
Microwave-Driven Synthesis of Iron-Oxide Nanoparticles for Molecular Imaging



Abstract
1. Introduction
1.1. Molecular Imaging
1.1.1. Optical Imaging
1.1.2. Photoacoustic Imaging
1.1.3. Nuclear Imaging
1.1.4. Magnetic Resonance Imaging
1.1.5. Magnetic Particle Imaging
1.2. Iron-Oxide Nanoparticles for Molecular Imaging
1.3. Traditional Synthesis of Iron-Oxide Nanoparticles
1.3.1. Coprecipitation
1.3.2. Thermal Decomposition
1.3.3. Hydrothermal and Solvothermal Synthesis
1.3.4. Pyrolysis
2. Microwave Synthesis of Nanoparticles
2.1. Metallic Nanostructures
2.2. Metal Oxides
2.3. Metal Chalcogenides
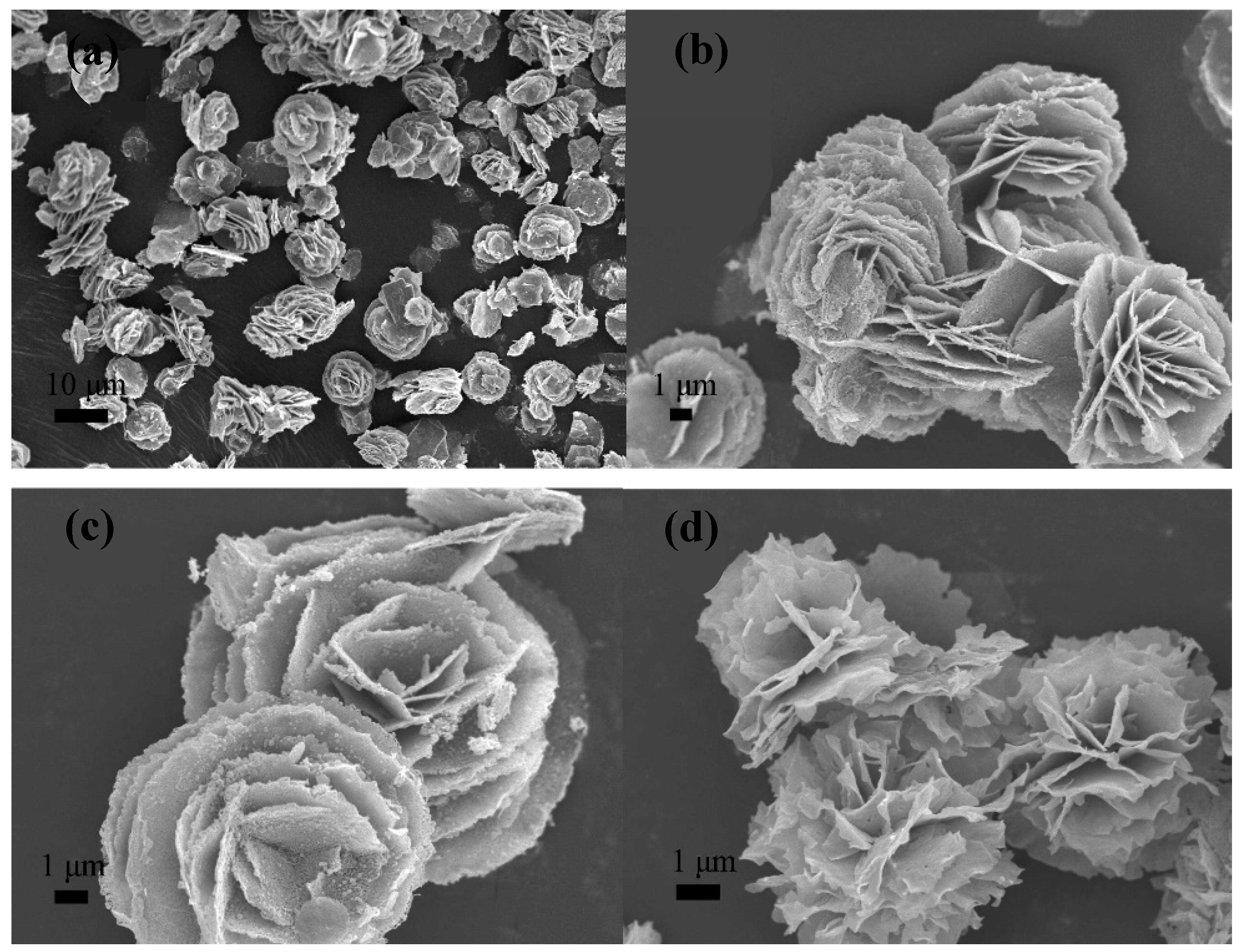
3. Microwave Synthesis of Iron-Oxide Nanoparticles
3.1. From Seconds to Less Than Five Minutes
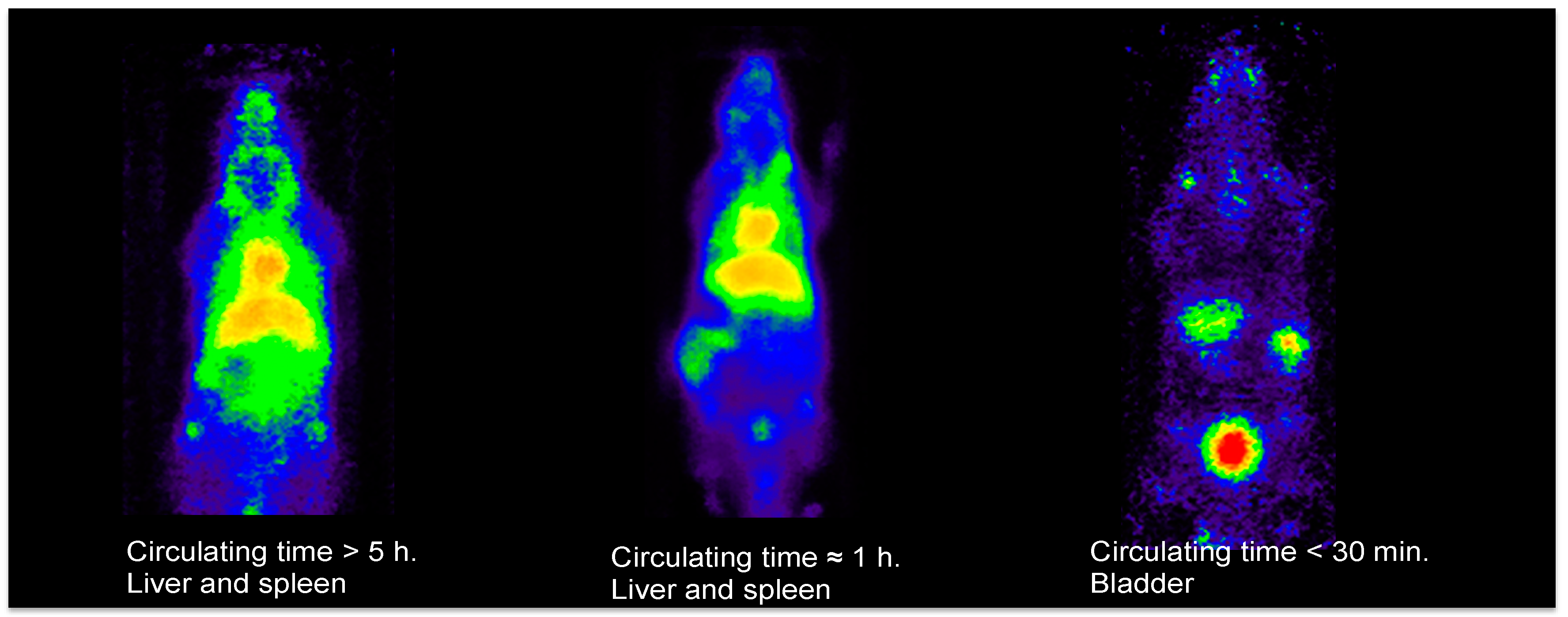
3.2. Less Than 30 Minutes
3.3. One Hour and More
4. Applications of Microwave-Produced Iron-Oxide Nanoparticles in Molecular Imaging
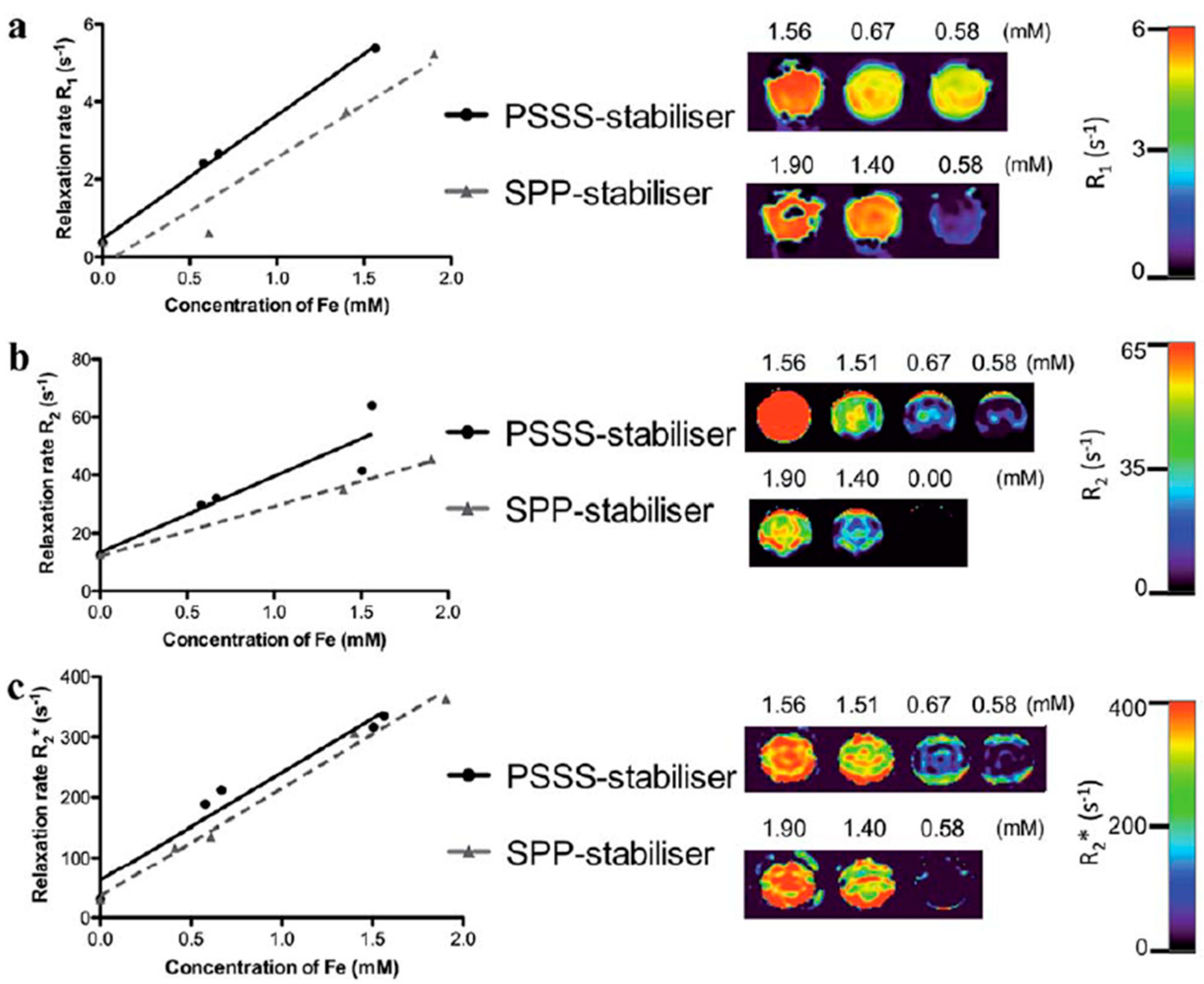
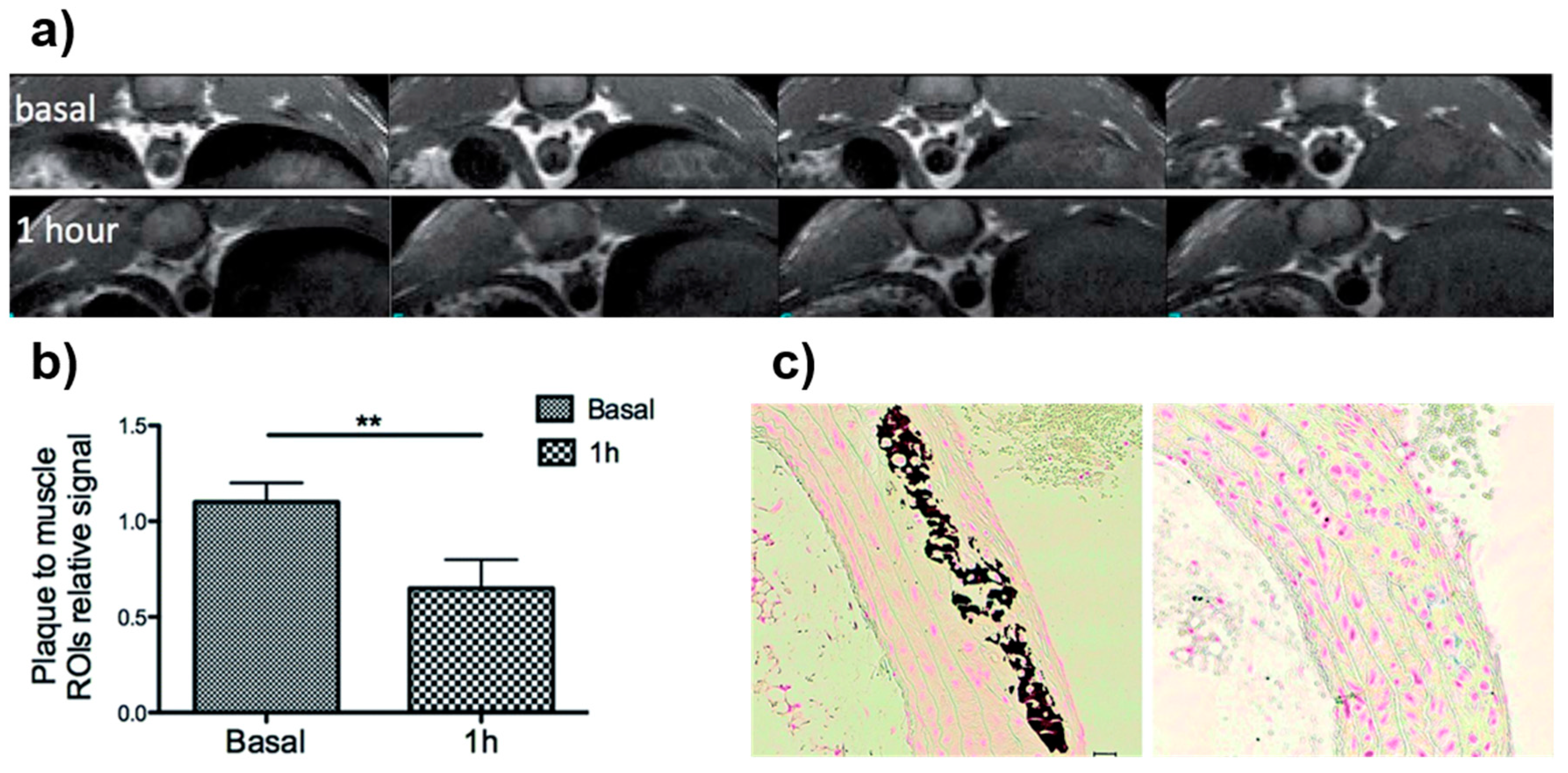
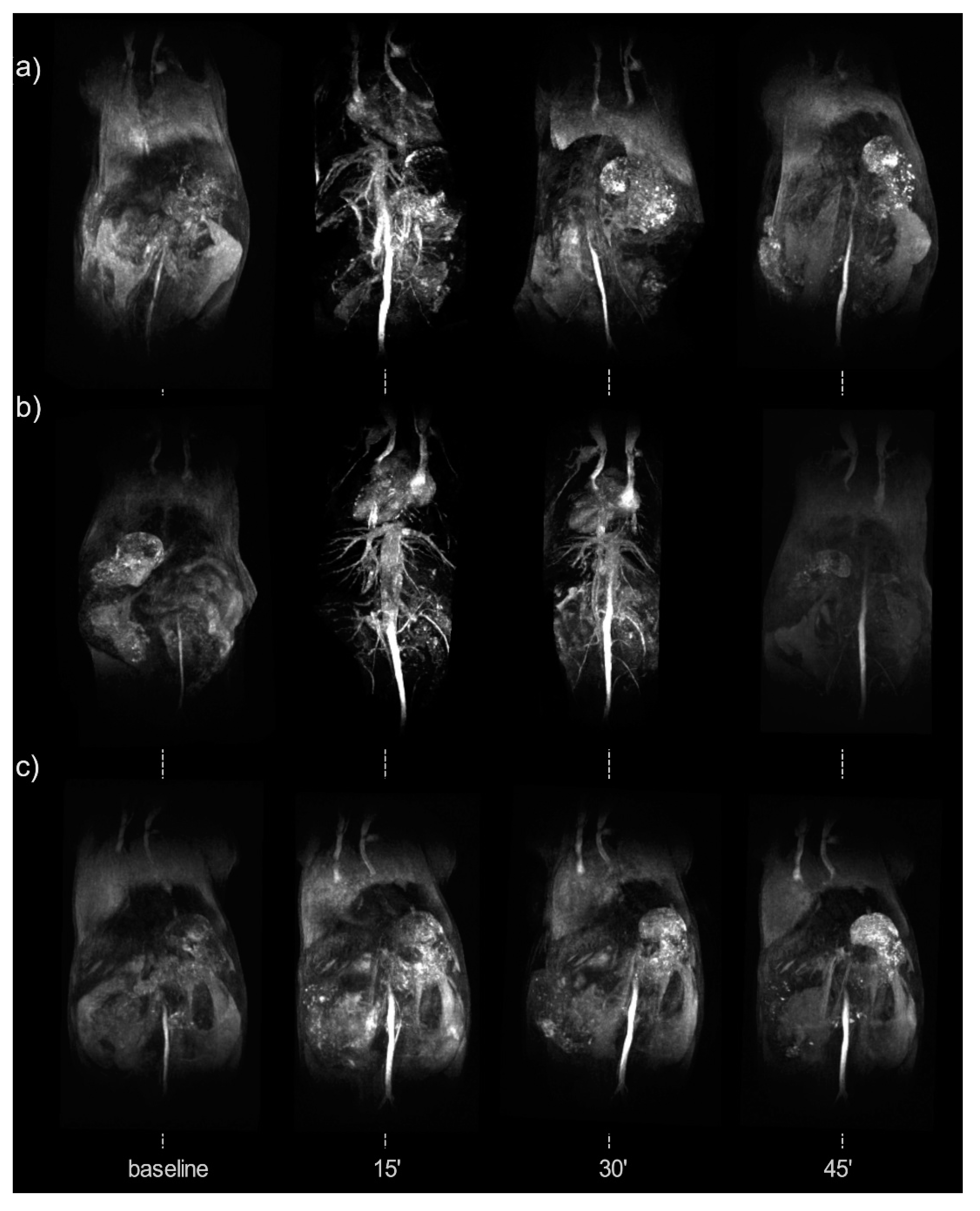
4.1. Iron-Oxide Nanoparticles as T2 Contrast Agents
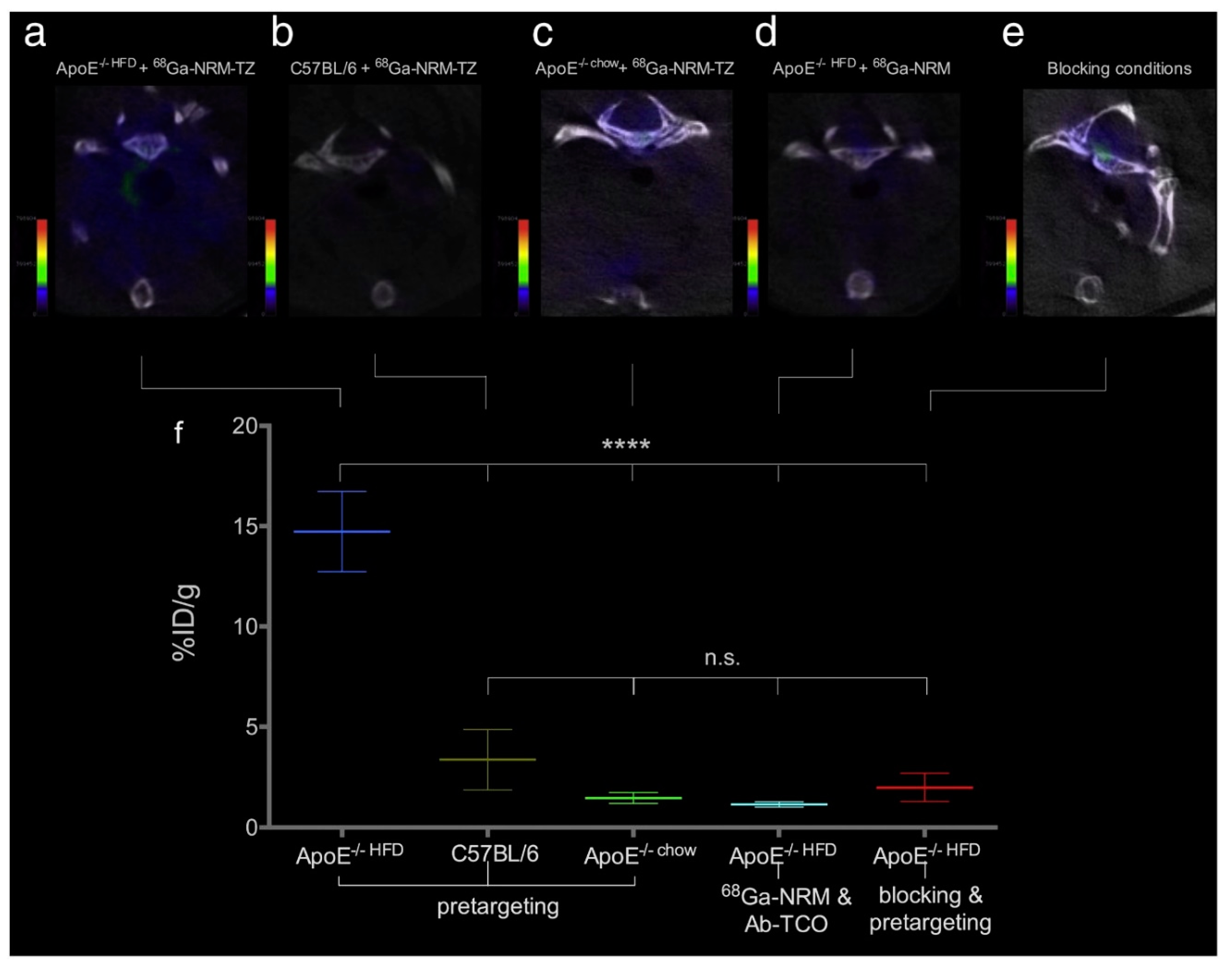
4.2. Iron-Oxide Nanoparticles as T1 Contrast Agents
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| α-Fe2O3 | Hematite |
| γ-Fe2O3 | Maghemite |
| [18F]FDG | 18F-fluorodeoxyglucose |
| AuNPs | Gold nanoparticles |
| CRD | Curdlan |
| CT | Computed tomography |
| DES | Deep eutectic solvent |
| DLS | Dynamic light scattering |
| DMSA | Dimercaptosuccinic acid |
| DMSO | Dimethyl sulfoxide |
| EDX | Energy-dispersive X-ray spectroscopy |
| FBS | Fetal bovine serum |
| Fe3O4 | Magnetite |
| FTIR | Fourier-transform infrared spectroscopy |
| IONPs | Iron-oxide nanoparticles |
| MI | Molecular imaging |
| MPI | Magnetic particle imaging |
| MRI | Magnetic resonance imaging |
| MW | Microwave |
| NPs | Nanoparticles |
| NRM | Nano-radiomaterial |
| NS | Nanospheres |
| OI | Optical imaging |
| PAI | Photoacoustic imaging |
| PET | Positron emission tomography |
