Systems Pharmacological Approach to Investigate the Mechanism of Ohwia caudata for Application to Alzheimer’s Disease


Abstract
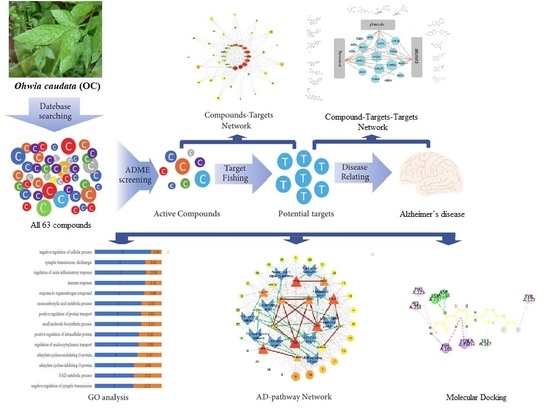
1. Introduction
2. Results
2.1. Identification of Active Compounds
2.2. Compound–Target Network
2.3. Compounds–Target–Target Network
2.4. GO Analysis
2.5. Compound–Target–Pathway Network
2.6. Kynurenine Pathway
2.7. Inflammation-Related Pathways
2.8. Molecular Docking
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. Establishment of Database
4.3. Extraction and Isolation
4.4. Prediction of Drug-Likeness, Oral Bioavailability, and Blood–Brain Barrier Permeability
4.5. Target Fishing
4.6. Compound–Target Network Construction
4.7. Compounds–Target–Target Network
4.8. Gene Ontology (GO) Analysis
4.9. Compound–Target–Pathway Network
4.10. Molecular Docking
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Targets Abbreviations
| Number | PDB ID | Targets | Abbreviations |
| 1 | 5F19 | Prostaglandin G/H synthase 2 | PTGS2 |
| 2 | 2HZP | Kynureninase | KYNU |
| 3 | 3UQU | Beta-secretase 1 | BACE1 |
| 4 | 3UON | Muscarinic acetylcholine receptor M2 | CHRM2 |
| 5 | 3O0G | Cyclin-dependent kinase 5 | CDK5 |
| 6 | 1aqw | Glutathione S-transferase | GSTP1 |
| 7 | 3DZU | Peroxisome proliferator activated receptor gamma | PPARG |
| 8 | 5X68 | Kynurenine 3-monooxygenase | KMO |
| 9 | 1H8F | Glycogen synthase kinase-3 beta | GSK3β |
| 10 | 4a79 | Monoamine oxidase B | MAOB |
| 11 | 5CXV | Muscarinic acetylcholine receptor M1 | CHRM1 |
| 12 | 1SAC | Serum amyloid P-component | APCS |
| 13 | 2YMD | 5-hydroxytryptamine 4 receptor | HTR4 |
| 14 | 2FSO | Mitogen-activated protein kinase 14 | MAPK14 |
| 15 | 1vzj | Acetylcholinesterase | ACHE |
| 16 | 3IVH | β-Amyloid precursor protein | APP |
References
- Chinthapalli, K. Alzheimer’s disease: Still a perplexing problem. BMJ 2014, 349, 4433. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; McWeeney, S.; Park, B.S.; Manczak, M.; Gutala, R.V.; Partovi, D.; Jung, Y.; Yau, V.; Searles, R.; Mori, M.; et al. Gene expression profiles of transcripts in amyloid precursor protein transgenic mice: Up-regulation of mitochondrial metabolism and apoptotic genes is an early cellular change in Alzheimer’s disease. Neurobiol. Aging 2004, 25, S160–S161. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, A.; Ekavali. A review on Alzheimer’s disease pathophysiology and its management: An update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef]
- Gaugler, J.; James, B.; Johnson, T.; Scholz, K.; Weuve, J. Alzheimer’s disease facts and figures. Alzheimers Dementia 2016, 12, 459–509. [Google Scholar]
- Anand, R.; Gill, K.D.; Mahdi, A.A. Therapeutics of Alzheimer’s disease: Past, present and future. Neuropharmacology 2014, 76, 27–50. [Google Scholar] [CrossRef]
- Laferla, F.M. Pathways linking Abeta and tau pathologies. Biochem. Soc. Trans. 2010, 38, 993–995. [Google Scholar] [CrossRef]
- Martin, C. Alzheimer’s disease: Strategies for disease modification. Nat. Rev. Drug Discov. 2010, 9, 387–398. [Google Scholar]
- Bhardwaj, D.; Mitra, C.; Narasimhulu, C.A.; Riad, A.; Doomra, M.; Parthasarathy, S. Alzheimer’s Disease-Current Status and Future Directions. J. Med. Food. 2017, 20. [Google Scholar] [CrossRef] [PubMed]
- Bajda, M.; Guzior, N.; Ignasik, M.; Malawska, B. Multi-target-directed ligands in Alzheimer’s disease treatment. Curr. Med. Chem. 2011, 18. [Google Scholar] [CrossRef]
- Gura, T. Hope in Alzheimer’s fight emerges from unexpected places. Nat. Med. 2008, 14, 894. [Google Scholar] [CrossRef]
