
Development of Highly Selective 1,2,3-Triazole-containing Peptidic Polo-like Kinase 1 Polo-box Domain-binding Inhibitors




Abstract
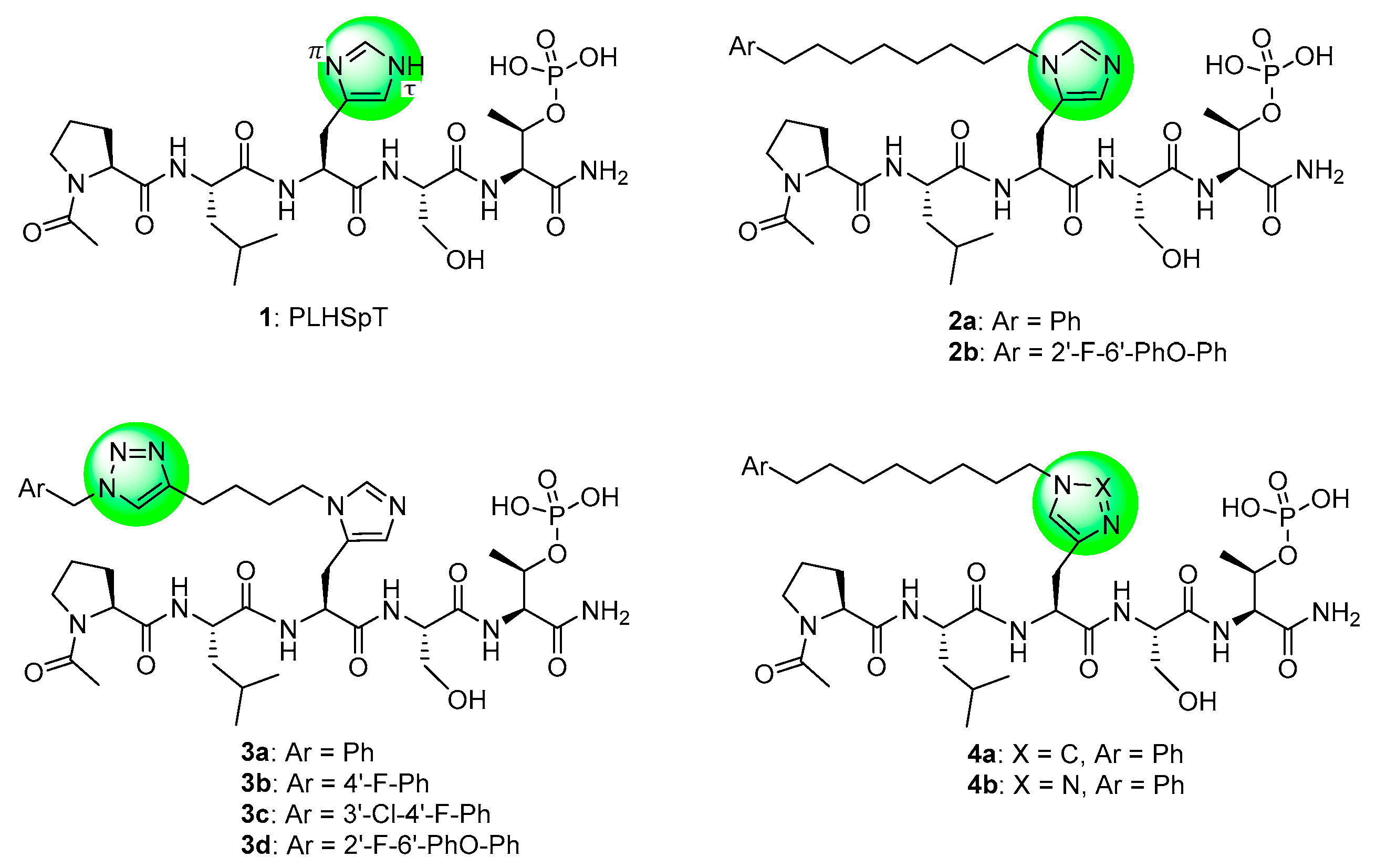
1. Introduction
2. Results and Discussion
2.1. Synthesis
2.2. Biological Evaluation
3. Experimental Section
3.1. Synthesis
3.1.1. General Procedures
3.1.2. Synthesis of 2-Fluoro-6-phenoxybenzaldehyde (6)
3.1.3. Synthesis of (2-Fluoro-6-phenoxyphenyl)methanol (7)
3.1.4. Synthesis of 2-(Bromomethyl)-1-fluoro-3-phenoxybenzene (8d)
3.1.5. General Procedure A for the Synthesis of Azides 9a–9d and 16
3.1.6. Synthesis of (Azidomethyl)benzene (9a)
3.1.7. Synthesis of 1-(Azidomethyl)-4-fluorobenzene (9b)
3.1.8. Synthesis of 4-(Azidomethyl)-2-chloro-1-fluorobenzene (9c)
3.1.9. Synthesis of 2-(Azidomethyl)-1-fluoro-3-phenoxybenzene (9d)
3.1.10. Synthesis of (8-Azidooctyl)benzene (16)
3.1.11. Synthesis of Nα-(((9H-Fluoren-9-yl)methoxy)carbonyl)-Nπ-(hex-5-yn-1-yl)-l-histidine (11)
3.2. Peptide Synthesis
3.2.1. General Solid-Phase Peptide Synthesis (SPPS)
3.2.2. Synthesis of Peptides 3a–3d and 4b Using On-resin Copper-catalyzed Alkyne-azide Cycloaddition Reaction (CuAAC)
3.2.3. Synthesis of Peptide 4b Using On-resin Ruthenium-catalyzed Alkyne-azide Cycloaddition Reaction (RuAAC)
3.2.4. Peptide Data
3.3. Determination of Binding Selectivity against the PBDs of Plks 1–3 Using Fluorescence Polarization
3.3.1. Expression and Purification of Isolated PBDs of Plks 1–3 for Fluorescence Polarization Assays
3.3.2. Evaluation of Binding Affinities against the Isolated PBDs of Plk1, Plk2 and Plk3 Using Fluorescence Polarization Assays
3.4. Evaluation of Binding Affinities against Full-length Plk1 Using ELISA Assays
3.4.1. Lysate Production for ELISA-based Inhibition Assay against Full Length Plk1
3.4.2. Determination of Inhibitory Potency in an ELISA Assay Using Full-Length Plk1
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zitouni, S.; Nabais, C.; Jana, S.C.; Guerrero, A.; Bettencourt-Dias, M. Polo-like kinases: Structural variations lead to multiple functions. Nat. Rev. Mol. Cell Biol. 2014, 15, 433–452. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-E.; Erikson, R.L.; Lee, K.S. Feed-forward mechanism of converting biochemical cooperativity to mitotic processes at the kinetochore plate. Proc. Natl. Acad. Sci. USA 2011, 108, 8200–8205. [Google Scholar] [CrossRef] [PubMed]
- Lowery, D.M.; Lim, D.; Yaffe, M.B. Structure and function of polo-like kinases. Oncogene 2005, 24, 248–259. [Google Scholar] [CrossRef] [PubMed]