| PDI | Polydispersity index |
| QDs | Quantum dots |
| r1 | Longitudinal relaxivity value |
| r2 | Transversal relaxivity value |
| RT | Room temperature |
| SWNT | Singe-walled carbon nanotube |
| SEM | Scanning electron microscopy |
| SPECT | Single-photon computed tomography |
| SQUID | Superconducting quantum interference device |
| STEM-HAADF | High-angle annular dark field scanning transmission electron microscopy |
| T1 | Longitudinal relaxation time |
| T2 | Transversal relaxation time |
| TCO | trans-cyclooctene |
| TGA | Thermogravimetric analysis |
| TEM | Transmission electron microscopy |
| VSM | Vibrating sample magnetometer |
| XRF | X-ray fluorescence spectroscopy |
| XPS | X-ray photoelectron spectroscopy |
| XRD | X-ray diffraction |
References
- Giguere, R.J.; Bray, T.L.; Duncan, S.M.; Majetich, G. Application of commercial microwave ovens to organic synthesis. Tetrahedron Lett. 1986, 27, 4945–4948. [Google Scholar] [CrossRef]
- Schmidt, B.; Riemer, M.; Karras, M. 2,2′-Biphenols via protecting group-free thermal or microwave-accelerated Suzuki–Miyaura coupling in water. J. Org. Chem. 2013, 78, 8680–8688. [Google Scholar] [CrossRef]
- Horikoshi, S.; Serpone, N. (Eds.) Microwaves in Nanoparticle Synthesis: Fundamentals and Applications; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar] [CrossRef]
- Koyama, Y.; Hama, Y.; Urano, Y.; Nguyen, D.M.; Choyke, P.L.; Kobayashi, H. Spectral fluorescence molecular imaging of lung metastases targeting HER2/neu. Clin. Cancer Res. 2007, 13, 2936–2945. [Google Scholar] [CrossRef]
- Bremer, C.; Bredow, S.; Mahmood, U.; Weissleder, R.; Tung, C.-H. Optical imaging of matrix metalloproteinase–2 activity in tumors: feasibility study in a mouse model. Radiology 2001, 221, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Raymond, S.B.; Skoch, J.; Hills, I.D.; Nesterov, E.E.; Swager, T.M.; Bacskai, B.J. Smart optical probes for near-infrared fluorescence imaging of Alzheimer’s disease pathology. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Maslov, K.; Zhang, H.F.; Hu, S.; Wang, L.V. Optical-resolution photoacoustic microscopy for in vivo imaging of single capillaries. Opt. Lett. 2008, 33, 929. [Google Scholar] [CrossRef] [PubMed]
