Oxytrodiflavanone A and Oxytrochalcoflavanones A,B: New Biflavonoids from Oxytropis chiliophylla


Abstract
:1. Introduction
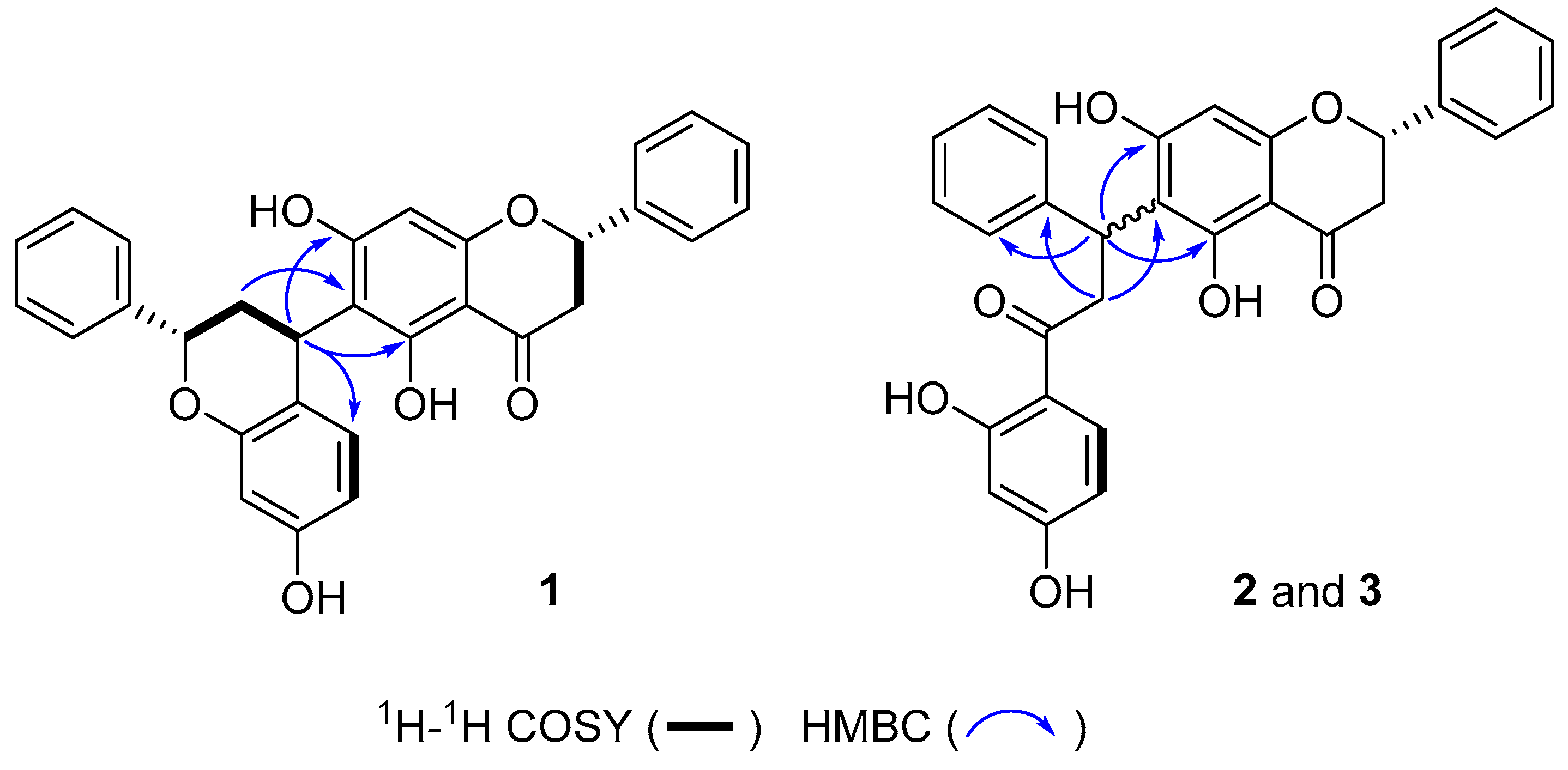
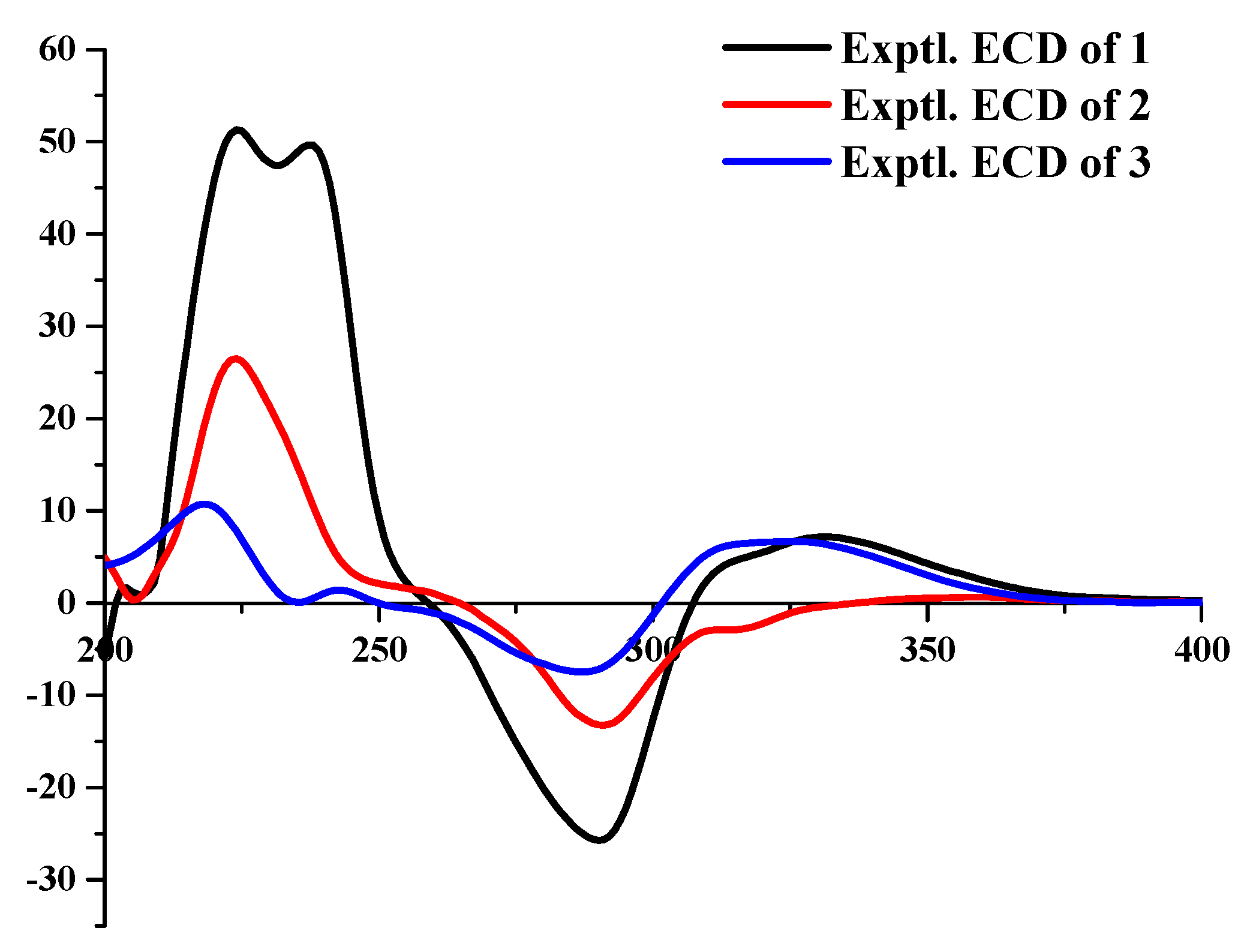
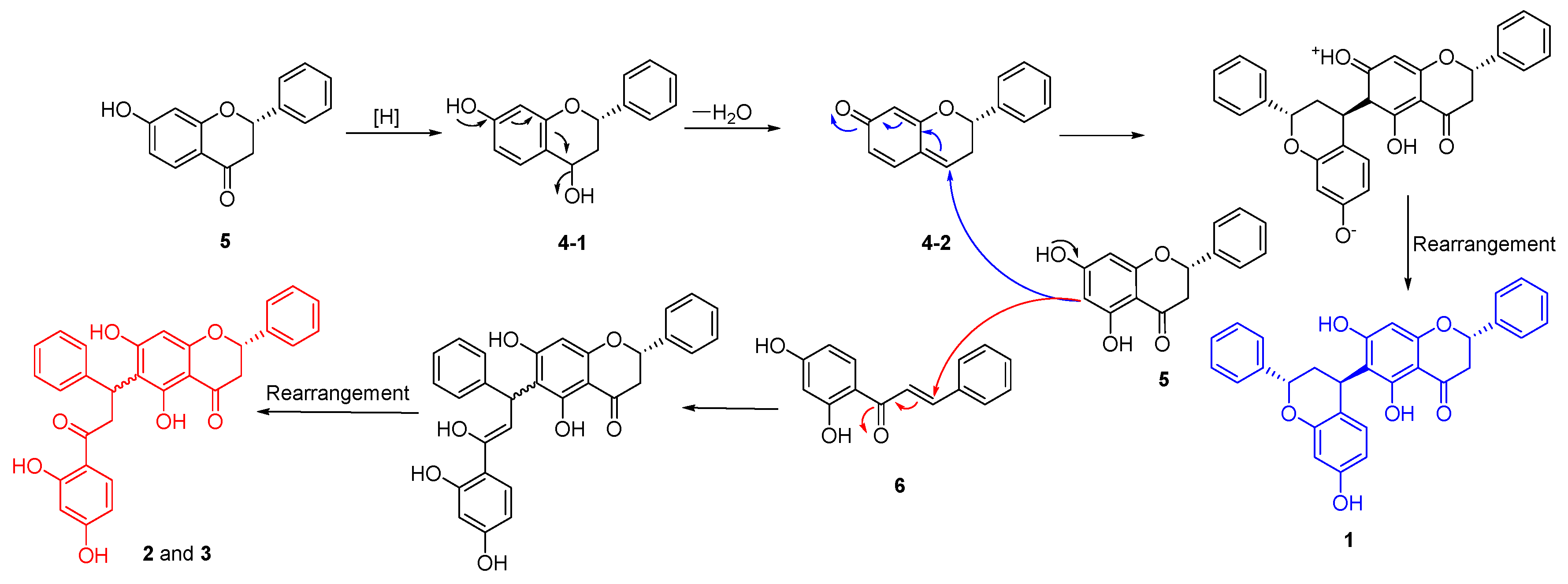
2. Results and Discussion
3. Experimental Section
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Cytotoxicity Assays against PC–3 Human Prostate Cancer Cells
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Committee of Chinese Pharmacopoeia. Chinese Pharmacopoeia; Chinese Medical Science and Technology Press: Beijing, China, 2015. [Google Scholar]
- Dimaer, D.Z.P.C. Jingzhu Materia Medica; Shanghai Science and Technology Press: Shanghai, China, 1986. [Google Scholar]
- The Chinese Academy of Sciences. Flora of China; Science Press: Beijing, China, 2005. [Google Scholar]
- Yao, S.; Ma, Y.; Tang, Y.; Chen, J.; Zhang, X. Chemical constituents of Oxytropis chiliophylla. Chin. J. Chin. Mater. Med. 2007, 32, 1660–1662. [Google Scholar]
- Kelsang, N.; Que, S.; Kelsang, P.; Zhang, X.; Samnor; Liang, H.; Zhang, Q. High-performance liquid chromatographic fingerprint analysis of Oxytropis falcata Bunge and Oxytropis chiliophylla Royle. J. Chin. Pharm. Sci. 2014, 23, 783–789. [Google Scholar]