- Strebhardt, K.; Ullrich, A. Targeting polo-like kinase 1 for cancer therapy. Nat. Rev. Cancer 2006, 6, 321–330. [Google Scholar] [CrossRef]
- Park, J.-E.; Soung, N.-K.; Johmura, Y.; Kang, Y.H.; Liao, C.; Lee, K.H.; Park, C.H.; Nicklaus, M.C.; Lee, K.S. Polo-box domain: A versatile mediator of polo-like kinase function. Cell. Mol. Life Sci. 2010, 67, 1957–1970. [Google Scholar] [CrossRef]
- Strebhardt, K. Multifaceted polo-like kinases: Drug targets and antitargets for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 643–660. [Google Scholar] [CrossRef]
- Lee, K.S.; Burke, T.R., Jr.; Park, J.-E.; Bang, J.K.; Lee, E. Recent advances and new strategies in targeting Plk1 for anticancer therapy. Trends Pharmacol. Sci. 2015, 36, 858–877. [Google Scholar] [CrossRef]
- Archambault, V.; Lepine, G.; Kachaner, D. Understanding the polo kinase machine. Oncogene 2015, 34, 4799–4807. [Google Scholar] [CrossRef]
- Jang, Y.-J.; Lin, C.-Y.; Ma, S.; Erikson, R.L. Functional studies on the role of the C-terminal domain of mammalian polo-like kinase. Proc. Nat. Acad. Sci. USA 2002, 99, 1984–1989. [Google Scholar] [CrossRef]
- Reindl, W.; Yuan, J.; Kraemer, A.; Strebhardt, K.; Berg, T. Inhibition of polo-like kinase 1 by blocking polo-box domain-dependent protein-protein interactions. Chem. Biol. 2008, 15, 459–466. [Google Scholar] [CrossRef]
- Reindl, W.; Graeber, M.; Strebhardt, K.; Berg, T. Development of high-throughput assays based on fluorescence polarization for inhibitors of the polo-box domains of polo-like kinases 2 and 3. Anal. Biochem. 2009, 395, 189–194. [Google Scholar] [CrossRef]
- Gjertsen, B.T.; Schoffski, P. Discovery and development of the polo-like kinase inhibitor volasertib in cancer therapy. Leukemia 2015, 29, 11–19. [Google Scholar] [CrossRef]
- Berg, A.; Berg, T. Inhibitors of the polo-box domain of polo-like kinase 1. ChemBioChem 2016, 17, 650–656. [Google Scholar] [CrossRef]
- Yun, S.-M.; Moulaei, T.; Lim, D.; Bang, J.K.; Park, J.-E.; Shenoy, S.R.; Liu, F.; Kang, Y.H.; Liao, C.; Soung, N.-K.; et al. Structural and functional analyses of minimal phosphopeptides targeting the polo-box domain of polo-like kinase 1. Nat. Struct. Mol. Biol. 2009, 16, 876–882. [Google Scholar] [CrossRef]
- Liu, F.; Park, J.-E.; Qian, W.-J.; Lim, D.; Graber, M.; Berg, T.; Yaffe, M.B.; Lee, K.S.; Burke, T.R., Jr. Serendipitous alkylation of a Plk1 ligand uncovers a new binding channel. Nat. Chem. Biol. 2011, 7, 595–601. [Google Scholar] [CrossRef]