- Zerda, A.D.L.; Liu, Z.; Bodapati, S.; Teed, R.; Vaithilingam, S.; Khuri-Yakub, B.T.; Chen, X.; Dai, H.; Gambhir, S.S. Ultrahigh sensitivity carbon nanotube agents for photoacoustic molecular imaging in living mice. Nano Lett. 2010, 10, 2168–2172. [Google Scholar] [CrossRef] [PubMed]
- Song, K.H.; Kim, C.; Cobley, C.M.; Xia, Y.; Wang, L.V. Noninvasive photoacoustic sentinel lymph node mapping using Au nanocages as a lymph node tracer in a rat model. In Proceedings of the Photons Plus Ultrasound: Imaging and Sensing, San Jose, CA, USA, 24–29 February 2009; Oraevsky, A.A., Wang, L.V., Eds.; SPIE BiOS: San Jose, CA, USA, 2009; p. 71772L. [Google Scholar] [CrossRef]
- Fernández-Barahona, I.; Muñoz-Hernando, M.; Pellico, J.; Ruiz-Cabello, J.; Herranz, F. Molecular imaging with 68Ga radio-nanomaterials: Shedding light on nanoparticles. Appl. Sci. 2018, 8, 1098. [Google Scholar] [CrossRef]
- Rowland, D.J.; Lewis, J.S.; Welch, M.J. Molecular imaging: The application of small animal positron emission tomography. J. Cell. Biochem. 2002, 87, 110–115. [Google Scholar] [CrossRef]
- Lin, T.-J.; Huang, C.-C.; Wang, I.-J.; Lin, J.-W.; Hung, K.-S.; Fan, L.; Tsao, H.-H.; Yang, N.-S.; Lin, K.-J. Validation of an Animal FDG PET Imaging System for Study of Glioblastoma Xenografted Mouse and Rat Models. He Zi Yi Xue Za Zhi 2010, 23, 77–83. [Google Scholar] [CrossRef]
- Lee, S.; Kang, S.-W.; Ryu, J.H.; Na, J.H.; Lee, D.-E.; Han, S.J.; Kang, C.M.; Choe, Y.S.; Lee, K.C.; Leary, J.F.; et al. Tumor-homing glycol chitosan-based optical/PET dual imaging nanoprobe for cancer diagnosis. Bioconjug. Chem. 2014, 25, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Leeper, N.J.; Park, S.