyen, A.; Kopper, L.; Kovalszky, I. Proteoglycans and tumor progression: Janus-faced molecules with contradictory functions in cancer. Semin. Cancer Biol. 2002, 12, 173–186. [Google Scholar] [CrossRef]
- Sasisekharan, R.; Shriver, Z.; Venkataraman, G.; Narayanasami, U. Roles of heparan-sulphate glycosaminoglycans in cancer. Nat. Rev. Cancer 2002, 2, 521–528. [Google Scholar] [CrossRef]
- Theunissen, J.W.; de Sauvage, F.J. Paracrine Hedgehog signaling in cancer. Cancer Res. 2009, 69, 6007–6010. [Google Scholar] [CrossRef]
- Yauch, R.L.; Gould, S.E.; Scales, S.J.; Tang, T.; Tian, H.; Ahn, C.P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D.; et al. A paracrine requirement for hedgehog signalling in cancer. Nature 2008, 455, 406–410. [Google Scholar] [CrossRef]
- Bailey, J.M.; Mohr, A.M.; Hollingsworth, M.A. Sonic hedgehog paracrine signaling regulates metastasis and lymphangiogenesis in pancreatic cancer. Oncogene 2009, 28, 3513–3525. [Google Scholar] [CrossRef]
- Dierks, C.; Beigi, R.; Guo, G.R.; Zirlik, K.; Stegert, M.R.; Manley, P.; Trussell, C.; Schmitt-Graeff, A.; Landwerlin, K.; Veelken, H.; et al. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell 2008, 14, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Berman, D.M.; Karhadkar, S.S.; Maitra, A.; Montes De Oca, R.; Gerstenblith, M.R.; Briggs, K.; Parker, A.R.; Shimada, Y.; Eshleman, J.R.; Watkins, D.N.; et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003, 425, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Chen, A.; Jamieson, C.H.; Fereshteh, M.; Abrahamsson, A.; Blum, J.; Kwon, H.Y.; Kim, J.; Chute, J.P.; Rizzieri, D.; et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 2009, 458, 776–779. [Google Scholar] [CrossRef]
- Iovine, V.; Mori, M.; Calcaterra, A.; Berardozzi, S.; Botta, B. One Hundred Faces of Cyclopamine. Curr. Pharm. Des. 2016, 22, 1658–1681. [Google Scholar] [CrossRef]
- Reifenberger, J.; Wolter, M.; Knobbe, C.B.; Kohler, B.; Schonicke, A.; Scharwachter, C.; Kumar, K.; Blaschke, B.; Ruzicka, T.; Reifenberger, G. Somatic mutations in the PTCH, SMOH, SUFUH and TP53 genes in sporadic basal cell carcinomas. Br. J. Dermatol. 2005, 152, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Watkins, D.N.; Berman, D.M.; Burkholder, S.G.; Wang, B.; Beachy, P.A.; Baylin, S.B. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature 2003, 422, 313–317. [Google Scholar] [CrossRef]
- Gulino, A.; Ferretti, E.; De Smaele, E. Hedgehog signalling in colon cancer and stem cells. EMBO Mol. Med. 2009, 1, 300–302. [Google Scholar] [CrossRef]
- Sanchez, P.; Hernandez, A.M.; Stecca, B.; Kahler, A.J.; DeGueme, A.M.; Barrett, A.; Beyna, M.; Datta, M.W.; Datta, S.; Ruiz i Altaba, A. Inhibition of prostate cancer proliferation by interference with SONIC HEDGEHOG-GLI1 signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 12561–12566. [Google Scholar] [CrossRef] [PubMed]
- Pak, E.; Segal, R.A. Hedgehog Signal Transduction: Key Players, Oncogenic Drivers, and Cancer Therapy. Dev. Cell 2016, 38, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Chahal, K.K.; Parle, M.; Abagyan, R. Hedgehog pathway and smoothened inhibitors in cancer therapies. Anticancer Drugs 2018, 29, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Sheng, T.; Li, C.; Zhang, X.; Chi, S.; He, N.; Chen, K.; McCormick, F.; Gatalica, Z.; Xie, J. Activation of the hedgehog pathway in advanced prostate cancer. Mol. Cancer 2004, 3, 29. [Google Scholar] [CrossRef]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef]
