
NNB-Type Tridentate Boryl Ligands Enabling a Highly Active Iridium Catalyst for C–H Borylation


Abstract
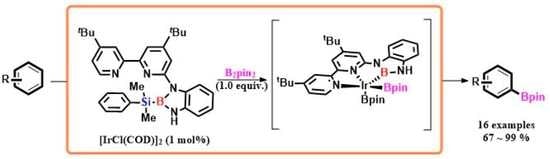
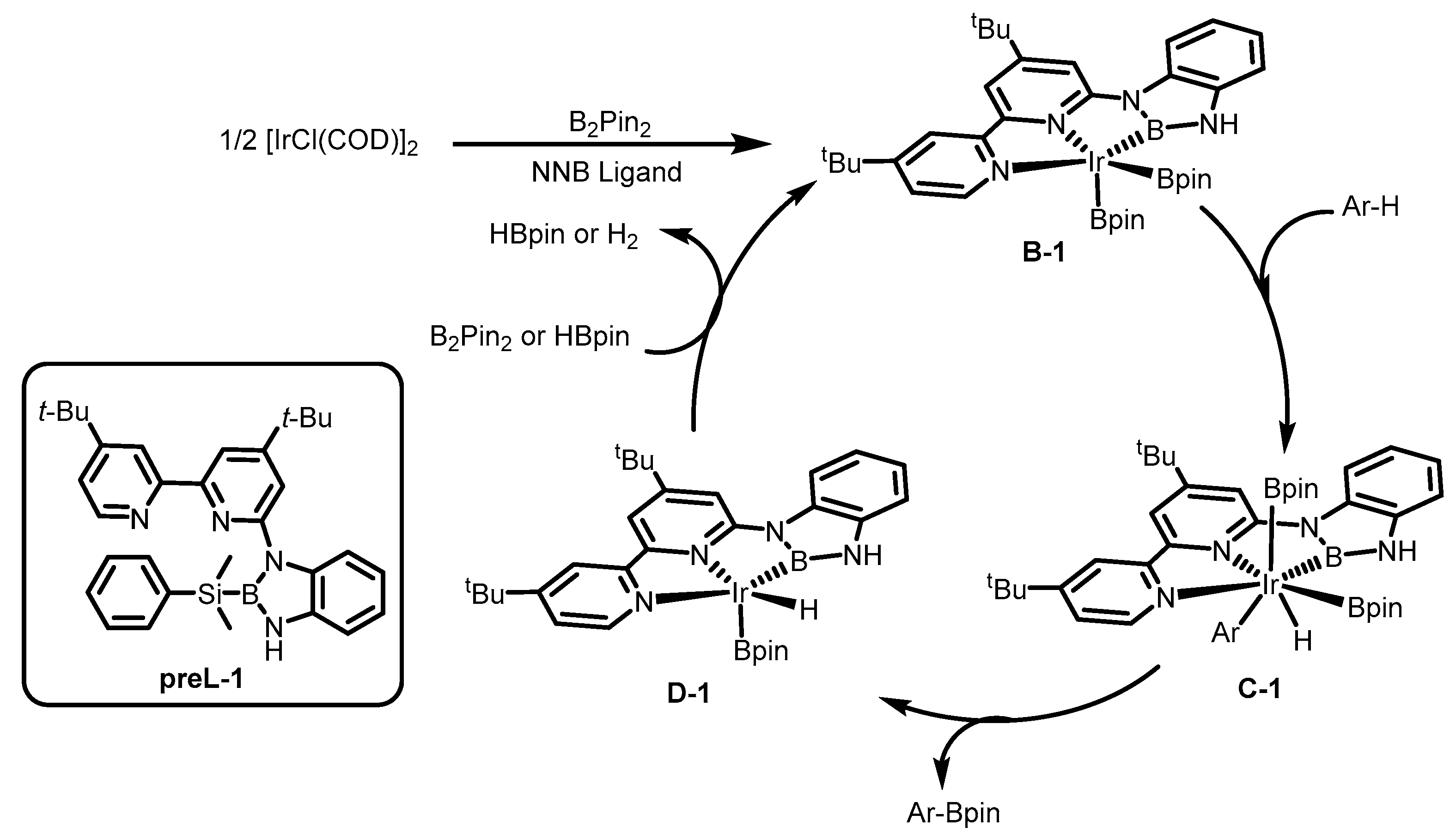
1. Introduction
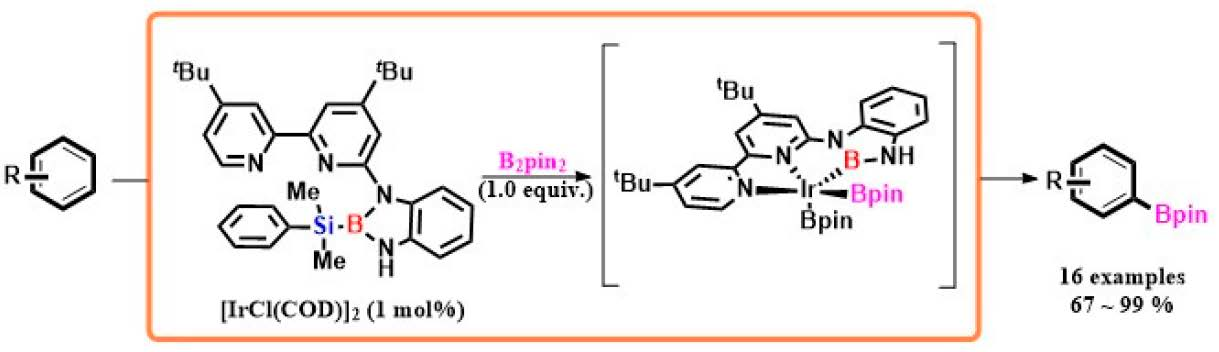
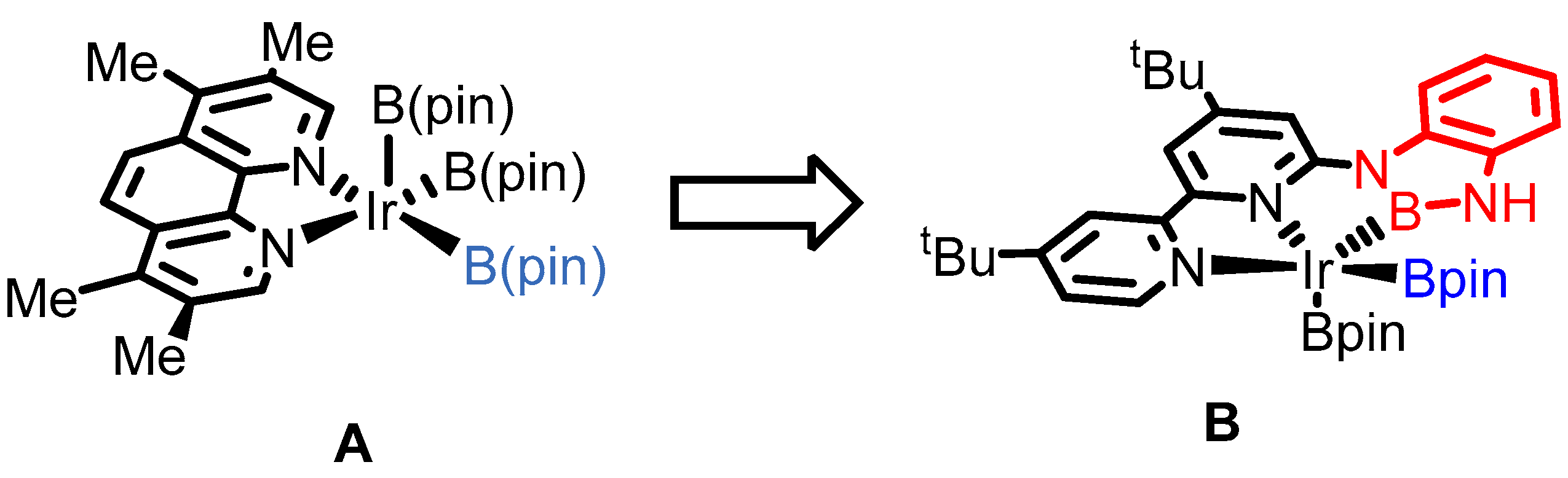
2. Results
3. Discussion
4. Materials and Methods
4.1. Methods and Material
4.1.1. General Information
4.1.2. Structural analysis
4.1.3. Materials
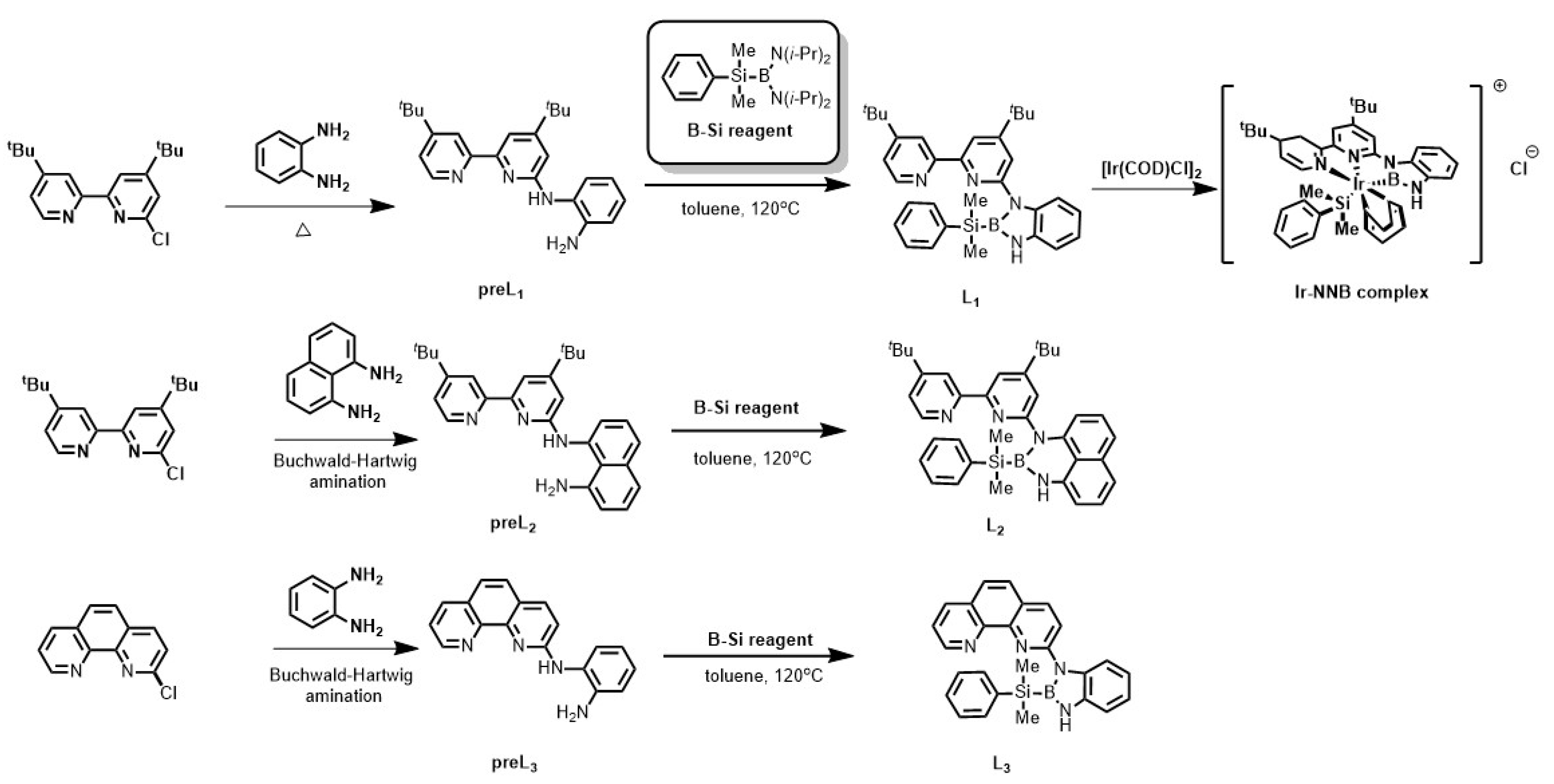
4.2. The Preparation of New NNB-Type Tridentate Boryl Anion Ligand
4.3. The Preparation of Iridium-NNB Ligand Complex
4.4. General Procedure for Iridium-Catalyzed arene C–H borylation Using NNB-type Ligand
Analytical Data of products 5 to 5p
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Irivine, G.J.; Rickard, C.E.F.; Roper, W.R.; Williamson, A.; Wright, L.J. A base-stabilized terminal borylene complex of osmium derived from reaction between a dichloroboryl complex and 8-aminoquinoline. Angew. Chem. Int. Ed. 2000, 39, 948–950. [Google Scholar] [CrossRef]
- Rickard, C.E.F.; Roper, W.R.; Williamson, A.; Wright, L.J. Tethered Osmium Boryl Complexes from the Reaction of Os(BCl2)Cl(CO)(PPh3)2 with 2-Hydroxypyridine. Organometallics 2002, 21, 1714–1718. [Google Scholar] [CrossRef]
- Rickard, C.E.F.; Roper, W.R.; Williamson, A.; Wright, L.J. Tethered Boryl and Base-Stabilized Borylene Osmium Complexes from the Reaction of Os(BCl2)Cl(CO)(PPh3)2 with 2-Aminopyridine. Organometallics 2002, 21, 4862–4872. [Google Scholar] [CrossRef]
- Braunschweig, H.; Kupfer, T.; Lutz, M.; Radacki, K.; Seeler, F.; Sigritz, R. Monofunctional Metathesis Polymers via Sacrificial Diblock Copolymers. Angew. Chem. Int. Ed. 2006, 45, 8048–8051. [Google Scholar] [CrossRef] [PubMed]