-M.; Smith, B.R. High-density lipoprotein nanoparticle imaging in atherosclerotic vascular disease central illustration schematic of HDL nanoparticles used for PET imaging. JACC Basic Transl. Sci. 2017, 2, 98–100. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Medina, C.; Binderup, T.; Lobatto, M.E.; Tang, J.; Calcagno, C.; Giesen, L.; Wessel, C.H.; Witjes, J.; Ishino, S.; Baxter, S.; et al. In vivo PET imaging of HDL in multiple atherosclerosis models. JACC Cardiovasc. Imaging 2016, 9, 950–961. [Google Scholar] [CrossRef]
- Pomper, M.; Lee, J. Small animal imaging in drug development. Curr. Pharm. Des. 2005, 11, 3247–3272. [Google Scholar] [CrossRef]
- Lechuga-Vieco, A.V.; Groult, H.; Pellico, J.; Mateo, J.; Enríquez, J.A.; Ruiz-Cabello, J.; Herranz, F. Protein corona and phospholipase activity drive selective accumulation of nanomicelles in atherosclerotic plaques. Nanomed. Nanotech. Biol. Med. 2018, 14, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Pellico, J.; Lechuga-Vieco, A.V.V.; Benito, M.; García-Segura, J.M.M.; Fuster, V.; Ruiz-Cabello, J.; Herranz, F. Microwave-driven synthesis of bisphosphonate nanoparticles allows in vivo visualisation of atherosclerotic plaque. RSC Adv. 2015, 5, 1661–1665. [Google Scholar] [CrossRef]
- Sun, C.; Veiseh, O.; Gunn, J.; Fang, C.; Hansen, S.; Lee, D.; Sze, R.; Ellenbogen, R.G.; Olson, J.; Zhang, M. In vivo MRI detection of gliomas by chlorotoxin-conjugated superparamagnetic nanoprobes. Small 2008, 4, 372–379. [Google Scholar] [CrossRef]
- Bauer, L.M.; Situ, S.F.; Griswold, M.A.; Samia, A.C.S. Magnetic particle imaging tracers: State-of-the-art and future directions. J. Phys. Chem. Lett. 2015, 6, 2509–2517. [Google Scholar] [CrossRef]
- Bulte, J.W.M.; Walczak, P.; Janowski, M.; Krishnan, K.M.; Arami, H.; Halkola, A.; Gleich, B.; Rahmer, J. Quantitative “hot-spot” imaging of transplanted stem cells using superparamagnetic tracers and magnetic particle imaging. Tomography 2015, 1, 91–97. [Google Scholar]
