
Quantitative Analysis of Tozadenant Using Liquid Chromatography-Mass Spectrometric Method in Rat Plasma and Its Human Pharmacokinetics Prediction Using Physiologically Based Pharmacokinetic Modeling



Abstract
:1. Introduction
2. Results
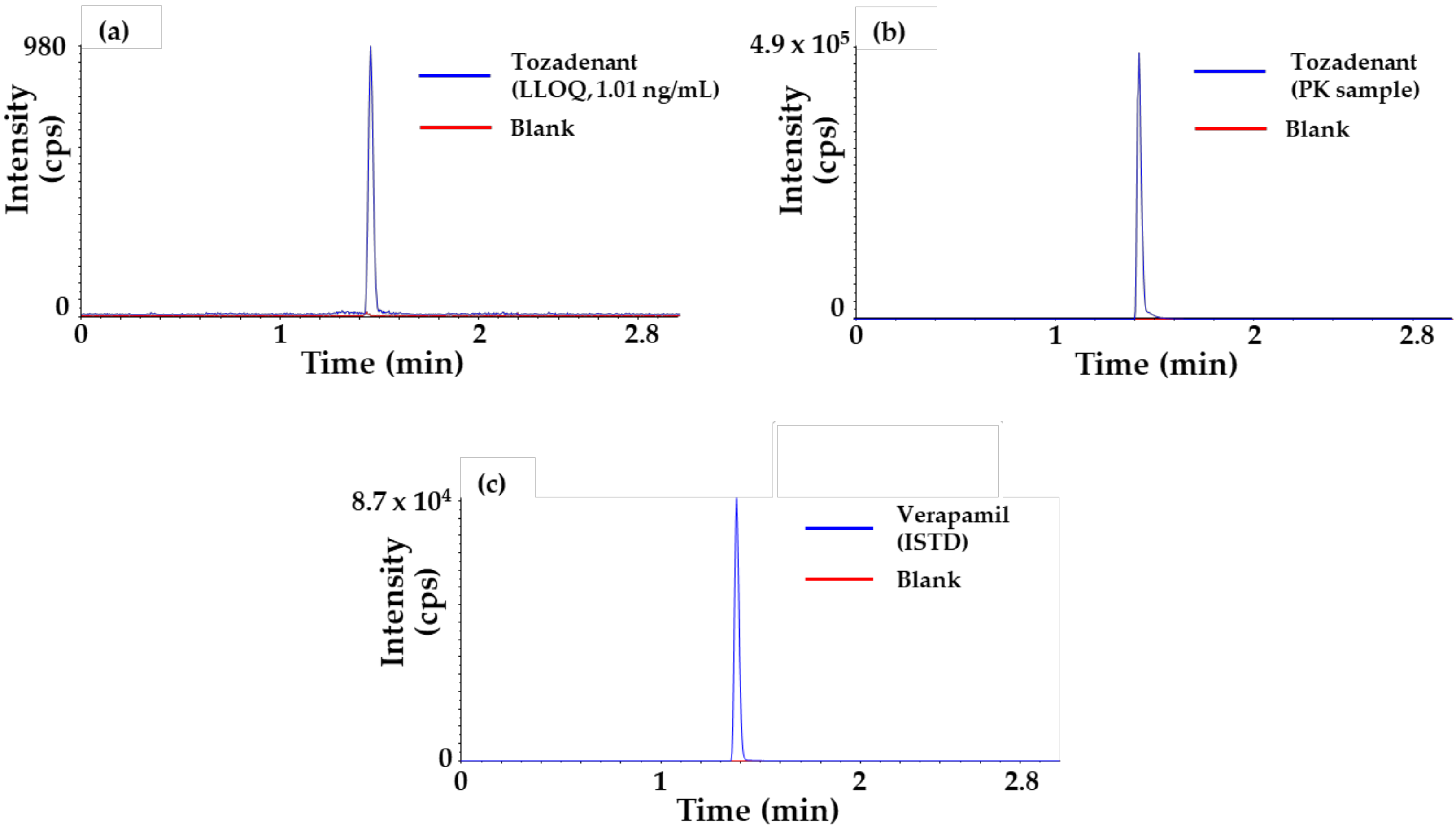
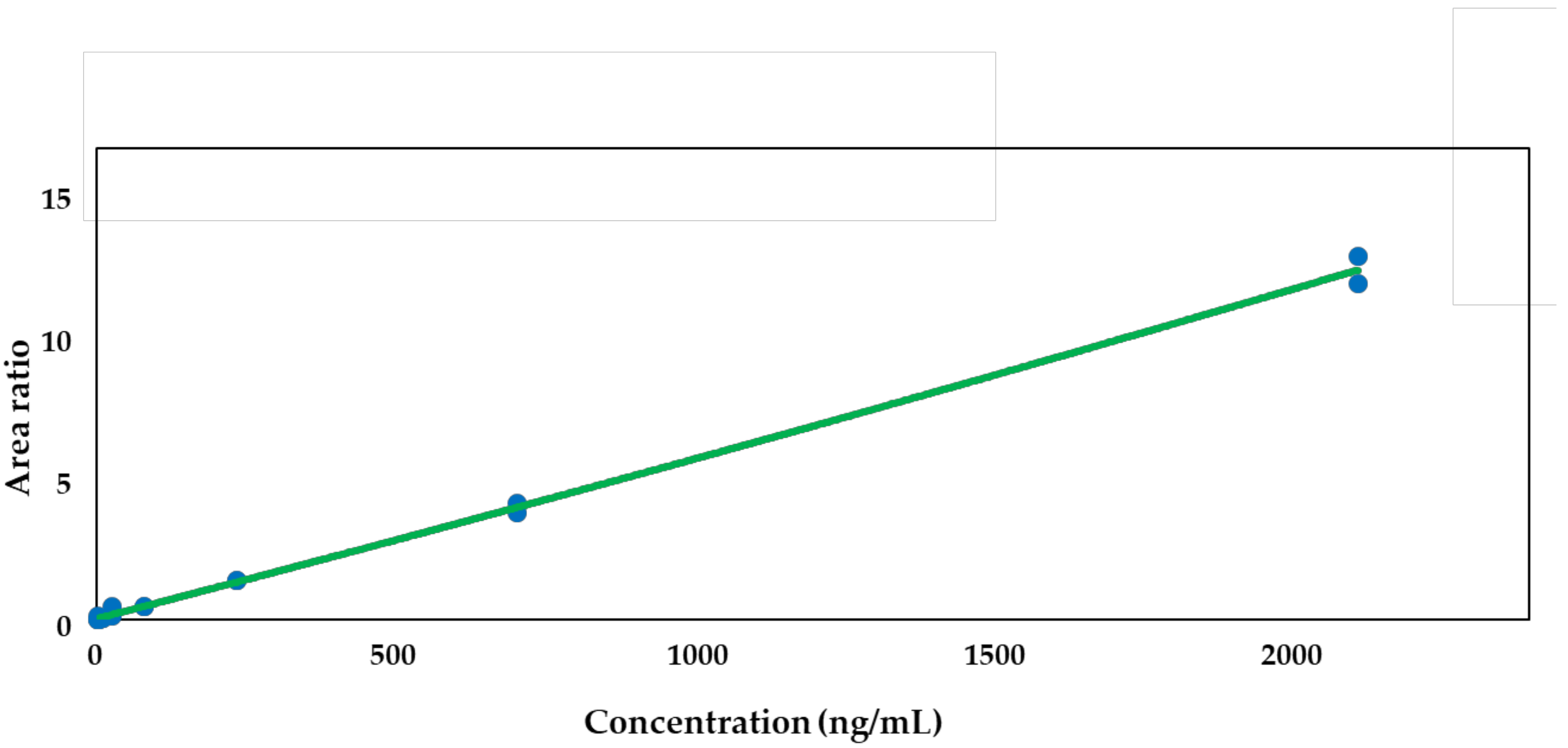
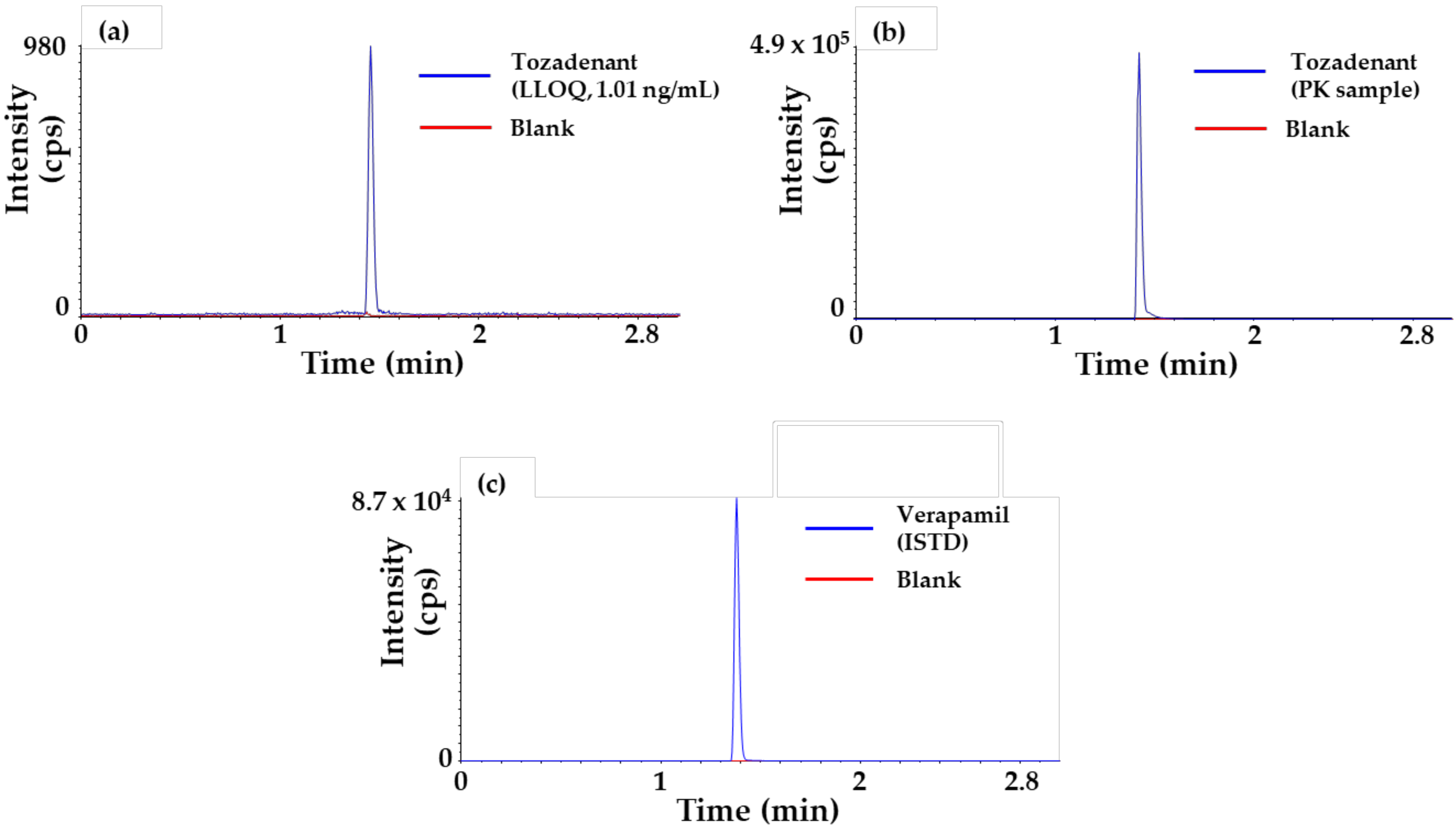
2.1. Method Qualification x
2.2. In Vitro Experiments
2.2.1. Plasma Protein Binding
2.2.2. Microsomal Metabolic Stability
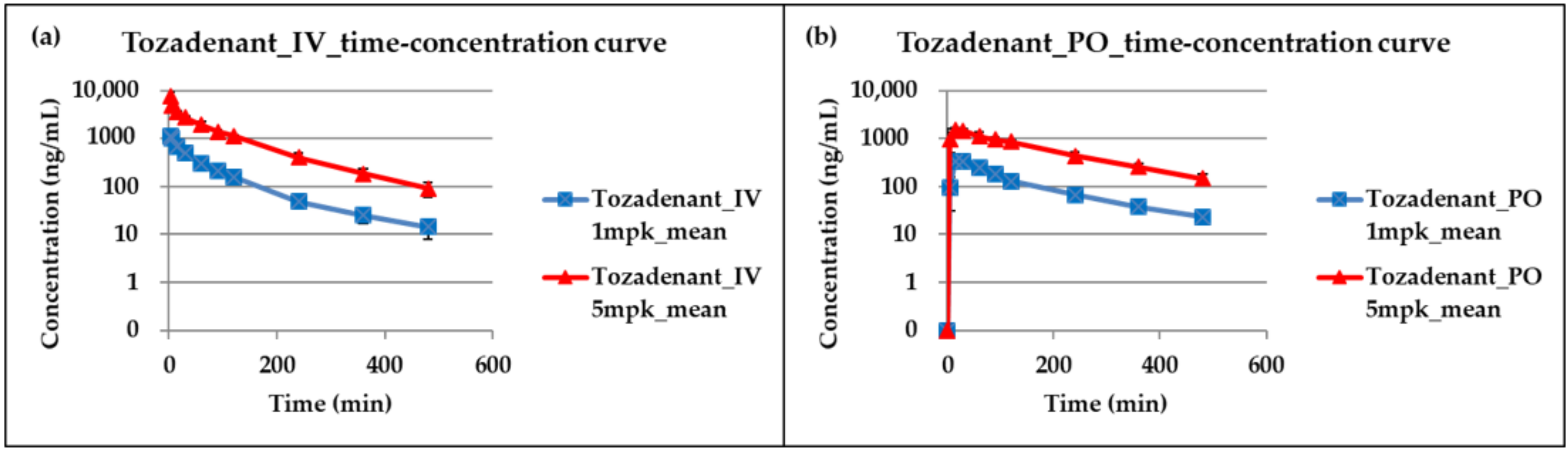
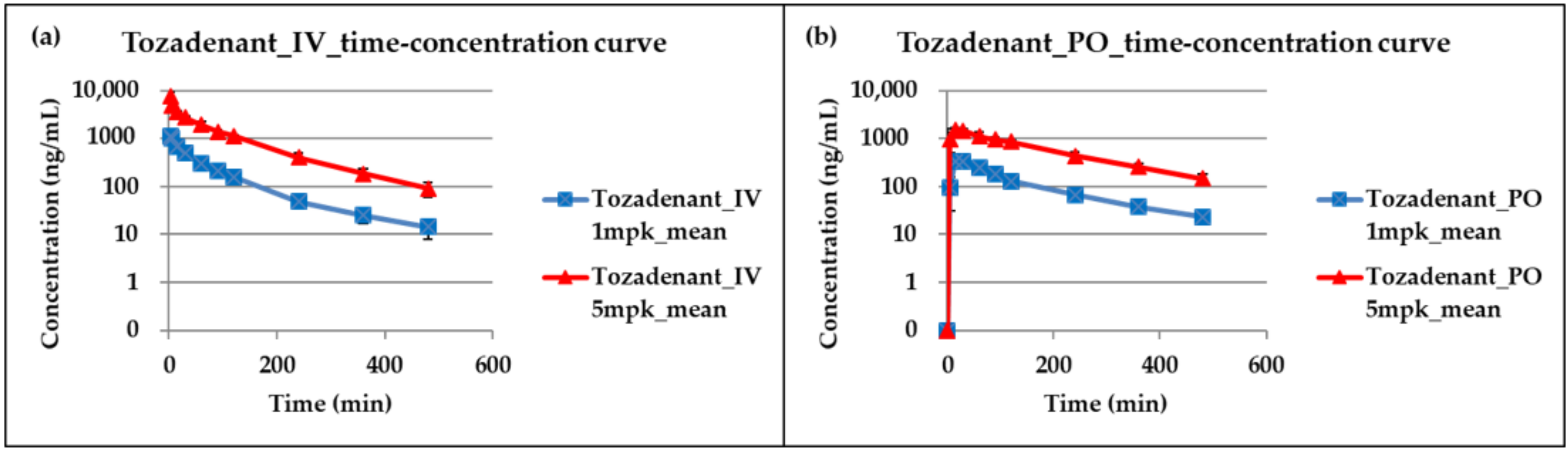
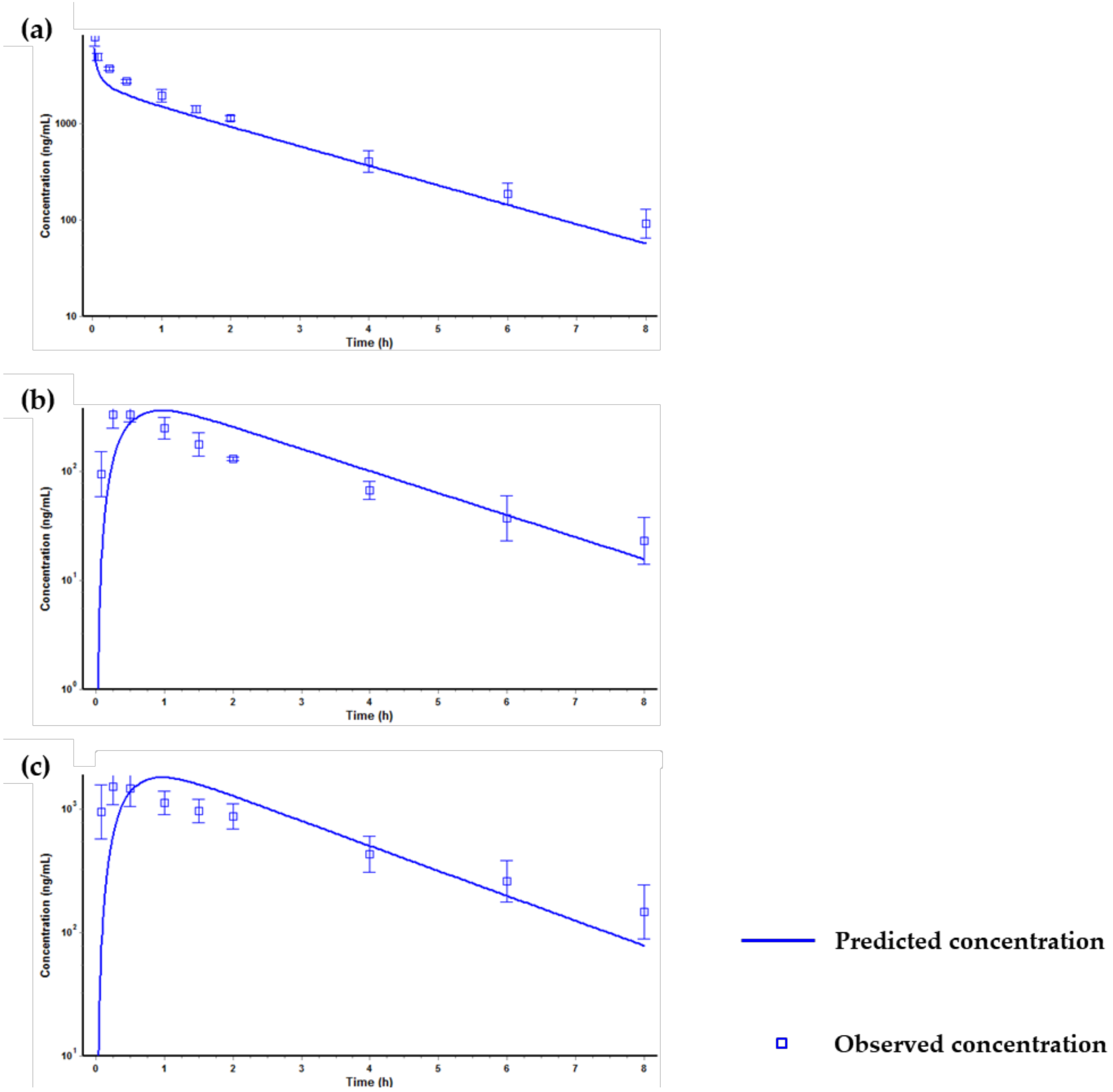
2.3. Application for a Pharmacokinetic Study in Rats
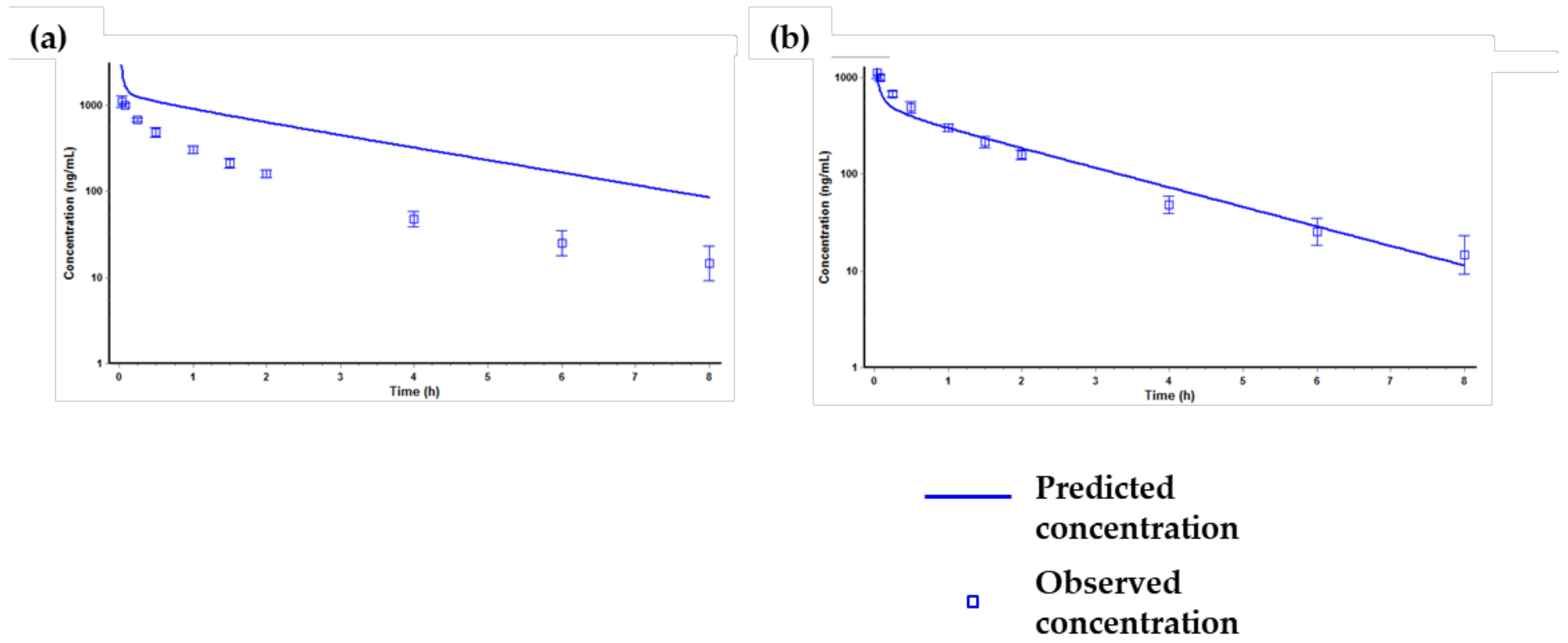
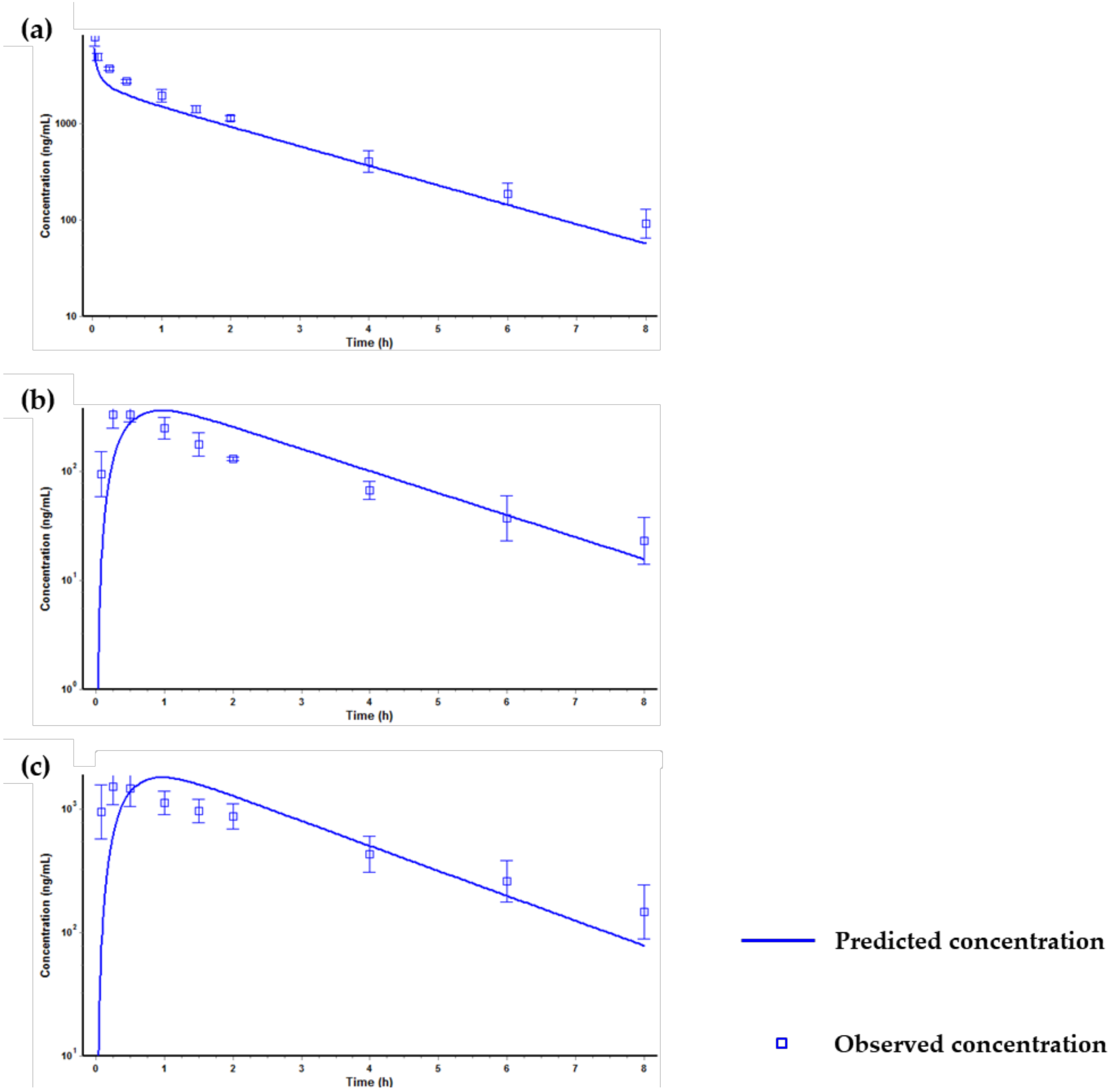
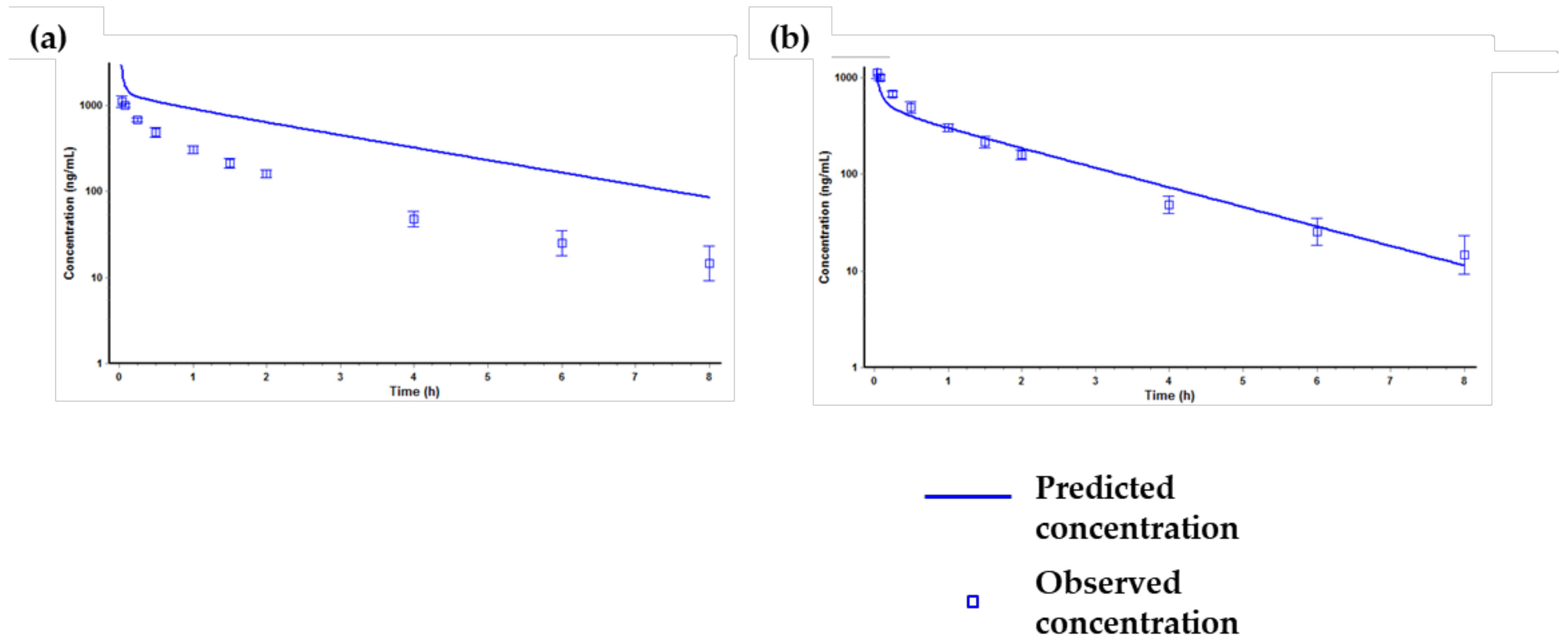
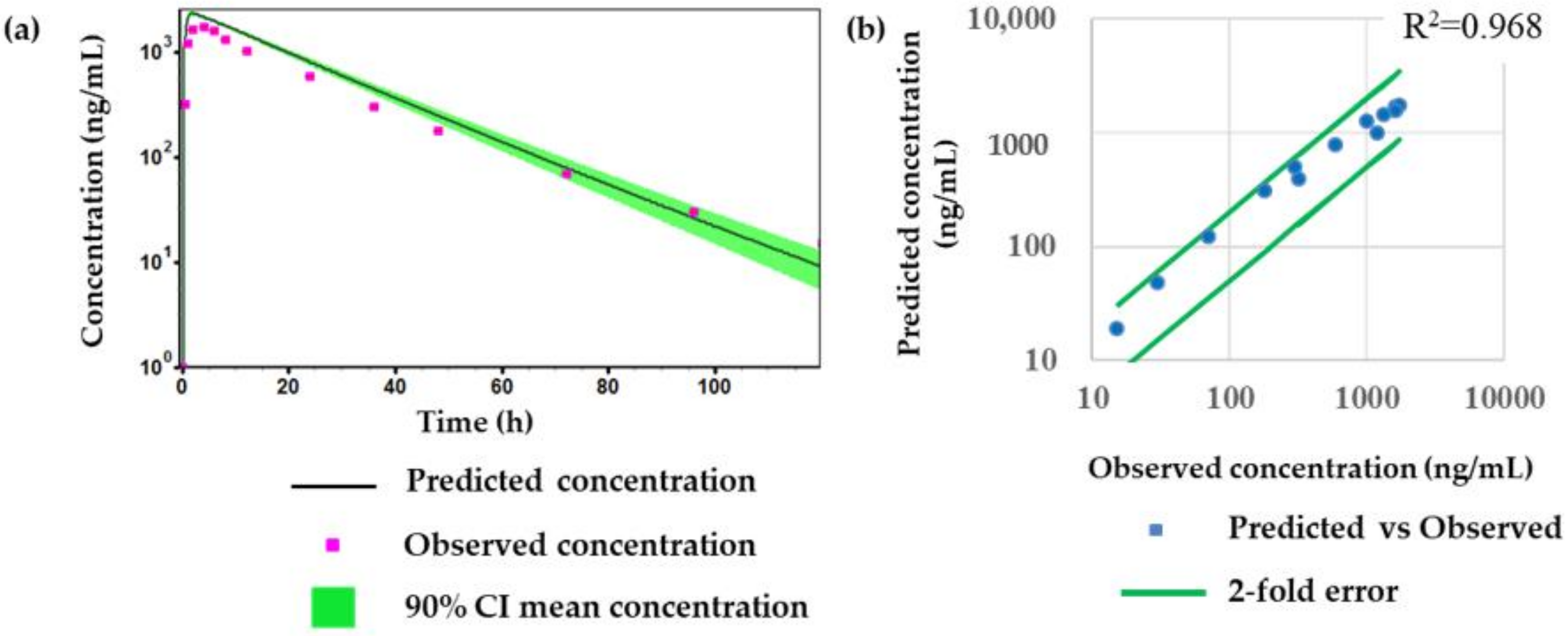
2.4. Prediction of Plasma Concentration-Time Profiles Using the PBPK Model
3. Discussion and Conclusions
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Preparation of Stock Solution, Calibration Standard, Qaulity Control, and Internal Standard
4.3. Sample Preparation
4.4. LC-MS/MS Conditions
4.5. Method Qualification
4.6. In Vitro Experiments
4.6.1. Plasma Protein Binding
4.6.2. Microsomal Metabolic Stability
4.7. Application for a Pharmacokinetic Study in Rat
4.8. Pharmacokinetic Data Analysis
4.9. Prediction of Plasma Concentration-Time Profiles Using the PBPK Model
Author Contributions
Funding
Conflicts of Interest
References
- Pinna, A.; Volpini, R.; Cristalli, G.; Morelli, M. New adenosine a2a receptor antagonists: Actions on Parkinson’s disease models. Eur. J. Pharmacol. 2005, 512, 157–164. [Google Scholar] [CrossRef]
- Richardson, P.J.; Kase, H.; Jenner, P.G. Adenosine a2a receptor antagonists as new agents for the treatment of Parkinson’s disease. Trends Pharmacol. Sci. 1997, 18, 338–344. [Google Scholar] [CrossRef]
- Hauser, R.A.; Olanow, C.W.; Kieburtz, K.D.; Pourcher, E.; Docu-Axelerad, A.; Lew, M.; Kozyolkin, O.; Neale, A.; Resburg, C.; Meya, U.; et al. Tozadenant (syn115) in patients with Parkinson’s disease who have motor fluctuations on levodopa: A phase 2b, double-blind, randomised trial. Lancet. Neurol. 2014, 13, 767–776. [Google Scholar] [CrossRef]