- Schubert, H.; Leis, W.; Mayer, H.A.; Wesemann, L. A bidentate boryl ligand: syntheses of platinum and iridium complexes. Chem. Commun. 2014, 50, 2738–2740. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.H.; Liang, X.; Li, P.F. Double N,B-Type Bidentate Boryl Ligands Enabling a Highly Active Iridium Catalyst for C–H Borylation. J. Am. Chem. Soc. 2015, 137, 8058–8061. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.H.; Liu, L.; Wang, H.; Ding, Y.S.; Zhou, J.; Mao, S.; Li, P.F. N,B-Bidentate Boryl Ligand-orted Iridium Catalyst for Efficient Functional-Group-Directed C–H Borylation. J. Am. Chem. Soc. 2016, 139, 91–94. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, S.; Li, P.F. Boron-selective reactions as powerful tools for modular synthesis of diverse complex molecules. Chem. Soc. Rev. 2015, 44, 8848–8858. [Google Scholar] [CrossRef]
- Zhu, J.; Lin, Z.Y.; Marder, T.B. Trans Influence of Boryl Ligands and Comparison with C, Si, and Sn Ligands. Inorg. Chem. 2005, 44, 9384–9390. [Google Scholar] [CrossRef]
- Segawa, Y.; Yamashita, M.; Nozaki, K. Boryl Anion Attacks Transition-Metal Chlorides To Form Boryl Complexes: Syntheses, Spectroscopic, and Structural Studies on Group 11 Borylmetal Complexes. Angew. Chem. Int. Ed. 2007, 46, 6710–6713. [Google Scholar] [CrossRef]
- Segawa, Y.; Yamashita, M.; Nozaki, K. Syntheses of PBP Pincer Iridium Complexes: A Supporting Boryl Ligand. J. Am. Chem. Soc. 2009, 131, 9201–9203. [Google Scholar] [CrossRef]
- Spokoyny, A.M.; Reuter, M.G.; Stern, C.L.; Ratner, M.A.; Seideman, T.; Mirkin, C.A. Carborane-Based Pincers: Synthesis and Structure of SeBSe and SBS Pd(II) Complexes. J. Am. Chem. Soc. 2009, 131, 9482–9483. [Google Scholar] [CrossRef]
- EI-Zaria, M.E.; Arii, H.; Nakamura, H. m-Carborane-based chiral NBN pincer-metal complexes: synthesis, structure, and application in asymmetric catalysis. Inorg. Chem. 2011, 50, 4149–4161. [Google Scholar] [CrossRef]
- Ogawa, H.; Yamashita, M. Platinum complexes bearing a boron-based PBP pincer ligand: synthesis, structure, and application as a catalyst for hydrosilylation of 1-decene. Dalton Trans. 2013, 42, 625–629. [Google Scholar] [CrossRef]