- Liu, F.; Park, J.-E.; Qian, W.-J.; Lim, D.; Scharow, A.; Berg, T.; Yaffe, M.B.; Lee, K.S.; Burke, T.R., Jr. Identification of high affinity polo-like kinase 1 (Plk1) polo-box domain binding peptides using oxime-based diversification. ACS Chem. Biol. 2012, 7, 805–810. [Google Scholar] [CrossRef]
- Liu, F.; Park, J.-E.; Qian, W.-J.; Lim, D.; Scharow, A.; Berg, T.; Yaffe, M.B.; Lee, K.S.; Burke, T.R., Jr. Peptoid-peptide hybrid ligands targeting the polo box domain of polo-like kinase 1. ChemBioChem 2012, 13, 1291–1296. [Google Scholar] [CrossRef]
- Qian, W.-J.; Park, J.-E.; Grant, R.; Lai, C.C.; Kelley, J.A.; Yaffe, M.B.; Lee, K.S.; Burke, T.R., Jr. Neighbor-directed histidine N(τ)–alkylation: A route to imidazolium-containing phosphopeptide macrocycles. Pept. Sci. 2015, 104, 663–673. [Google Scholar] [CrossRef]
- Hymel, D.; Grant, R.A.; Tsuji, K.; Yaffe, M.B.; Burke, T.R., Jr. Histidine N(τ)-cyclized macrocycles as a new genre of polo-like kinase 1 polo-box domain-binding inhibitors. Bioorg. Med. Chem. Lett. 2018, 28, 3202–3205. [Google Scholar] [CrossRef]
- Sledz, P.; Stubbs, C.J.; Lang, S.; Yang, Y.-Q.; McKenzie, G.J.; Venkitaraman, A.R.; Hyvoenen, M.; Abell, C. From crystal packing to molecular recognition: Prediction and discovery of a binding site on the surface of polo-like kinase 1. Angew. Chem. Int. Ed. Engl. 2011, 50, 4003–4006. [Google Scholar] [CrossRef]
- Tan, Y.S.; Śledź, P.; Lang, S.; Stubbs, C.J.; Spring, D.R.; Abell, C.; Best, R.B. Using ligand-mapping simulations to design a ligand selectively trgeting a cryptic surface pocket of polo-like kinase 1. Angew. Chem. Int. Ed. Engl. 2012, 51, 10078–10081. [Google Scholar] [CrossRef]
- Śledź, P.; Lang, S.; Stubbs, C.J.; Abell, C. High-throughput interrogation of ligand binding mode using a fluorescence-based assay. Angew. Chem. Int. Ed. Engl. 2012, 51, 7680–7683. [Google Scholar] [CrossRef]
- Elia, A.E.; Rellos, P.; Haire, L.F.; Chao, J.W.; Ivins, F.J.; Hoepker, K.; Mohammad, D.; Cantley, L.C.; Smerdon, S.J.; Yaffe, M.B. The molecular basis for phosphodependent substrate targeting and regulation of Plks by the polo-box domain. Cell 2003, 115, 83–95. [Google Scholar] [CrossRef]
- Qian, W.-J.; Park, J.-E.; Lee, K.S.; Burke, T.R., Jr. Non-proteinogenic amino acids in the pThr-2 position of a pentamer peptide that confer high binding affinity for the polo box domain (PBD) of polo-like kinase 1 (Plk1). Bioorg. Med. Chem. Lett. 2012, 22, 7306–7308. [Google Scholar] [CrossRef]