- Coccurello, R.; Breysse, N.; Amalric, M. Simultaneous blockade of adenosine a2a and metabotropic glutamate mglu5 receptors increase their efficacy in reversing parkinsonian deficits in rats. Neuropsychopharmacology 2004, 29, 1451–1461. [Google Scholar] [CrossRef]
- Da Prada, M.; Keller, H.H.; Pieri, L.; Kettler, R.; Haefely, W.E. The pharmacology of Parkinson’s disease: Basic aspects and recent advances. Experientia 1984, 40, 1165–1172. [Google Scholar] [CrossRef]
- Hurley, M.J.; Mash, D.C.; Jenner, P. Adenosine a(2a) receptor mrna expression in Parkinson’s disease. Neurosci. Lett. 2000, 291, 54–58. [Google Scholar] [CrossRef]
- Black, K.J.; Koller, J.M.; Campbell, M.C.; Gusnard, D.A.; Bandak, S.I. Quantification of indirect pathway inhibition by the adenosine a2a antagonist syn115 in Parkinson disease. J. Neurosci. 2010, 30, 16284–16292. [Google Scholar] [CrossRef]
- De Lera Ruiz, M.; Lim, Y.H.; Zheng, J. Adenosine a2a receptor as a drug discovery target. J. Med. Chem. 2014, 57, 3623–3650. [Google Scholar] [CrossRef] [PubMed]
- Fuxe, K.; Ferre, S.; Genedani, S.; Franco, R.; Agnati, L.F. Adenosine receptor-dopamine receptor interactions in the basal ganglia and their relevance for brain function. Physiol. Behav. 2007, 92, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Grondin, R.; Bedard, P.J.; Hadj Tahar, A.; Gregoire, L.; Mori, A.; Kase, H. Antiparkinsonian effect of a new selective adenosine a2a receptor antagonist in mptp-treated monkeys. Neurology 1999, 52, 1673–1677. [Google Scholar] [CrossRef]
- Jenner, P.; Mori, A.; Hauser, R.; Morelli, M.; Fredholm, B.B.; Chen, J.F. Adenosine, adenosine a 2a antagonists, and Parkinson’s disease. Parkinsonism Relat. Disord. 2009, 15, 406–413. [Google Scholar] [CrossRef]
- Kanda, T.; Jackson, M.J.; Smith, L.A.; Pearce, R.K.; Nakamura, J.; Kase, H.; Kuwana, Y.; Jenner, P. Adenosine a2a antagonist: A novel antiparkinsonian agent that does not provoke dyskinesia in parkinsonian monkeys. Ann. Neurol. 1998, 43, 507–513. [Google Scholar] [CrossRef]
- Mancel, V.; Mathy, F.X.; Boulanger, P.; English, S.; Croft, M.; Kenney, C.; Knott, T.; Stockis, A.; Bani, M. Pharmacokinetics and metabolism of [(14)c]-tozadenant (syn-115), a novel a2a receptor antagonist ligand, in healthy volunteers. Xenobiotica 2017, 47, 705–718. [Google Scholar] [CrossRef]