- Sun, Y.; Zhu, R.X.; Ye, H.; Tang, K.L.; Zhao, J.; Chen, Y.J.; Liu, Q.; Cao, Z.W. Towards a bioinformatics analysis of anti-Alzheimer’s herbal medicines from a target network perspective. Brief. Bioinf. 2013, 14, 327–343. [Google Scholar] [CrossRef]
- Pang, X.C.; Kang, D.; Fang, J.S.; Zhao, Y.; Xu, L.J.; Lian, W.W.; Liu, A.L.; Du, G.H. Network pharmacology-based analysis of Chinese herbalNaodesheng formula for application to Alzheimer’s disease. Chin. J. Nat. Med. 2018, 16, 53–62. [Google Scholar]
- Jung-Han, L.; Si-Young, L.; Kyung-Seok, L.; Hyun-Jung, J.; Kyung-Ho, L.; Tae-Ryong, H.; Young-Sook, P. Prolyl endopeptidase inhibitors from the leaves of Ginkgo biloba. Planta Med. 2004, 70, 1228–1230. [Google Scholar]
- Hisako, S.; Hirofumi, S.; Kiyoshi, I.; Yoshihisa, T.; Yoshiki, K. Prenylated flavonoids from the stems and leaves of Desmodium caudatum and evaluation of their inhibitory activity against the film-forming growth of Zygosaccharomyces rouxii F51. J. Agric. Food Chem. 2004, 62, 6345–6353. [Google Scholar]
- Xie, W.Z.; Fan, C.S.; Zhou, Z.Y. Quan Guo Zhong Cao Yao Hui Bian; People’s Medical Publishing House: Beijing, China, 1996; p. 97. [Google Scholar]
- Guo, J.; Feng, X.; Zhou, S.; Yan, W.; Meng, D. Potential anti-Alzheimer’s disease activities of the roots of Desmodium caudatum. Ind. Crops Prod. 2016, 90, 94–99. [Google Scholar] [CrossRef]
- Tattersall, M.H.; Sodergren, J.E.; Dengupta, S.K.; Trites, D.H.; Modest, E.J.; Rd, F.E. Pharmacokinetics of actinoymcin Din patients with malignant melanoma. Clin. Pharmacol. Ther. 1975, 17, 701. [Google Scholar] [CrossRef]
- Bhowmik, A.; Khan, R.; Ghosh, M.K. Blood Brain Barrier: A Challenge for Effectual Therapy of Brain Tumors. BioMedRes. Int. 2015. [Google Scholar] [CrossRef]
- Nixon, R.A. Amyloid precursor protein and endosomal-lysosomal dysfunction in Alzheimer’s disease: Inseparable partners in a multifactorial disease. FASEB J. 2017, 31, 2729. [Google Scholar] [CrossRef]
- Gong, C.X.; Liu, F.; Iqbal, K. Multifactorial Hypothesis and Multi-Targets for Alzheimer’s Disease. J. Alzheimers Dis. 2018, 64, S107–S117. [Google Scholar] [CrossRef]
- Lima, S.; Kurnar, S.; Gawandi, V.; Momany, C.; Phillips, R. Crystal Structure of the Homo sapiens Kynureninase-3-Hydroxyhippuric Acid Inhibitor Complex: Insights into the Molecular Basis of Kynureninase Substrate Specificity. J. Med. Chem. 2009, 52, 389–396. [Google Scholar]
- Guillemin, G.J.; Brew, B.J.; Noonan, C.E.; Takikawa, O.; Cullen, K.M. Indoleamine 2,3 dioxygenase and quinolinic acid immunoreactivity in alzheimer’s disease hippocampus. Neuropathol. Appl. Neurobiol. 2005, 31, 395–404. [Google Scholar]
- Tang, N.; Zhang, Y.P.; Ji, L.; Tam, C. Association of prostaglandin-endoperoxide synthase 2 (PTGS2) polymorphisms and Alzheimer’s disease in Chinese. Neurobiol. Aging 2008, 29, 856–860. [Google Scholar]
- Fang, J.; Liu, C.; Wang, Q.; Lin, P.; Cheng, F. In silico polypharmacology of natural products. Brief. Bioinf. 2017. [Google Scholar] [CrossRef]
- Luo, Y.; Qi, W.; Zhang, Y. A systems pharmacology approach to decipher the mechanism of danggui-shaoyao-san decoction for the treatment of neurodegenerative diseases. J. Ethnopharmacol. 2016, 8, 66–81. [Google Scholar] [CrossRef]
- Shinu, C.; Subir, S. Novel Thiosemicarbazide Hybrids with Amino Acids and Peptides Against Hepatocellular Carcinoma: A Molecular Designing Approach Towards Multikinase Inhibitor. Curr. Comput. Aided Drug Des. 2015, 11, 279–290. [Google Scholar]
- QikProp; v 3.0; Schrodinger, LLC: New York, NY, USA, 2018.