- Zhao, X.Z.; Hymel, D.; Burke, T.R., Jr. Application of oxime-diversification to optimize ligand interactions within a cryptic pocket of the polo-like kinase 1 polo-box domain. Bioorg. Med. Chem. Lett. 2016, 26, 5009–5012. [Google Scholar] [CrossRef][Green Version]
- Zhao, X.Z.; Hymel, D.; Burke, T.R., Jr. Enhancing polo-like kinase 1 selectivity of polo-box domain-binding peptides. Bioorg. Med. Chem. 2017, 25, 5041–5049. [Google Scholar] [CrossRef]
- Angell, Y.L.; Burgess, K. Peptidomimetics via copper-catalyzed azide-alkyne cycloadditions. Chem. Soc. Rev. 2007, 36, 1674–1689. [Google Scholar] [CrossRef]
- Yoo, B.; Shin, S.B.Y.; Huang, M.L.; Kirshenbaum, K. Peptoid macrocycles: Making the rounds with peptidomimetic oligomers. Chem. Eur. J. 2010, 16, 5528–5537. [Google Scholar] [CrossRef]
- Kappe, C.O.; Van der Eycken, E. Click chemistry under non-classical reaction conditions. Chem. Soc. Rev. 2010, 39, 1280–1290. [Google Scholar] [CrossRef]
- Pedersen, D.S.; Abell, A. 1,2,3-Triazoles in peptidomimetic chemistry. Eur. J. Org. Chem. 2011, 13, 2399–2411. [Google Scholar] [CrossRef]
- Ahmad Fuaad, A.A.; Azmi, F.; Skwarczynski, M.; Toth, I. Peptide conjugation via CuAAC ‘click’ chemistry. Molecules 2013, 18, 13148–13174. [Google Scholar] [CrossRef]
- Das, R.; Majumdar, N.; Lahiri, A. A review on 1,3-dipolar cycloaddition reactions in bioconjugation and it’s importance in pharmaceutical chemistry. Int. J. Res. Pharm. Chem. 2014, 4, 467–472. [Google Scholar] [CrossRef]
- Fehlhammer, W.P.; Beck, W. Azide chemistry—An inorganic perspective, part II [3+2]-cycloaddition reactions of metal azides and related systems. Z. Anorg. Allg. Chem. 2015, 641, 1599–1678. [Google Scholar] [CrossRef]
- Johansson, J.R.; Beke-Somfai, T.; Said Stålsmeden, A.; Kann, N. Ruthenium-catalyzed azide alkyne cycloaddition reaction: Scope, mechanism, and applications. Chem. Rev. 2016, 116, 14726–14768. [Google Scholar] [CrossRef] [PubMed]
- Barlow, T.M.A.; Tourwé, D.; Ballet, S. Cyclisation to form small, medium and large rings by use of catalysed and uncatalysed azide–alkyne cycloadditions (AACs). Eur. J. Org. Chem. 2017, 32, 4678–4694. [Google Scholar] [CrossRef]
- Buysse, K.; Farard, J.; Nikolaou, A.; Vanderheyden, P.; Vauquelin, G.; Sejer Pedersen, D.; Tourwé, D.; Ballet, S. Amino triazolo diazepines (Ata) as constrained histidine mimics. Org. Lett. 2011, 13, 6468–6471. [Google Scholar] [CrossRef]