- Morelli, M.; Wardas, J. Adenosine a(2a) receptor antagonists: Potential therapeutic and neuroprotective effects in Parkinson’s disease. Neurotox Res. 2001, 3, 545–556. [Google Scholar] [CrossRef]
- Stromberg, I.; Popoli, P.; Muller, C.E.; Ferre, S.; Fuxe, K. Electrophysiological and behavioural evidence for an antagonistic modulatory role of adenosine a2a receptors in dopamine d2 receptor regulation in the rat dopamine-denervated striatum. Eur. J. Neurosci. 2000, 12, 4033–4037. [Google Scholar] [CrossRef]
- Torvinen, M.; Marcellino, D.; Canals, M.; Agnati, L.F.; Lluis, C.; Franco, R.; Fuxe, K. Adenosine a2a receptor and dopamine d3 receptor interactions: Evidence of functional a2a/d3 heteromeric complexes. Mol. Pharmacol. 2005, 67, 400–407. [Google Scholar] [CrossRef]
- Dasgupta, S.; Ferre, S.; Kull, B.; Hedlund, P.B.; Finnman, U.B.; Ahlberg, S.; Arenas, E.; Fredholm, B.B.; Fuxe, K. Adenosine a2a receptors modulate the binding characteristics of dopamine d2 receptors in stably cotransfected fibroblast cells. Eur. J. Pharmacol. 1996, 316, 325–331. [Google Scholar] [CrossRef]
- Ferre, S.; von Euler, G.; Johansson, B.; Fredholm, B.B.; Fuxe, K. Stimulation of high-affinity adenosine a2 receptors decreases the affinity of dopamine d2 receptors in rat striatal membranes. Proc. Natl. Acad. Sci. USA 1991, 88, 7238–7241. [Google Scholar] [CrossRef]
- Tanganelli, S.; Sandager Nielsen, K.; Ferraro, L.; Antonelli, T.; Kehr, J.; Franco, R.; Ferre, S.; Agnati, L.F.; Fuxe, K.; Scheel-Kruger, J. Striatal plasticity at the network level. Focus on adenosine a2a and d2 interactions in models of Parkinson’s disease. Parkinsonism Relat. Disord. 2004, 10, 273–280. [Google Scholar] [CrossRef]
- Hauser, R.A.; Hubble, J.P.; Truong, D.D.; Istradefylline, U.S.S.G. Randomized trial of the adenosine a(2a) receptor antagonist istradefylline in advanced pd. Neurology 2003, 61, 297–303. [Google Scholar] [CrossRef]
- Chen, J.F.; Xu, K.; Petzer, J.P.; Staal, R.; Xu, Y.H.; Beilstein, M.; Sonsalla, P.K.; Castagnoli, K.; Castagnoli, N., Jr.; Schwarzschild, M.A. Neuroprotection by caffeine and a(2a) adenosine receptor inactivation in a model of Parkinson’s disease. J. Neurosci. 2001, 21, RC143. [Google Scholar] [CrossRef]