- Wu, F.; Zhang, Y.; Sun, B.; McMahon, A.P.; Wang, Y. Hedgehog Signaling: From Basic Biology to Cancer Therapy. Cell Chem. Biol. 2017, 24, 252–280. [Google Scholar] [CrossRef]
- Hall, T.M.; Porter, J.A.; Beachy, P.A.; Leahy, D.J. A potential catalytic site revealed by the 1.7-A crystal structure of the amino-terminal signalling domain of Sonic hedgehog. Nature 1995, 378, 212–216. [Google Scholar] [CrossRef]
- Rebollido-Rios, R.; Bandari, S.; Wilms, C.; Jakuschev, S.; Vortkamp, A.; Grobe, K.; Hoffmann, D. Signaling domain of Sonic Hedgehog as cannibalistic calcium-regulated zinc-peptidase. PLoS Comput. Biol. 2014, 10, e1003707. [Google Scholar] [CrossRef]
- Gong, X.; Qian, H.; Cao, P.; Zhao, X.; Zhou, Q.; Lei, J.; Yan, N. Structural basis for the recognition of Sonic Hedgehog by human Patched1. Science 2018, 361. [Google Scholar] [CrossRef]
- Bosanac, I.; Maun, H.R.; Scales, S.J.; Wen, X.; Lingel, A.; Bazan, J.F.; de Sauvage, F.J.; Hymowitz, S.G.; Lazarus, R.A. The structure of SHH in complex with HHIP reveals a recognition role for the Shh pseudo active site in signaling. Nat. Struct. Mol. Biol. 2009, 16, 691–697. [Google Scholar] [CrossRef]
- Bishop, B.; Aricescu, A.R.; Harlos, K.; O’Callaghan, C.A.; Jones, E.Y.; Siebold, C. Structural insights into hedgehog ligand sequestration by the human hedgehog-interacting protein HHIP. Nat. Struct. Mol. Biol. 2009, 16, 698–703. [Google Scholar] [CrossRef]
- Maun, H.R.; Wen, X.; Lingel, A.; de Sauvage, F.J.; Lazarus, R.A.; Scales, S.J.; Hymowitz, S.G. The hedgehog pathway antagonist 5E1 binds hedgehog at the pseudo-active site. J. Biol. Chem. 2010, 285, 26570–26580. [Google Scholar] [CrossRef]
- Pepinsky, R.B.; Rayhorn, P.; Day, E.S.; Dergay, A.; Williams, K.P.; Galdes, A.; Taylor, F.R.; Boriack-Sjodin, P.A.; Garber, E.A. Mapping sonic hedgehog-receptor interactions by steric interference. J. Biol. Chem. 2000, 275, 10995–11001. [Google Scholar] [CrossRef]
- Tian, H.; Callahan, C.A.; DuPree, K.J.; Darbonne, W.C.; Ahn, C.P.; Scales, S.J.; de Sauvage, F.J. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 4254–4259. [Google Scholar] [CrossRef]
- Curran, T.; Ng, J.M. Cancer: Hedgehog’s other great trick. Nature 2008, 455, 293–294. [Google Scholar] [CrossRef]
- Jakobs, P.; Schulz, P.; Ortmann, C.; Schurmann, S.; Exner, S.; Rebollido-Rios, R.; Dreier, R.; Seidler, D.G.; Grobe, K. Bridging the gap: Heparan sulfate and Scube2 assemble Sonic hedgehog release complexes at the surface of producing cells. Sci. Rep. 2016, 6, 26435. [Google Scholar] [CrossRef] [PubMed]
- Jakobs, P.; Schulz, P.; Schurmann, S.; Niland, S.; Exner, S.; Rebollido-Rios, R.; Manikowski, D.; Hoffmann, D.; Seidler, D.G.; Grobe, K. Ca(2+) coordination controls sonic hedgehog structure and its Scube2-regulated release. J. Cell Sci. 2017, 130, 3261–3271. [Google Scholar] [CrossRef]
- Brand, A.H.; Perrimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 1993, 118, 401–415. [Google Scholar]
- Tabata, T.; Kornberg, T.B. Hedgehog is a signaling protein with a key role in patterning Drosophila imaginal discs. Cell 1994, 76, 89–102. [Google Scholar] [CrossRef]
- Burke, R.; Nellen, D.; Bellotto, M.; Hafen, E.; Senti, K.A.; Dickson, B.J.; Basler, K. Dispatched, a novel sterol-sensing domain protein dedicated to the release of cholesterol-modified hedgehog from signaling cells. Cell 1999, 99, 803–815. [Google Scholar] [CrossRef]
- Caspary, T.; Garcia-Garcia, M.J.; Huangfu, D.; Eggenschwiler, J.T.; Wyler, M.R.; Rakeman, A.S.; Alcorn, H.L.; Anderson, K.V. Mouse Dispatched homolog1 is required for long-range, but not juxtacrine, Hh signaling. Curr. Biol. 2002, 12, 1628–1632. [Google Scholar] [CrossRef]