- Chen, Y.; Li, Z.; Liu, Y.; Lin, T.; Sun, H.; Yang, D.; Jiang, C. Identification of novel and selective non-peptide inhibitors targeting the polo-box domain of polo-like kinase 1. Bioorg. Chem. 2018, 81, 278–288. [Google Scholar] [CrossRef]
- Appel, R. Tertiary phosphane/tetrachloromethane, a versatile reagent for chlorination, dehydration, and phosphorus-nitrogen linkage. Angew. Chem. Int. Ed. Engl. 1975, 14, 801–811. [Google Scholar] [CrossRef]
- Qian, W.; Liu, F.; Burke, T.R., Jr. Investigation of unanticipated alkylation at the N(pi) position of a histidyl residue under Mitsunobu conditions and synthesis of orthogonally protected histidine analogues. J. Org. Chem. 2011, 76, 8885–8890. [Google Scholar] [CrossRef]
- Tornoe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise Huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. Engl. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Tron, G.C.; Pirali, T.; Billington, R.A.; Canonico, P.L.; Sorba, G.; Genazzani, A.A. Click chemistry reactions in medicinal chemistry: Applications of the 1,3-dipolar cycloaddition between azides and alkynes. Med. Res. Rev. 2008, 28, 278–308. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, X.; Xue, P.; Sun, H.H.Y.; Williams, I.D.; Sharpless, K.B.; Fokin, V.V.; Jia, G. Ruthenium-catalyzed cycloaddition of alkynes and organic azides. J. Am. Chem. Soc. 2005, 127, 15998–15999. [Google Scholar] [CrossRef]
- Rasmussen, L.K.; Boren, B.C.; Fokin, V.V. Ruthenium-catalyzed cycloaddition of aryl azides and alkynes. Org. Lett. 2007, 9, 5337–5339. [Google Scholar] [CrossRef]
- Creary, X.; Anderson, A.; Brophy, C.; Crowell, F.; Funk, Z. Method for assigning structure of 1,2,3-triazoles. J. Org. Chem. 2012, 77, 8756–8761. [Google Scholar] [CrossRef]
- Xu, J.; Shen, C.; Wang, T.; Quan, J. Structural basis for the inhibition of polo-like kinase 1. Nat. Struct. Mol. Biol. 2013, 20, 1047–1053. [Google Scholar] [CrossRef]
- Fish, P.V.; Ryckmans, T.; Stobie, A.; Wakenhut, F. [4-(Phenoxy)pyridin-3-yl]methylamines: A new class of selective noradrenaline reuptake inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 1795–1798. [Google Scholar] [CrossRef]
- Siebertz, K.D.; Hackenberger, C.P.R. Chemoselective triazole-phosphonamidate conjugates suitable for photorelease. Chem. Commun. 2018, 54, 763–766. [Google Scholar] [CrossRef]
- Boren, B.C.; Narayan, S.; Rasmussen, L.K.; Zhang, L.; Zhao, H.; Lin, Z.; Jia, G.; Fokin, V.V. Ruthenium-catalyzed azide−alkyne cycloaddition: Scope and mechanism. J. Am. Chem. Soc. 2008, 130, 8923–8930. [Google Scholar] [CrossRef]