- Zheng, B.; Von See, M.P.; Yu, E.; Gunel, B.; Lu, K.; Vazin, T.; Schaffer, D.V.; Goodwill, P.W.; Conolly, S.M. Quantitative magnetic particle imaging monitors the transplantation, biodistribution, and clearance of stem cells in vivo. Theranostics 2016, 6, 291–301. [Google Scholar] [CrossRef]
- Ferguson, R.M.; Khandhar, A.P.; Kemp, S.J.; Arami, H.; Saritas, E.U.; Croft, L.R.; Konkle, J.; Goodwill, P.W.; Halkola, A.; Rahmer, J.; et al. Magnetic particle imaging with tailored iron oxide nanoparticle tracers. IEEE Trans. Med. Imaging 2015, 34, 1077–1084. [Google Scholar] [CrossRef]
- Evertsson, M.; Kjellman, P.; Cinthio, M.; Andersson, R.; Tran, T.A.; in’t Zandt, R.; Grafström, G.; Toftevall, H.; Fredriksson, S.; Ingvar, C.; et al. Combined magnetomotive ultrasound, PET/CT, and MR imaging of 68Ga-labelled superparamagnetic iron oxide nanoparticles in rat sentinel lymph nodes in vivo. Sci. Rep. 2017, 7, 4824. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-Y.; Li, Z.; Chen, K.; Hsu, A.R.; Xu, C.; Xie, J.; Sun, S.; Chen, X. PET/MRI dual-modality tumor imaging using arginine-glycine-aspartic (RGD)–conjugated radiolabeled iron oxide nanoparticles. J. Nucl. Med. 2008, 49, 1371–1379. [Google Scholar] [CrossRef]
- De-Zhi, L.; Hong-Da, C.; Feng, B.; Zhen-Xin, W. Progress of multimodal molecular imaging technology in diagnosis of tumor. Chin. J. Anal. Chem. 2016, 44, 1609–1618. [Google Scholar]
- Santamaria, F.; Montella, S.; Camera, L.; Palumbo, C.; Greco, L.; Boner, A.L. Lung structure abnormalities, but normal lung function in pediatric bronchiectasis. Chest 2006, 130, 480–486. [Google Scholar] [CrossRef]
- Hu, S.; Maslov, K.; Wang, L.V. Noninvasive label-free imaging of microhemodynamics by optical-resolution photoacoustic microscopy. Opt. Express 2009, 17, 7688–7693. [Google Scholar] [CrossRef]
- Zheng, M.; Ruan, S.; Liu, S.; Sun, T.; Qu, D.; Zhao, H.; Xie, Z.; Gao, H.; Jing, X.; Sun, Z. Self-targeting fluorescent carbon dots for diagnosis of brain cancer cells. ACS Nano 2015, 9, 11455–11461. [Google Scholar] [CrossRef]