- Wang, J.; Liu, Y.; Kelsang, N.; Zeng, K.; Liang, H.; Zhang, Q.; Tu, P. Rhamnocitrin glycosides from Oxytropis chiliophylla. Phytochem. Lett. 2017, 19, 50–54. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, X.; Kelsang, N.; Tu, G.; Kong, D.; Lu, J.; Zhang, Y.; Liang, H.; Tu, P.; Zhang, Q. Structurally diverse cytotoxic dimeric chalcones from Oxytropis chiliophylla. J. Nat. Prod. 2018, 81, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Seo, E.; Silva, G.; Chai, H.; Chagwedera, T.; Farnsworth, N.; Cordell, G.; Pezzuto, J.; Kinghorn, A. Cytotoxic prenylated flavanones from Monotes engler. Phytochemistry 1997, 45, 509–515. [Google Scholar] [CrossRef]
- Meesakul, P.; Pudhom, K.; Pyne, S.; Laphookhieo, S. Hybrid flavan-flavanones from Friesodielsia desmoids and their inhibitory activities against nitric oxide production. RSC Adv. 2017, 7, 17545–17550. [Google Scholar] [CrossRef]
- Bajgai, S.; Prachyawarakorn, V.; Mahidol, C.; Ruchirawat, S.; Kittakoop, P. Hybrid flavan-chalcones, aromatase and lipoxygenase inhibitors, from Desmos cochinchinensis. Phytochemistry 2011, 72, 2062–2067. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Li, X.; Ferreira, D. 4-Arylflavan-3-ols as proanthocyanidin models: Absolute configuration via density functional calculation of electronic circular dichroism. J. Nat. Prod. 2010, 73, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Pesca, M.S.; Piaz, F.D.; Sanogo, R.; Vassallo, A.; De Abreu, M.B.; Rapisarda, A.; Germanó, M.P.; Certo, G.; Falco, S.D.; De Tommasi, N.; et al. Bioassay-guided isolation of proanthocyanidins with antiangiogenic activies. J. Nat. Prod. 2013, 76, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Coetzee, J.; Steynberg, J.P.; Steynberg, P.J.; Brandt, E.V.; Ferreira, D. Oligomeric flavonoids. Part 18. Dimeric prorobinetinidins from Robinia pseudacacia. Tetrahedron 1995, 51, 2339–2352. [Google Scholar] [CrossRef]
- Liu, Y.; Nair, M.G. An efficient and economical MTT assay for determining the antioxidant activity of plant natural product extracts and pure compounds. J. Nat. Prod. 2010, 73, 1193–1195. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1H-NMR a | 13C-NMR | 1H-NMR b |
|---|---|---|---|
| 1 | - | - | - |
| 2 | 5.42 (dd, 13.1, 2.5) | 79.17 | 5.56 (dd, 13.2, 3.0) |
| 3α | 3.09 (dd, 17.2, 13.1) | 43.69 | 17.02 (dd, 17.0, 13.2) |
| 3β | 2.84 (dd, 17.2, 2.5) | 2.89 (17.0, 3.0) | |
| 4 | - | 196.11 | - |
| 5 | - | 162.07 | - |
| 6 | - | 109.96 | - |
| 7 | - | 164.13 | - |
| 8 | 6.00 (s) | 96.99 | 6.44 (s) |
| 9 | - | 161.47 | - |
| 10 | - | 103.12 | - |
| 1′ | - | 138.57 | - |
| 2′/6′ | 7.43–7.46 c | 126.26 | 7.24–7.42 c |
| 3′/5′ | 7.43–7.46 c | 129.03 | 7.24–7.42 c |