- Ma, Y.; Erkner, A.; Gong, R.; Yao, S.; Taipale, J.; Basler, K.; Beachy, P.A. Hedgehog-mediated patterning of the mammalian embryo requires transporter-like function of dispatched. Cell 2002, 111, 63–75. [Google Scholar] [CrossRef]
- Nakamura, T.; Aikawa, T.; Iwamoto-Enomoto, M.; Iwamoto, M.; Higuchi, Y.; Pacifici, M.; Kinto, N.; Yamaguchi, A.; Noji, S.; Kurisu, K.; et al. Induction of osteogenic differentiation by hedgehog proteins. Biochem. Biophys. Res. Commun. 1997, 237, 465–469. [Google Scholar] [CrossRef]
- Ericson, J.; Morton, S.; Kawakami, A.; Roelink, H.; Jessell, T.M. Two critical periods of Sonic Hedgehog signaling required for the specification of motor neuron identity. Cell 1996, 87, 661–673. [Google Scholar] [CrossRef]
- Cooper, M.K.; Porter, J.A.; Young, K.E.; Beachy, P.A. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science 1998, 280, 1603–1607. [Google Scholar] [CrossRef]
- Esko, J.D.; Lindahl, U. Molecular diversity of heparan sulfate. J. Clin. Investig. 2001, 108, 169–173. [Google Scholar] [CrossRef]
- Kusche-Gullberg, M.; Nybakken, K.; Perrimon, N.; Lindahl, U. Drosophila heparan sulfate, a novel design. J. Biol. Chem. 2012, 287, 21950–21956. [Google Scholar] [CrossRef]
- Farshi, P.; Ohlig, S.; Pickhinke, U.; Hoing, S.; Jochmann, K.; Lawrence, R.; Dreier, R.; Dierker, T.; Grobe, K. Dual roles of the cardin-weintraub motif in multimeric sonic hedgehog. J. Biol. Chem. 2011, 286, 23608–23619. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Walker, J.; Zhang, J.; Ding, S.; Schultz, P.G. Purmorphamine induces osteogenesis by activation of the hedgehog signaling pathway. Chem. Biol. 2004, 11, 1229–1238. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Hsieh, P.H.; Liu, X.; Zhou, W.; Zhang, X.; Zhao, J.; Xu, Y.; Zhang, F.; Linhardt, R.J.; Liu, J. Construction and characterisation of a heparan sulphate heptasaccharide microarray. Chem. Commun. 2017, 53, 1743–1746. [Google Scholar] [CrossRef] [PubMed]
- Stanton, B.Z.; Peng, L.F.; Maloof, N.; Nakai, K.; Wang, X.; Duffner, J.L.; Taveras, K.M.; Hyman, J.M.; Lee, S.W.; Koehler, A.N.; et al. A small molecule that binds Hedgehog and blocks its signaling in human cells. Nat. Chem. Biol. 2009, 5, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Hitzenberger, M.; Schuster, D.; Hofer, T.S. The Binding Mode of the Sonic Hedgehog Inhibitor Robotnikinin, a Combined Docking and QM/MM MD Study. Front. Chem. 2017, 5, 76. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; McLellan, J.S.; Ayala, A.M.; Leahy, D.J.; Linhardt, R.J. Kinetic and structural studies on interactions between heparin or heparan sulfate and proteins of the hedgehog signaling pathway. Biochemistry 2007, 46, 3933–3941. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.F.; Shriver, Z.; Gi, Y.W.; Venkataraman, G.; Sasisekharan, R. Dynamic regulation of tumor growth and metastasis by heparan sulfate glycosaminoglycans. Semin. Thromb. Hemost. 2002, 28, 67–78. [Google Scholar] [CrossRef]
- Varki, N.M.; Varki, A. Heparin inhibition of selectin-mediated interactions during the hematogenous phase of carcinoma metastasis: Rationale for clinical studies in humans. Semin. Thromb. Hemost. 2002, 28, 53–66. [Google Scholar] [CrossRef]
- Mundhenke, C.; Meyer, K.; Drew, S.; Friedl, A. Heparan sulfate proteoglycans as regulators of fibroblast growth factor-2 receptor binding in breast carcinomas. Am. J. Pathol. 2002, 160, 185–194. [Google Scholar] [CrossRef]