- Schwarzschild, M.A.; Agnati, L.; Fuxe, K.; Chen, J.F.; Morelli, M. Targeting adenosine a2a receptors in Parkinson’s disease. Trends Neurosci. 2006, 29, 647–654. [Google Scholar] [CrossRef]
- LeWitt, P.A.; Guttman, M.; Tetrud, J.W.; Tuite, P.J.; Mori, A.; Chaikin, P.; Sussman, N.M.; Group, U.S.S. Adenosine a2a receptor antagonist istradefylline (kw-6002) reduces “off” time in Parkinson’s disease: A double-blind, randomized, multicenter clinical trial (6002-us-005). Ann. Neurol. 2008, 63, 295–302. [Google Scholar] [CrossRef]
- Nonaka, H.; Ichimura, M.; Takeda, M.; Nonaka, Y.; Shimada, J.; Suzuki, F.; Yamaguchi, K.; Kase, H. Kf17837 ((e)-8-(3,4-dimethoxystyryl)-1,3-dipropyl-7-methylxanthine), a potent and selective adenosine a2 receptor antagonist. Eur. J. Pharmacol. 1994, 267, 335–341. [Google Scholar] [CrossRef]
- Ongini, E.; Fredholm, B.B. Pharmacology of adenosine a2a receptors. Trends Pharmacol. Sci. 1996, 17, 364–372. [Google Scholar] [CrossRef]
- Shimada, J.; Suzuki, F.; Nonaka, H.; Ishii, A.; Ichikawa, S. (e)-1,3-dialkyl-7-methyl-8-(3,4,5-trimethoxystyryl)xanthines: Potent and selective adenosine a2 antagonists. J. Med. Chem. 1992, 35, 2342–2345. [Google Scholar] [CrossRef]
- Jenner, P. An overview of adenosine a2a receptor antagonists in Parkinson’s disease. Int. Rev. Neurobiol. 2014, 119, 71–86. [Google Scholar]
- Barret, O.; Hannestad, J.; Alagille, D.; Vala, C.; Tavares, A.; Papin, C.; Morley, T.; Fowles, K.; Lee, H.; Seibyl, J.; et al. Adenosine 2a receptor occupancy by tozadenant and preladenant in rhesus monkeys. J. Nucl. Med. 2014, 55, 1712–1718. [Google Scholar] [CrossRef]
- Chen, Y.; Jin, J.Y.; Mukadam, S.; Malhi, V.; Kenny, J.R. Application of ivive and pbpk modeling in prospective prediction of clinical pharmacokinetics: Strategy and approach during the drug discovery phase with four case studies. Biopharm. Drug Dispos. 2012, 33, 85–98. [Google Scholar] [CrossRef]
- Jones, H.M.; Gardner, I.B.; Watson, K.J. Modelling and pbpk simulation in drug discovery. AAPS J. 2009, 11, 155–166. [Google Scholar] [CrossRef]
- Lave, T.; Parrott, N.; Grimm, H.P.; Fleury, A.; Reddy, M. Challenges and opportunities with modelling and simulation in drug discovery and drug development. Xenobiotica 2007, 37, 1295–1310. [Google Scholar] [CrossRef]