- Lin, T.P.; Peters, J.C. Boryl-Mediated Reversible H2 Activation at Cobalt: Catalytic Hydrogenation, Dehydrogenation, and Transfer Hydrogenation. J. Am. Chem. Soc. 2013, 135, 15310–15313. [Google Scholar] [CrossRef]
- Lin, T.P.; Peters, J.C. Boryl–Metal Bonds Facilitate Cobalt/Nickel-Catalyzed Olefin Hydrogenation. J. Am. Chem. Soc. 2014, 136, 13672–13683. [Google Scholar] [CrossRef]
- Noguchi, H.; Hojo, K.; Suginome, M. Boron-Masking Strategy for the Selective Synthesis of Oligoarenes via Iterative Suzuki−Miyaura Coupling. J. Am. Chem. Soc. 2007, 129, 758–759. [Google Scholar] [CrossRef]
- Gillis, E.P.; Burke, M.D. A Simple and Modular Strategy for Small Molecule Synthesis: Iterative Suzuki−Miyaura Coupling of B-Protected Haloboronic Acid Building Blocks. J. Am. Chem. Soc. 2007, 129, 6716–6717. [Google Scholar] [CrossRef]
- Wang, C.; Glorius, F. Controlled Iterative Cross-Coupling: On the Way to the Automation of Organic Synthesis. Angew. Chem. Int. Ed. 2009, 48, 5240–5244. [Google Scholar] [CrossRef]
- Hall, D. Boronic Acids: Preparation and Application in Organic Synthesis, Medicine and Materials, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2011. [Google Scholar]
- Xu, L.; Li, P.F. Differentiated Di- and Polyboron Compounds: Synthesis and Application in Successive Suzuki–Miyaura Coupling. Synlett 2014, 25, 1799–1802. [Google Scholar]
- Xu, L.; Wang, G.H.; Zhang, S.; Wang, H.; Wang, L.H.; Liu, L.; Jiao, J.; Li, P.F. Recent advances in catalytic C−H borylation reactions. Tetrahedron 2017, 73, 7123–7157. [Google Scholar] [CrossRef]
- Iverson, C.N.; Smith, M.R. Stoichiometric and Catalytic B−C Bond Formation from Unactivated Hydrocarbons and Boranes. J. Am. Chem. Soc. 1999, 121, 7696–7697. [Google Scholar] [CrossRef]
- Cho, J.Y.; Iverson, C.N.; Smith, M.R. Steric and Chelate Directing Effects in Aromatic Borylation. J. Am. Chem. Soc. 2000, 122, 12868–12869. [Google Scholar] [CrossRef]