- Pellico, J.; Ruiz-Cabello, J.; Saiz-Alía, M.; del Rosario, G.; Caja, S.; Montoya, M.; Fernández de Manuel, L.; Morales, M.P.P.; Gutiérrez, L.; Galiana, B.; et al. Fast synthesis and bioconjugation of 68Ga core-doped extremely small iron oxide nanoparticles for PET/MR imaging. Contrast Media Mol. Imaging 2016, 11, 203–210. [Google Scholar] [CrossRef]
- Sarigiannis, Y.; Kolokithas-Ntoukas, A.; Beziere, N.; Zbořil, R.; Papadimitriou, E.; Avgoustakis, K.; Lamprou, M.; Medrikova, Z.; Rousalis, E.; Ntziachristos, V.; et al. Synthesis and evaluation of condensed magnetic nanocrystal clusters with in vivo multispectral optoacoustic tomography for tumour targeting. Biomaterials 2016, 91, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Perlman, O.; Azhari, H. Ultrasonic computed tomography imaging of iron oxide nanoparticles. Phys. Med. Biol. 2017, 62, 825–842. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.M.; Gilbert, D.A.; Liu, K.; Louie, A.Y. Rapid size-controlled synthesis of dextran-coated, 64Cu-doped iron oxide nanoparticles. ACS Nano 2012, 6, 3461–3467. [Google Scholar] [CrossRef]
- Blanco-Andujar, C.; Walter, A.; Cotin, G.; Bordeianu, C.; Mertz, D.; Felder-Flesch, D.; Begin-Colin, S. Design of iron oxide-based nanoparticles for MRI and magnetic hyperthermia. Nanomedicine 2016, 11, 1889–1910. [Google Scholar] [CrossRef] [PubMed]
- Pellico, J.; Ruiz-Cabello, J.; Fernández-Barahona, I.; Gutiérrez, L.; Lechuga-Vieco, A.V.; Enríquez, J.A.; Morales, M.P.; Herranz, F. One-step fast synthesis of nanoparticles for MRI: Coating chemistry as the key variable determining positive or negative contrast. Langmuir 2017, 33, 10239–10247. [Google Scholar] [CrossRef]
- Bulte, J.W.M. Superparamagnetic iron oxides as MPI tracers: A primer and review of early applications. Adv. Drug Deliv. Rev. 2018. [Google Scholar] [CrossRef]
- Xia, B.; Li, J.; Shi, J.; Zhang, Y.; Zhang, Q.; Chen, Z.; Wang, B. Biodegradable and magnetic-fluorescent porous silicon@iron oxide nanocomposites for fluorescence/magnetic resonance bimodal imaging of tumor in vivo. ACS Biomater. Sci. Eng. 2017, 3, 2579–2587. [Google Scholar] [CrossRef]