- Rowland, M.; Peck, C.; Tucker, G. Physiologically-based pharmacokinetics in drug development and regulatory science. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 45–73. [Google Scholar] [CrossRef]
- Poulin, P.; Jones, R.D.; Jones, H.M.; Gibson, C.R.; Rowland, M.; Chien, J.Y.; Ring, B.J.; Adkison, K.K.; Ku, M.S.; He, H.; et al. Phrma cpcdc initiative on predictive models of human pharmacokinetics, part 5: Prediction of plasma concentration-time profiles in human by using the physiologically-based pharmacokinetic modeling approach. J. Pharm. Sci. 2011, 100, 4127–4157. [Google Scholar] [CrossRef]
- Shardlow, C.E.; Generaux, G.T.; Patel, A.H.; Tai, G.; Tran, T.; Bloomer, J.C. Impact of physiologically based pharmacokinetic modeling and simulation in drug development. Drug Metab. Dispos. 2013, 41, 1994–2003. [Google Scholar] [CrossRef]
- Yang, F.; Wang, B.; Liu, Z.; Xia, X.; Wang, W.; Yin, D.; Sheng, L.; Li, Y. Prediction of a therapeutic dose for buagafuran, a potent anxiolytic agent by physiologically based pharmacokinetic/pharmacodynamic modeling starting from pharmacokinetics in rats and human. Front. Pharmacol. 2017, 8, 683. [Google Scholar] [CrossRef]
- Allan, G.; Davis, J.; Dickins, M.; Gardner, I.; Jenkins, T.; Jones, H.; Webster, R.; Westgate, H. Pre-clinical pharmacokinetics of uk-453,061, a novel non-nucleoside reverse transcriptase inhibitor (nnrti), and use of in silico physiologically based prediction tools to predict the oral pharmacokinetics of uk-453,061 in man. Xenobiotica 2008, 38, 620–640. [Google Scholar] [CrossRef]
- Brochot, A.; Zamacona, M.; Stockis, A. Physiologically based pharmacokinetic/pharmacodynamic animal-to-man prediction of therapeutic dose in a model of epilepsy. Basic Clin. Pharmacol. Toxicol. 2010, 106, 256–262. [Google Scholar] [CrossRef]
- Bungay, P.J.; Tweedy, S.; Howe, D.C.; Gibson, K.R.; Jones, H.M.; Mount, N.M. Preclinical and clinical pharmacokinetics of pf-02413873, a nonsteroidal progesterone receptor antagonist. Drug Metab. Dispos. 2011, 39, 1396–1405. [Google Scholar] [CrossRef]
- De Buck, S.S.; Sinha, V.K.; Fenu, L.A.; Nijsen, M.J.; Mackie, C.E.; Gilissen, R.A. Prediction of human pharmacokinetics using physiologically based modeling: A retrospective analysis of 26 clinically tested drugs. Drug Metab. Dispos. 2007, 35, 1766–1780. [Google Scholar] [CrossRef]