- Nguyen, P.; Blom, H.P.; Westcott, S.A.; Taylor, N.J.; Marder, T.B. Synthesis and structures of the first transition-metal tris(boryl) complexes: iridium complexes (.eta.6-arene)Ir(BO2C6H4)3. J. Am. Chem. Soc. 1993, 115, 9329–9330. [Google Scholar] [CrossRef]
- Cho, J.Y.; Tse, M.K.; Holmes, D.; Maleczka, R.E.; Smith, M.R. Remarkably selective iridium catalysts for the elaboration of aromatic C-H bonds. Science 2002, 295, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Ishiyama, T.; Takagi, J.; Ishida, K.; Miyaura, N. Mild Iridium-Catalyzed Borylation of Arenes. High Turnover Numbers, Room Temperature Reactions, and Isolation of a Potential Intermediate. J. Am. Chem. Soc. 2002, 124, 390–391. [Google Scholar] [CrossRef]
- Ishiyama, T.; Takagi, J.; Hartwig, J.F.; Miyaura, N. A Stoichiometric Aromatic C-H Borylation Catalyzed by Iridium(I)/2,2′-Bipyridine Complexes at Room Temperature. Angew. Chem. Int. Ed. 2002, 41, 3056–3058. [Google Scholar] [CrossRef]
- Takagi, J.; Sato, K.; Hartwig, J.F.; Ishiyama, T.; Miyaure, N. Iridium-catalyzed C–H coupling reaction of heteroaromatic compounds with bis(pinacolato)diboron: regioselective synthesis of heteroarylboronates. Tetrahedron Lett. 2002, 43, 5649–5651. [Google Scholar] [CrossRef]
- Mkhalid, I.A.I.; Coventry, D.N.; Albesa-Jove, D.; Batsanov, A.S.; Howard, J.A.K.; Marder, T.B.; Perutz, R.N. Ir-Catalyzed Borylation of C-H Bonds in N-Containing Heterocycles: Regioselectivity in the Synthesis of Heteroaryl Boronate Esters. Angew. Chem. Int. Ed. 2006, 45, 489–491. [Google Scholar] [CrossRef]
- Ishiyama, T.; Nobuta, Y.; Hartwig, J.F.; Miyaure, N. Room temperature borylation of arenes and heteroarenes using stoichiometric amounts of pinacolborane catalyzed by iridium complexes in an inert solvent. Chem. Commun. 2003, 23, 2924–2925. [Google Scholar] [CrossRef]