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Mering, C.V.; Jensen, L.J.; Snel, B.; Hooper, S.D.; Krupp, M.; Foglierini, M.; Jouffre, N.; Huynen, M.A.; Bork, P. STRING: Known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2005, 33, 433–437. [Google Scholar] [CrossRef]
- Peng, J.; Wang, H.G.; Lu, J.Y.; Hui, W.W.; Wang, Y.D.; Shang, X.Q. Identifying term relations cross different gene ontology categories. BMC Bioinf. 2017, 18, 573. [Google Scholar] [CrossRef]
- Liu, H.; Wang, L.; Lv, M.; Pei, R.; Li, P.; Pei, Z.; Wang, Y.; Su, W.; Xie, X.Q. Alzplatform: Analzheimer’s disease domain-specific chemogenomics knowledgebase for polypharmacology and target identification research. J. Chem. Inf. Model. 2014, 54, 1050–1060. [Google Scholar] [CrossRef]
- Anwar, R. New tips for structure prediction by comparative modeling. Bioinformation 2009, 3, 263–267. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Comp. 1 δC | Comp. 1 δH | Comp. 2 δC | Comp. 2 δH |
|---|---|---|---|---|
| 2 | 145.2 | 78.4 | 5.43 (1H, dd, J = 12.2, 2.8 Hz) | |
| 3 | 137.0 | 42.0 | 3.21 (1H, dd, J = 12.2, 17.0 Hz) 2.76 (1H, dd, J = 2.8, 17.0 Hz) | |
| 4 | 172.3 | 197.2 | ||
| 5 | 123.4 | 7.80 (1H, d, J = 8.8 Hz) | 159.5 | |
| 6 | 113.9 | 6.97 (1H, d, J = 8.8Hz) | 104.0 | |
| 7 | 159.4 | 158.9 | ||
| 8 | 114.4 | 102.0 | ||
| 9 | 154.1 | 154.7 | ||
| 10 | 114.5 | 101.0 | ||
| 1′ | 122.6 | 129.5 | ||
| 2′ | 129.3 | 8.03 (1H, d, J = 8.9 Hz) | 115.4 | 6.75 (1H, s) |
| 3′ | 115.5 | 6.93 (1H, d, J = 8.9Hz) | 145.3 | |
| 4′ | 158.8 | 117.7 | 6.90 (1H, s) | |
| 5′ | 115.5 | 6.93 (1H, d, J = 8.9Hz) | 145.7 | |
| 6′ | 129.3 | 8.03 (1H, d, J = 8.9 Hz) | 114.2 | 6.75 (1H, s) |
| 1″ | 21.9 | 3.56 (2H, d, J = 6.6 Hz) | 115.3 | 6.44 (1H, d, J = 10.0 Hz) |
| 2″ | 122.1 | 5.22 (1H, t, J = 6.6 Hz) | 126.7 | 5.63 (1H, d, J = 10.0 Hz) |
| 3″ | 131.5 | 77.9 | ||
| 4″ | 25.9 | 1.77 (s, 3H) | 28.0 | 1.38 (s, 3H) |
| 5″ | 17.9 | 1.63(s, 3H) | 27.9 | 1.41((s, 3H) |
| 6-Me | 7.4 | 1.89 (s, 3H) |
| Comp. | QPlogS | QPPCaco | QPlogBB | QPP MDCK | Percent Human Oral Absorption | Rule of Five | Rule of Three |
|---|---|---|---|---|---|---|---|
| C1 | −4.993 | 287.747 | −1.28 | 128.708 | 86.403 | 0 | 0 |
| C2 | −5.733 | 178.660 | −1.35 | 76.892 | 84.863 | 0 | 1 |
| C3 | −3.294 | 4887.536 | 0.23 | 2749 | 100 | 0 | 0 |
| C4 | −5.055 | 337.098 | −1.34 | 152.725 | 90.744 | 0 | 0 |
| C5 | −0.393 | 514.138 | −0.52 | 241.025 | 73.539 | 0 | 0 |
| C6 | −1.542 | 328.798 | −0.76 | 148.665 | 78.23 | 0 | 0 |
| C7 | −1.08 | 2646.027 | −0.133 | 1416.199 | 100 | 0 | 0 |