- Jones, H.M.; Parrott, N.; Jorga, K.; Lave, T. A novel strategy for physiologically based predictions of human pharmacokinetics. Clin. Pharmacokinet. 2006, 45, 511–542. [Google Scholar] [CrossRef]
- Wang, B.; Liu, Z.; Li, D.; Yang, S.; Hu, J.; Chen, H.; Sheng, L.; Li, Y. Application of physiologically based pharmacokinetic modeling in the prediction of pharmacokinetics of bicyclol controlled-release formulation in human. Eur. J. Pharm. Sci. 2015, 77, 265–272. [Google Scholar] [CrossRef]
- Gao, Z.W.; Zhu, Y.T.; Yu, M.M.; Zan, B.; Liu, J.; Zhang, Y.F.; Chen, X.Y.; Li, X.N.; Zhong, D.F. Preclinical pharmacokinetics of tpn729ma, a novel pde5 inhibitor, and prediction of its human pharmacokinetics using a pbpk model. Acta Pharmacol. Sin. 2015, 36, 1528–1536. [Google Scholar] [CrossRef]
- Shou, M. Prediction of pharmacokinetics and drug-drug interactions from in vitro metabolism data. Curr. Opin. Drug Discov. Devel. 2005, 8, 66–77. [Google Scholar]
- Jones, H.M.; Dickins, M.; Youdim, K.; Gosset, J.R.; Attkins, N.J.; Hay, T.L.; Gurrell, I.K.; Logan, Y.R.; Bungay, P.J.; Jones, B.C.; et al. Application of pbpk modelling in drug discovery and development at pfizer. Xenobiotica 2012, 42, 94–106. [Google Scholar] [CrossRef]
- Sinha, V.K.; Snoeys, J.; Osselaer, N.V.; Peer, A.V.; Mackie, C.; Heald, D. From preclinical to human--prediction of oral absorption and drug-drug interaction potential using physiologically based pharmacokinetic (pbpk) modeling approach in an industrial setting: A workflow by using case example. Biopharm. Drug Dispos. 2012, 33, 111–121. [Google Scholar] [CrossRef]
- Naritomi, Y.; Terashita, S.; Kimura, S.; Suzuki, A.; Kagayama, A.; Sugiyama, Y. Prediction of human hepatic clearance from in vivo animal experiments and in vitro metabolic studies with liver microsomes from animals and humans. Drug Metab. Dispos. 2001, 29, 1316–1324. [Google Scholar]
- Obach, R.S. Nonspecific binding to microsomes: Impact on scale-up of in vitro intrinsic clearance to hepatic clearance as assessed through examination of warfarin, imipramine, and propranolol. Drug Metab. Dispos. 1997, 25, 1359–1369. [Google Scholar]