- Paul, S.; Chotana, G.A.; Holmes, D.; Reichle, R.C., Jr.; Maleczka, R.E.; Smith, M.R., III. Ir-Catalyzed Functionalization of 2-Substituted Indoles at the 7-Position: Nitrogen-Directed Aromatic Borylation. J. Am. Chem. Soc. 2006, 128, 15552–15553. [Google Scholar] [CrossRef]
- Lo, W.F.; Kaiser, H.M.; Spannenberg, A.; Beller, M.; Tse, M.K. A highly selective Ir-catalyzed borylation of 2-substituted indoles: a new access to 2,7- and 2,4,7-substituted indoles. Tetrahedron Lett. 2007, 48, 371–375. [Google Scholar] [CrossRef]
- Datta, A.; Kollhofer, A.; Plenio, H. Ir-catalyzed C–H activation in the synthesis of borylated ferrocenes and half sandwich compounds. Chem. Commun. 2004, 35, 1508–1509. [Google Scholar] [CrossRef]
- Anderson, A.G., Jr.; Nelson, J.A.; Tazuma, J.J. Azulene. III. Electrophilic Substitution1–3. J. Am. Chem. Soc. 1953, 75, 4980–4989. [Google Scholar] [CrossRef]
- Coventry, D.V.; Batsanov, A.S.; Goeta, A.E.; Howard, J.A.K.; Marder, T.B. Selective Ir-catalysed borylation of polycyclic aromatic hydrocarbons: structures of naphthalene-2,6-bis(boronate), pyrene-2,7-bis(boronate) and perylene-2,5,8,11-tetra(boronate) esters. Chem. Commun. 2005, 36, 2172–2174. [Google Scholar] [CrossRef]
- Kimoto, T.; Tanaka, K.; Sakai, Y.; Ohno, A.; Yoza, K.; Kobayashi, K. 2,8- and 2,9-Diboryltetracenes as Useful Building Blocks for Extended π-Conjugated Tetracenes. Org. Lett. 2009, 11, 3658–3661. [Google Scholar] [CrossRef]
- Chen, H.Y.; Schlecht, S.; Semple, T.C.; Hartwig, J.F. Thermal, Catalytic, Regiospecific Functionalization of Alkanes. Science 2000, 287, 1995–1997. [Google Scholar] [CrossRef]
- Wang, W.J.; Chuang, K.S.; Luo, C.F.; Liu, H.Y. An efficient one-pot route to an aza-bridged bis-phenanthroline macrocyclic compound. Tetrahedron Lett. 2000, 41, 8565–8568. [Google Scholar] [CrossRef]
- Wang, W.J.; Sengul, A.; Luo, C.F.; Kao, H.C.; Cheng, Y.H. Facile one-step synthesis of a thia-bridged bis-1,10-phenanthroline macrocycle. Tetrahedron Lett. 2003, 44, 7099–7101. [Google Scholar] [CrossRef]