- Lin, Y.C.; Lee, Y.C.; Li, L.H.; Cheng, C.J.; Yang, R.B. Tumor suppressor SCUBE2 inhibits breast-cancer cell migration and invasion through the reversal of epithelial-mesenchymal transition. J. Cell Sci. 2014, 127, 85–100. [Google Scholar] [CrossRef]
- Cheng, C.J.; Lin, Y.C.; Tsai, M.T.; Chen, C.S.; Hsieh, M.C.; Chen, C.L.; Yang, R.B. SCUBE2 suppresses breast tumor cell proliferation and confers a favorable prognosis in invasive breast cancer. Cancer Res. 2009, 69, 3634–3641. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Sullivan, R.J.; Lauffenburger, D.A. Molecular Pathways: Receptor Ectodomain Shedding in Treatment, Resistance, and Monitoring of Cancer. Clin. Cancer Res. 2017, 23, 623–629. [Google Scholar] [CrossRef]
- Vyas, N.; Goswami, D.; Manonmani, A.; Sharma, P.; Ranganath, H.A.; VijayRaghavan, K.; Shashidhara, L.S.; Sowdhamini, R.; Mayor, S. Nanoscale organization of hedgehog is essential for long-range signaling. Cell 2008, 133, 1214–1227. [Google Scholar] [CrossRef]
- Guglier, S.; Hricovini, M.; Raman, R.; Polito, L.; Torri, G.; Casu, B.; Sasisekharan, R.; Guerrini, M. Minimum FGF2 binding structural requirements of heparin and heparan sulfate oligosaccharides as determined by NMR spectroscopy. Biochemistry 2008, 47, 13862–13869. [Google Scholar] [CrossRef] [PubMed]
- Faham, S.; Hileman, R.E.; Fromm, J.R.; Linhardt, R.J.; Rees, D.C. Heparin structure and interactions with basic fibroblast growth factor. Science 1996, 271, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Shriver, Z.; Raman, R.; Venkataraman, G.; Drummond, K.; Turnbull, J.; Toida, T.; Linhardt, R.; Biemann, K.; Sasisekharan, R. Sequencing of 3-O sulfate containing heparin decasaccharides with a partial antithrombin III binding site. Proc. Natl. Acad. Sci. USA 2000, 97, 10359–10364. [Google Scholar] [CrossRef] [PubMed]
- Bateman, J.R.; Lee, A.M.; Wu, C.T. Site-specific transformation of Drosophila via phiC31 integrase-mediated cassette exchange. Genetics 2006, 173, 769–777. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Transgenic fly lines and vectors for the coupled expression of Shh and Hh acyltransferase (resulting in quantitative Shh N-palmitoylation) are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disp1 | Ptc | Shh | Gli1 | Gli2 | Gli3 | Gpc1 | Gp2 | Gp3 | Gpc4 | Gpc5 | Gpc6 | A10 | A12 | A17 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Panc1 | +++ | n.a. | + | - | - | + | +++ | - | - | - | - | + | ++ | - | +++ |
| C3H10T1/2 | + | + | n.a. | - | + | ++ | + | - | - | ++ | - | ++ | n.a. | n.a. | n.a. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manikowski, D.; Jakobs, P.; Jboor, H.; Grobe, K. Soluble Heparin and Heparan Sulfate Glycosaminoglycans Interfere with Sonic Hedgehog Solubilization and Receptor Binding. Molecules 2019, 24, 1607. https://doi.org/10.3390/molecules24081607
Manikowski D, Jakobs P, Jboor H, Grobe K. Soluble Heparin and Heparan Sulfate Glycosaminoglycans Interfere with Sonic Hedgehog Solubilization and Receptor Binding. Molecules. 2019; 24(8):1607. https://doi.org/10.3390/molecules24081607
Chicago/Turabian StyleManikowski, Dominique, Petra Jakobs, Hamodah Jboor, and Kay Grobe. 2019. "Soluble Heparin and Heparan Sulfate Glycosaminoglycans Interfere with Sonic Hedgehog Solubilization and Receptor Binding" Molecules 24, no. 8: 1607. https://doi.org/10.3390/molecules24081607
APA StyleManikowski, D., Jakobs, P., Jboor, H., & Grobe, K. (2019). Soluble Heparin and Heparan Sulfate Glycosaminoglycans Interfere with Sonic Hedgehog Solubilization and Receptor Binding. Molecules, 24(8), 1607. https://doi.org/10.3390/molecules24081607