- Obach, R.S. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: An examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab. Dispos. 1999, 27, 1350–1359. [Google Scholar]
Sample Availability: Samples of the compounds are commercially available at MedChem Express (Monmouth Junction, NJ, USA) or ther vendors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Run Number | Statistics | QC Low (15.04 ng/mL) | QC Medium (165.46 ng/mL) | QC High (1820 ng/mL) |
| 1 | Mean n % Acc % CV | 15.94 3 106.01 6.81 | 171.91 3 103.90 1.32 | 1802.27 3 99.03 4.62 |
| 2 | Mean n % Acc % CV | 14.95 3 99.38 6.19 | 157.76 3 95.34 1.13 | 1684.91 3 92.58 0.24 |
| 3 | Mean n % Acc % CV | 15.32 3 101.88 4.34 | 163.48 3 98.80 1.78 | 1807.70 3 99.32 0.51 |
| Inter-run | Mean n % Acc % CV | 15.40 9 102.42 5.84 | 164.38 9 99.35 3.95 | 1764.96 9 96.98 4.15 |
| Statistics | Dilution QC (6600 ng/mL) |
|---|---|
| Mean | 6268.4 |
| n | 3 |
| % Acc | 94.98 |
| % CV | 4.3 |
| Assessment | Statistics | QC Low (15.04 ng/mL) | QC Medium (165.46 ng/mL) | QC High (1820 ng/mL) |
|---|---|---|---|---|
| Short-term (room temperature, 12 h) | Mean n % Acc % CV | 16.48 3 109.6 5.9 | 165.6 3 100.09 1.91 | 1804.08 3 99.13 2.9 |
| Long-term (−20 °C, 14 days) | Mean n % Acc % CV | 14.87 3 98.87 2.1 | 156.13 3 94.36 3.87 | 1648.15 3 90.56 0.34 |
| Freeze-thaw (−20 °C, 3 cycles) | Mean n % Acc % CV | 14.68 3 97.63 6.64 | 155.64 3 94.07 6.18 | 1733.81 3 95.26 1.47 |
| Stock (−20 °C, 28 days) | Mean n % Acc % CV | 15.65 3 104.08 4.41 | 175.44 3 106.03 2.68 | 1814.65 3 99.71 4.31 |
| Species | Clint, in vitro (mL/min/mg) | Clint (mL/min/mg) | ClH (mL/min/mg) |
|---|---|---|---|
| Rat | 0.0021 ±0.0003 | 3.78 ±0.67 | 3.53 ±0.58 |
| Human | 0.0008 ±0.0002 | 0.99 ±0.23 | 0.95 ±0.20 |
| PK (Pharmokinetic) Study | Dose (mg/kg) | T1/2 (min) | Tmax (min) | Cmax (ng/mL) | AUClast (min × ng/mL) | AUCINF (min × ng/mL) | Clearancev(Cl) (mL/min/kg) | Volume of Distribution(Vd) (L/kg) |
|---|---|---|---|---|---|---|---|---|
| IV | 1 | 139.26 ±40.05 | 2 | 1118.92 ±164.61 | 67142.34 ±6521.56 | 70342.35 ±7723.57 | 14.36 ±1.71 | 1.63 ±0.33 |
| 5 | 99.69 ±19.07 | 2 | 7820.27 ±1627.22 | 431241.43 ±38843.52 | 445005.24 ±45762.56 | 11.31 ±1.13 | 1.28 ±0.09 | |
| PO | 1 | 147.91 ±57.85 | 22.5 ±8.66 | 368.97 ±60.06 | 48958.98 ±5784.28 | 54665.08 ±10852.83 | ||