- Zajac, M.A. An Application of Borane As a Protecting Group for Pyridine. J. Org. Chem. 2008, 73, 6899–6901. [Google Scholar] [CrossRef]
- Biaco, S.; Musetti, C.; Waldeck, A.; Sparapan, S.; Seitz, J.D.; Krapcho, A.P.; Palumbo, M.; Sissi, C. Bis-phenanthroline derivatives as suitable scaffolds for effective G-quadruplex recognition. Dalton Trans. 2010, 39, 5833–5841. [Google Scholar]
- Chavant, P.Y.; Vaultiev, M. Preparation of some organo-bis(diisopropylamino)boranes and their application to the synthesis of oxazaborolidines. J. Organomet. Chem. 1993, 455, 37–46. [Google Scholar] [CrossRef]
- Buynak, J.D.; Geng, B. Synthesis and Reactivity of Silylboranes. Organometallics 1995, 14, 3112–3115. [Google Scholar] [CrossRef]
- Matsumoto, A.; Ito, Y. New Generation of Organosilyl Radicals by Photochemically Induced Homolytic Cleavage of Silicon−Boron Bonds. J. Org. Chem. 2000, 65, 5707–5711. [Google Scholar] [CrossRef]
- Hennessy, E.J.; Buchwald, S.L. Synthesis of 4,5-Dianilinophthalimide and Related Analogues for Potential Treatment of Alzheimer’s Disease via Palladium-Catalyzed Amination. J. Org. Chem. 2005, 70, 7371–7375. [Google Scholar] [CrossRef]
- Shekhar, S.; Ryberg, P.; Hartwig, J.F.; Mathew, J.S.; Blackmond, D.G.; Strieter, E.R.; Buchwald, S.L. Reevaluation of the Mechanism of the Amination of Aryl Halides Catalyzed by BINAP-Ligated Palladium Complexes. J. Am. Chem. Soc. 2006, 128, 3584–3591. [Google Scholar] [CrossRef]
- Ge, S.Z.; Green, R.A.; Hartwig, J.F. Controlling First-Row Catalysts: Amination of Aryl and Heteroaryl Chlorides and Bromides with Primary Aliphatic Amines Catalyzed by a BINAP-Ligated Single-Component Ni(0) Complex. J. Am. Chem. Soc. 2014, 136, 1617–1627. [Google Scholar] [CrossRef]