- Hymel, D.; Burke, T.R., Jr. Phosphatase-stable phosphoamino acid mimetics that enhance binding affinities with the polo-box domain of polo-like kinase 1. ChemMedChem 2017, 12, 202–206. [Google Scholar] [CrossRef]
- Hanisch, A.; Wehner, A.; Nigg, E.A.; Sillje, H.H.W. Different Plk1 functions show distinct dependencies on polo-Box domain-mediated targeting. Mol. Biol. Cell 2006, 17, 448–459. [Google Scholar] [CrossRef]
- Golsteyn, R.M.; Schultz, S.J.; Bartek, J.; Ziemiecki, A.; Ried, T.; Nigg, E.A. Cell cycle analysis and chromosomal localization of human Plk1, a putative homologue of the mitotic kinases Drosophila polo and Saccharomyces cerevisiae Cdc5. J. Cell Sci. 1994, 107, 1509–1517. [Google Scholar]
Sample Availability: Samples of select peptides may be available from the authors in limited quantities. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Peptide | R | IC50 (nM) | ||
|---|---|---|---|---|
| PLK1 PBD | PLK2 PBD | PLK3 PBD | ||
| 1i |  | 650 ± 39 | ND iv | ND |
| 17 |  | 1000 ± 140 | ND | ND |
| 13 |  | 1100 ± 45 | ND | ND |
| 2aii |  | 15 ± 0.94 | 220 ± 15 | 1100 ± 230 |
| 2biii |  | 15 ± 0.33 | 180 ± 14 | 450 ± 87 |
| 3a |  | 110 ± 6.6 | ND | ND |
| 3b |  | 100 ± 19 | ND | ND |
| 3c |  | 130 ± 9.8 | ND | ND |
| 3d |  | 25 ± 1.6 | 5900 ± 420 | 9900 ± 2200 |
| 4b |  | 17 ± 0.17 | 690 ± 20 | 3400 ± 130 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, X.Z.; Tsuji, K.; Hymel, D.; Burke, T.R., Jr. Development of Highly Selective 1,2,3-Triazole-containing Peptidic Polo-like Kinase 1 Polo-box Domain-binding Inhibitors. Molecules 2019, 24, 1488. https://doi.org/10.3390/molecules24081488
Zhao XZ, Tsuji K, Hymel D, Burke TR Jr. Development of Highly Selective 1,2,3-Triazole-containing Peptidic Polo-like Kinase 1 Polo-box Domain-binding Inhibitors. Molecules. 2019; 24(8):1488. https://doi.org/10.3390/molecules24081488
Chicago/Turabian StyleZhao, Xue Zhi, Kohei Tsuji, David Hymel, and Terrence R. Burke, Jr. 2019. "Development of Highly Selective 1,2,3-Triazole-containing Peptidic Polo-like Kinase 1 Polo-box Domain-binding Inhibitors" Molecules 24, no. 8: 1488. https://doi.org/10.3390/molecules24081488
APA StyleZhao, X. Z., Tsuji, K., Hymel, D., & Burke, T. R., Jr. (2019). Development of Highly Selective 1,2,3-Triazole-containing Peptidic Polo-like Kinase 1 Polo-box Domain-binding Inhibitors. Molecules, 24(8), 1488. https://doi.org/10.3390/molecules24081488