- Duan, Y.; Xu, Y.; Mao, D.; Liew, W.H.; Guo, B.; Wang, S.; Cai, X.; Thakor, N.; Yao, K.; Zhang, C.-J.; et al. Photoacoustic and magnetic resonance imaging bimodal contrast agent displaying amplified photoacoustic signal. Small 2018, 14, e1800652. [Google Scholar] [CrossRef]
- Pham, T.N.H.; Lengkeek, N.A.; Greguric, I.; Kim, B.J.; Pellegrini, P.A.; Bickley, S.A.; Tanudji, M.R.; Jones, S.K.; Hawkett, B.S.; Pham, B.T. Tunable and noncytotoxic PET/SPECT-MRI multimodality imaging probes using colloidally stable ligand-free superparamagnetic iron oxide nanoparticles. Int. J. Nanomed. 2017, 12, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wang, L.; Lin, R.; Wang, A.Y.; Yang, L.; Kuang, M.; Qian, W.; Mao, H. Casein-coated iron oxide nanoparticles for high MRI contrast enhancement and efficient cell targeting. ACS Appl. Mater. Interfaces 2013, 5, 4632–4639. [Google Scholar] [CrossRef]
- Fernández-Barahona, I.; Gutiérrez, L.; Veintemillas-Verdaguer, S.; Pellico, J.; Morales, M.d.P.; Catala, M.; del Pozo, M.A.; Ruiz-Cabello, J.; Herranz, F. Cu-doped extremely small iron oxide nanoparticles with large longitudinal relaxivity: one-pot synthesis and in vivo targeted molecular imaging. ACS Omega 2019, 4, 2719–2727. [Google Scholar]
- Salimi, M.; Sarkar, S.; Saber, R.; Delavari, H.; Alizadeh, A.M.; Mulder, H.T. Magnetic hyperthermia of breast cancer cells and MRI relaxometry with dendrimer-coated iron-oxide nanoparticles. Cancer Nanotechnol. 2018, 9, 7. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Han, J.; Hao, Q. Gram-scale preparation of iron oxide nanoparticles with renal clearance properties for enhanced t 1 -weighted magnetic resonance imaging. ACS Appl. Bio Mater. 2018, 1, 1389–1397. [Google Scholar] [CrossRef]
- Hurley, K.R.; Ring, H.L.; Etheridge, M.; Zhang, J.; Gao, Z.; Shao, Q.; Klein, N.D.; Szlag, V.M.; Chung, C.; Reineke, T.M.; et al. Predictable heating and positive mri contrast from a mesoporous silica-coated iron oxide nanoparticle. Mol. Pharm. 2016, 13, 2172–2183. [Google Scholar] [CrossRef]