- Ramírez-López, P.; Ros, A.; Romero-Arenas, A.; Iglesias-Sigüenza, J.; Fernández, R.; Lassaletta, J.M. Synthesis of IAN-type N,N-Ligands via Dynamic Kinetic Asymmetric Buchwald–Hartwig Amination. J. Am. Chem. Soc. 2016, 138, 12053–12056. [Google Scholar] [CrossRef]
- Ruiz-Castillo, P.; Buchwald, S.L. Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions. Chem. Rev. 2016, 116, 12564–12649. [Google Scholar] [CrossRef]
- Noguchi, H.; Shioda, T.; Chou, C.M.; Suginome, M. Differentially Protected Benzenediboronic Acids: Divalent Cross-Coupling Modules for the Efficient Synthesis of Boron-Substituted Oligoarenes. Org. Lett. 2008, 10, 377–380. [Google Scholar] [CrossRef]
- Iwadate, N.; Suginome, Y. Differentially Protected Diboron for Regioselective Diboration of Alkynes: Internal-Selective Cross-Coupling of 1-Alkene-1,2-diboronic Acid Derivatives. J. Am. Chem. Soc. 2010, 132, 2548–2549. [Google Scholar] [CrossRef]
- Xu, L.; Ding, S.Y.; Li, P.F. Site-Differentiated Polyboron Arenes Prepared by Direct C–H Borylation and Their Highly Selective Suzuki–Miyaura Cross-Coupling Reactions. Angew. Chem. Int. Ed. 2014, 53, 1822–1826. [Google Scholar] [CrossRef]
- Lilio, A.M.; Grice, K.A.; Kubiak, C.P. A series of dinuclear copper complexes bridged by phosphanylbipyridine ligands: synthesis, structural characterization and electrochemistry. Eur. J. Inorg. Chem. 2013, 23, 4016–4023. [Google Scholar] [CrossRef]
- Engel, Y.; Dahan, A.; Rozenshine-Kemelmakher, E.; Gozin, M. Tweezer-type ratiometric chemosensor for ureas and uranium salts. J. Org. Chem. 2007, 72, 2318–2328. [Google Scholar] [CrossRef]
- Preshlock, S.M.; Ghaffari, B.; Maligres, P.E.; Krska, S.W.; Maleczka, R.E.; Smith, M.R. High-throughput optimization of Ir-catalyzed C-H borylation: A tutorial for pratical applications. J. Am. Chem. Soc. 2013, 135, 7572–7582. [Google Scholar] [CrossRef]
- Tang, W.; Keshipeddy, S.; Zhang, Y.; Wei, X.; Savoie, J.; Patel, N.; Yee, N.K.; Senanayake, C.H. Efficient monophosphorus ligands for palladium-catalyzed miyaura borylation. Org. Lett. 2011, 13, 1366–1369. [Google Scholar] [CrossRef]