| 4′ | 7.43–7.46 c | 129.03 | 7.24–7.42 c |
| 5-OH | 12.58 (s) | - | 13.19 |
| 1″ | - | - | - |
| 2″ | 5.30 (t, 5.0) | 75.76 | 5.99 (dd, 6.0, 3.6) |
| 3″α | 2.40 (t, 5.9) | 34.94 | 3.27 (ddd, 13.8, 8.4, 3.6) |
| 3″β | 2.40 (t, 5.9) | 2.65 (dt, 13.8, 6.0) | |
| 4″ | 4.52 (t, 5.9) | 28.25 | 5.07 (t, 7.2) |
| 5″ | 6.98 (d, 8.4) | 130.73 | 7.67 (d, 7.8) |
| 6″ | 6.45 (dd, 8.4, 2.1) | 104.61 | 6.85 (dd, 8.4, 2.4) |
| 7″-OH | - | 156.76 | 11.35 |
| 8″ | 6.55 (d, 2.1) | 109.82 | 7.15 (d, 2.4) |
| 9″ | - | 156.25 | - |
| 10″ | - | 112.98 | - |
| 1‴ | - | 140.81 | - |
| 2‴/6‴ | 7.35–7.42 c | 126.00 | 7.24–7.42 c |
| 3‴/5‴ | 7.35–7.42 c | 128.69 | 7.24–7.42 c |
| 4‴ | 7.35–7.42 c | 127.93 | 7.24–7.42 c |
| No. | 1H-NMR | 13C-NMR | No. | 1H-NMR | 13C-NMR |
|---|---|---|---|---|---|
| 1 | - | - | H-β | 5.10 (dd, 9.4, 4.6) | 34.51 |
| 2 | 5.35 (dd, 13.2, 2.8) | 79.22 | H-α a | 4.25 (dd, 17.5, 9.4) | 40.03 |
| 3α | 3.09 (dd, 17.1, 13.2) | 43.59 | H-α b | 3.73 (dd, 17.5, 4.6) | |
| 3β | 2.84 (dd, 17.1, 2.8) | 1″ | - | 142.36 | |
| 4 | - | 196.19 | 2″/6″ | 7.37–7.42 a | 127.86 |
| 5 | - | 162.23 | 3″/5″ | 7.37–7.42 a | 128.37 |
| 6 | - | 111.05 | 4″ | 7.37–7.42 a | 126.46 |
| 7 | - | 163.20 | 1‴ | - | 114.07 |
| 8 | 6.05 (s) | 96.89 | 2‴ | - | 165.34 |
| 9 | - | 161.51 | 3‴ | 6.34 (d, 1.0) | 103.68 |
| 10 | - | 103.48 | 4‴ | - | 163.23 |