| C8 | −1.08 | 2646.103 | −0.13 | 1416.243 | 100 | 0 | 0 |
| C9 | −1.881 | 1220.263 | 0.50 | 678.697 | 96.387 | 0 | 0 |
| C10 | −0.22 | 497.093 | 0.61 | 516.978 | 80.741 | 0 | 0 |
| C11 | 1.168 | 1710.644 | 0.17 | 1785.53 | 90.664 | 0 | 0 |
| C12 | −1.756 | 1202 | −0.22 | 603.556 | 87.474 | 0 | 0 |
| C13 | −1.554 | 116.972 | −0.62 | 61.872 | 77.047 | 0 | 0 |
| C14 | −1.694 | 97.467 | −0.99 | 50.8 | 70.752 | 0 | 0 |
| C15 | −1.171 | 192.969 | −1.44 | 83.57 | 62.799 | 0 | 0 |
| C16 | −1.228 | 223.474 | −1.47 | 97.936 | 64.567 | 0 | 1 |
| C17 | −4.368 | 125.867 | −1.72 | 52.658 | 79.335 | 0 | 1 |
| C18 | −5.121 | 216.221 | −1.26 | 94.505 | 86.586 | 0 | 0 |
| C19 | −4.656 | 159.964 | −1.65 | 68.233 | 82.202 | 0 | 1 |
| C20 | −3.685 | 127.628 | −1.52 | 53.454 | 78.142 | 0 | 1 |
| C21 | −4.863 | 196.428 | −1.26 | 85.19 | 85.179 | 0 | 0 |
| C22 | −3.737 | 457.384 | −0.95 | 212.4 | 90.08 | 0 | 1 |
| C23 | −4.444 | 220.66 | −1.23 | 96.604 | 84.383 | 0 | 0 |
| C24 | −4.489 | 232.352 | −1.23 | 102.148 | 83.481 | 0 | 0 |
| C25 | −4.637 | 1114.163 | −0.481 | 556.027 | 100 | 0 | 0 |
| C26 | −5.976 | 1562.723 | −0.49 | 801.518 | 100 | 1 | 1 |
| C27 | −6.127 | 1015.613 | −0.60 | 503.063 | 100 | 0 | 1 |
| C28 | −3.871 | 640.678 | −1.09 | 305.739 | 93.583 | 0 | 1 |
| No. | Name | Structure | No. | Name | Structure |
|---|---|---|---|---|---|
| C1 | compound 1 |  | C15 | 4-hydroxy-3-methoxyphenyl-β-d-glucopyranoside |  |
| C2 | compound 2 |  | C16 | koaburaside |  |
| C3 | harmine |  | C17 | noranhydroicaritin |  |
| C4 | 4,4′-diphenylmethane-bislmethy carbamate |  | C18 | desmodin B |  |
| C5 | nicotinamide |  | C19 | cudraflavanone B |  |
| C6 | 5-hydroxy-indole-3-aldehyde |  | C20 | leachianone G |  |
| C7 | N-chloromethyl-N,N-dimethyltryptamine |  | C21 | desmodol |  |
| C8 | N,N-dimethyltryptamine N12-oxide |  | C22 | caudatan C |  |
| C9 | N,N-dimethyltryptamine |  | C23 | citrusinol |  |
| C10 | nicotinic acid |  | C24 | yukovanol |  |
| C11 | ammothamnine |  | C25 | caudatan A |  |
| C12 | loliolide |  | C26 | 3β-12-ene-3, 23, 28-triol |  |
| C13 | salicylic acid |  | C27 | soyasapogenel B |  |
| C14 | ferulic acid |  | C28 | (+)-5′-methoxyisolariciresinol-9-O-β-d-glucopyranoside |  |
| Compound | PTGS2 | KYNU | BACE1 | CDK5 | CHRM2 |
|---|---|---|---|---|---|
| Reference | −279.275 | −71.135 | −269.837 | −157.913 | −130.289 |
| 1 | −109.826 | −82.362 | −131.327 | −100.114 | −124.923 |
| 2 | −109.851 | −81.0566 | −125.875 | −103.123 | −113.16c |
| 3 | −101.917 | −90.059 | −97.622 | −81.515 | −102.699 |