- Boebel, T.A.; Hartwig, J.F. Iiridium-catalyzed preparation of silylboranes by silane borylation and their use in the catalytic borylation of arenes. Organometallics 2008, 27, 6013–6019. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Ligand | B2pin2 | Temperature | Time | Yield(NMR) b |
|---|---|---|---|---|---|
| 1 | dtbpy | 1.0 | 100 | 3h | 72% |
| 2 | 1,10-ph | 1.0 | 100 | 3h | 90% |
| 3 | tmphen | 1.o | 100 | 3h | 94% |
| 4 | L1 | 1.0 | 100 | 3h | 94% (90% c) |
| 5 d | Ir-NNB complex | 1.0 | 100 | 3h | 92% |
| 6 | L1 | 0.5 | 100 | 3 h | 64% |
| 7 | L1 | 0.6 | 100 | 3 h | 68% |
| 8 | L1 | 0.7 | 100 | 3 h | 71% |
| 9 | L1 | 0.8 | 100 | 3 h | 82% |
| 10 | L1 | 1.1 | 100 | 3 h | 86% |
| 11 | L1 | 1.0 | 100 | 0.5 h | 25% |
| 12 | L1 | 1.0 | 100 | 1 h | 81% |
| 13 | L1 | 1.0 | 100 | 2 h | 89% |
| 14 | L1 | 1.0 | 80 | 3 h | 66% |
| 15 | L1 | 1.0 | 90 | 3 h | 72% |
| 16 | L2 | 1.0 | 100 | 3 h | 90% |
| 17 | L3 | 1.0 | 100 | 3 h | 82% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, S.; Wang, L.; Miao, Z.; Li, P. NNB-Type Tridentate Boryl Ligands Enabling a Highly Active Iridium Catalyst for C–H Borylation. Molecules 2019, 24, 1434. https://doi.org/10.3390/molecules24071434
Ding S, Wang L, Miao Z, Li P. NNB-Type Tridentate Boryl Ligands Enabling a Highly Active Iridium Catalyst for C–H Borylation. Molecules. 2019; 24(7):1434. https://doi.org/10.3390/molecules24071434
Chicago/Turabian StyleDing, Siyi, Linghua Wang, Zongcheng Miao, and Pengfei Li. 2019. "NNB-Type Tridentate Boryl Ligands Enabling a Highly Active Iridium Catalyst for C–H Borylation" Molecules 24, no. 7: 1434. https://doi.org/10.3390/molecules24071434
APA StyleDing, S., Wang, L., Miao, Z., & Li, P. (2019). NNB-Type Tridentate Boryl Ligands Enabling a Highly Active Iridium Catalyst for C–H Borylation. Molecules, 24(7), 1434. https://doi.org/10.3390/molecules24071434