| 5 | 144.84 ±42.76 | 27.5 ±23.98 | 1666.3 ±448.52 | 279450.32 ±64941.07 | 313549.48 ±88341.03 |
| Parameters | Values |
|---|---|
| Molecular weight (g/mol) | 406.5 |
| pKa 1 | 3.28, 4.7, 10.81 |
| Log P 1 | 1.96 |
| Permeability (cm2/s) 1 | 1.62 |
| Solubility at pH 7 (mg/mL) 1 | 0.28 |
| Blood/plasma concentration ratio (Rbp) 1 in rat and human 1 | 0.82 |
| Unbound fraction (Fup) in rat and human (%) 2 | 26.63, 26.72 |
| Clint in vitro in rat and human (mL/min/mg) 2 | 0.0021, 0.0008 |
| PK Parameter | AUClast (µg × h/mL) | Cmax (µg/mL) | Tmax (h) | T1/2 (h) |
|---|---|---|---|---|
| Observed | 35.0 | 1.74 | 4 | 15 |
| Predicted | 49.6 | 1.8 | 2.8 | 17.4 |
| Prediction fold error | 1.4 | 1.0 | 0.7 | 1.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, B.i.; Park, M.-H.; Shin, S.-H.; Byeon, J.-J.; Park, Y.; Kim, N.; Choi, J.; Shin, Y.G. Quantitative Analysis of Tozadenant Using Liquid Chromatography-Mass Spectrometric Method in Rat Plasma and Its Human Pharmacokinetics Prediction Using Physiologically Based Pharmacokinetic Modeling. Molecules 2019, 24, 1295. https://doi.org/10.3390/molecules24071295
Lee Bi, Park M-H, Shin S-H, Byeon J-J, Park Y, Kim N, Choi J, Shin YG. Quantitative Analysis of Tozadenant Using Liquid Chromatography-Mass Spectrometric Method in Rat Plasma and Its Human Pharmacokinetics Prediction Using Physiologically Based Pharmacokinetic Modeling. Molecules. 2019; 24(7):1295. https://doi.org/10.3390/molecules24071295
Chicago/Turabian StyleLee, Byeong ill, Min-Ho Park, Seok-Ho Shin, Jin-Ju Byeon, Yuri Park, Nahye Kim, Jangmi Choi, and Young G. Shin. 2019. "Quantitative Analysis of Tozadenant Using Liquid Chromatography-Mass Spectrometric Method in Rat Plasma and Its Human Pharmacokinetics Prediction Using Physiologically Based Pharmacokinetic Modeling" Molecules 24, no. 7: 1295. https://doi.org/10.3390/molecules24071295
APA StyleLee, B. i., Park, M.-H., Shin, S.-H., Byeon, J.-J., Park, Y., Kim, N., Choi, J., & Shin, Y. G. (2019). Quantitative Analysis of Tozadenant Using Liquid Chromatography-Mass Spectrometric Method in Rat Plasma and Its Human Pharmacokinetics Prediction Using Physiologically Based Pharmacokinetic Modeling. Molecules, 24(7), 1295. https://doi.org/10.3390/molecules24071295