| 1′ | - | 138.51 | 5‴ | 6.40 (dd, 8.9, 1.0) | 108.21 |
| 2′/6′ | 7.37–7.42 a | 126.27 | 6‴ | 7.83 (d, 8.9) | 132.74 |
| 3′/5′ | 7.37–7.42 a | 129.01 | 5-OH | 12.46 (s) | |
| 4′ | 7.37–7.42 a | 129.01 | 2‴-OH | 12.40 (s) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Kelsang, N.; Lu, J.; Zhang, Y.; Liang, H.; Tu, P.; Kong, D.; Zhang, Q. Oxytrodiflavanone A and Oxytrochalcoflavanones A,B: New Biflavonoids from Oxytropis chiliophylla. Molecules 2019, 24, 1468. https://doi.org/10.3390/molecules24081468
Liu Y, Kelsang N, Lu J, Zhang Y, Liang H, Tu P, Kong D, Zhang Q. Oxytrodiflavanone A and Oxytrochalcoflavanones A,B: New Biflavonoids from Oxytropis chiliophylla. Molecules. 2019; 24(8):1468. https://doi.org/10.3390/molecules24081468
Chicago/Turabian StyleLiu, Yang, Norbo Kelsang, Jianghai Lu, Yingtao Zhang, Hong Liang, Pengfei Tu, Dexin Kong, and Qingying Zhang. 2019. "Oxytrodiflavanone A and Oxytrochalcoflavanones A,B: New Biflavonoids from Oxytropis chiliophylla" Molecules 24, no. 8: 1468. https://doi.org/10.3390/molecules24081468
APA StyleLiu, Y., Kelsang, N., Lu, J., Zhang, Y., Liang, H., Tu, P., Kong, D., & Zhang, Q. (2019). Oxytrodiflavanone A and Oxytrochalcoflavanones A,B: New Biflavonoids from Oxytropis chiliophylla. Molecules, 24(8), 1468. https://doi.org/10.3390/molecules24081468