| 4 | −116.921 | −112.357 | −147.607 | −125.264 | −120.443 |
| 5 | −66.7496 | −59.008 | −61.787 | −83.45 | −64.733 |
| 6 | −87.8179 | −85.553 | −89.709 | −71.066 | −87.349 |
| 7 | −97.7527 | −80.036 | −104.692 | −87.507 | −113.467 |
| 8 | −96.806 | −90.1924 | −101.014 | −82.776 | −103.683 |
| 9 | −95.682 | −79.2415 | −94.954 | −83.71 | −98.256 |
| 10 | −87.768 | −47.2166 | −96.669 | −73.057 | −93.652 |
| 11 | −83.11 | −30.309 | −78.291 | −58.436 | −77.146 |
| 12 | −100.12 | −61.922 | −87.5 | −77.206 | −87.393 |
| 13 | −74.49 | −66.975 | −67.683 | −53.451 | −66.144 |
| 14 | −101.205 | −89.905 | −103.201 | −84.782 | −96.424 |
| 15 | −91.99 | −65.687 | −101.422 | −80.586 | −103.666 |
| 16 | −97.489 | −64.878 | −107.982 | −84.402 | −107.419 |
| 17 | −113.147 | −79.252 | −131.154 | −103.973 | −126.566 |
| 18 | −105.505 | −82.359 | −122.934 | −97.775 | −109.951 |
| 19 | −121.982 | −94.5721 | −138.589 | −113.973 | −109.814 |
| 20 | −118.492 | −95.216 | −128.598 | −104.169 | −124.998 |
| 21 | −105.56 | −80.545 | −127.578 | −102.149 | −113.791 |
| 22 | −101.112 | −63.776 | −126.693 | −83.45 | −117.44 |
| 23 | −100.213 | −64.122 | −122.083 | −96.788 | −111.528 |
| 24 | −99.225 | −63.626 | −119.269 | −91.528 | −110.426 |
| 25 | −88.806 | −55.989 | −69.709 | −73.446 | −116.637 |
| 26 | −89.21 | −74.515 | −95.219 | −98.8 | −67.588 |
| 27 | −90.942 | −52.395 | −83.509 | −101.319 | −68.174 |
| 28 | −118.408 | −72.669 | −136.226 | −107.887 | −133.31 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Y.-w.; Wang, Y.; Guo, Z.-f.; Du, K.-c.; Meng, D.-l. Systems Pharmacological Approach to Investigate the Mechanism of Ohwia caudata for Application to Alzheimer’s Disease. Molecules 2019, 24, 1499. https://doi.org/10.3390/molecules24081499
Sun Y-w, Wang Y, Guo Z-f, Du K-c, Meng D-l. Systems Pharmacological Approach to Investigate the Mechanism of Ohwia caudata for Application to Alzheimer’s Disease. Molecules. 2019; 24(8):1499. https://doi.org/10.3390/molecules24081499
Chicago/Turabian StyleSun, Yi-wei, Yue Wang, Zi-feng Guo, Kai-cheng Du, and Da-li Meng. 2019. "Systems Pharmacological Approach to Investigate the Mechanism of Ohwia caudata for Application to Alzheimer’s Disease" Molecules 24, no. 8: 1499. https://doi.org/10.3390/molecules24081499
APA StyleSun, Y.-w., Wang, Y., Guo, Z.-f., Du, K.-c., & Meng, D.-l. (2019). Systems Pharmacological Approach to Investigate the Mechanism of Ohwia caudata for Application to Alzheimer’s Disease. Molecules, 24(8), 1499. https://doi.org/10.3390/molecules24081499