- Bhavesh, R.; Lechuga-Vieco, A.; Ruiz-Cabello, J.; Herranz, F. T1-MRI fluorescent iron oxide nanoparticles by microwave assisted synthesis. Nanomaterials 2015, 5, 1880–1890. [Google Scholar] [CrossRef]
- Arias, L.; Pessan, J.; Vieira, A.; Lima, T.; Delbem, A.; Monteiro, D. Iron oxide nanoparticles for biomedical applications: A perspective on synthesis, drugs, antimicrobial activity, and toxicity. Antibiotics 2018, 7, 46. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.K.; Zhang, Y.; Voit, W.; Rao, K.V.; Muhammed, M. Synthesis and characterization of surfactant-coated superparamagnetic monodispersed iron oxide nanoparticles. J. Magn. Magn. Mater. 2001, 225, 30–36. [Google Scholar] [CrossRef]
- Gazeau, F.; Shilov, V.; Bacri, J.C.; Dubois, E.; Gendron, F.; Perzynski, R.; Raikher, Y.L.; Stepanov, V.I. Magnetic resonance of nanoparticles in a ferrofluid: evidence of thermofluctuational effects. J. Magn. Magn. Mater. 1999, 202, 535–546. [Google Scholar] [CrossRef]
- Gazeau, F.; Bacri, J.C.; Gendron, F.; Perzynski, R.; Raikher, Y.; Stepanov, V.I.; Dubois, E. Magnetic resonance of ferrite nanoparticles: Evidence of surface effects. J. Magn. Magn. Mater. 1998, 186, 175–187. [Google Scholar] [CrossRef]
- Jolivet, J.-P.; Henry, M.; Livage, J. Metal Oxide Chemistry and Synthesis: From Solution to Solid State; John Wiley & Sons: Hoboken, NJ, USA, 2000. [Google Scholar]
- Laurent, S.; Forge, D.; Port, M.; Roch, A.; Robic, C.; Vander Elst, L.; Muller, R.N. Magnetic iron oxide nanoparticles: Synthesis, stabilization, vectorization, physicochemical characterizations, and biological applications. Chem. Rev. 2008, 108, 2064–2110. [Google Scholar] [CrossRef] [PubMed]
- Shieh, D.-B.; Cheng, F.-Y.; Su, C.-H.; Yeh, C.-S.; Wu, M.-T.; Wu, Y.-N.; Tsai, C.-Y.; Wu, C.-L.; Chen, D.-H.; Chou, C.-H. Aqueous dispersions of magnetite nanoparticles with NH3+ surfaces for magnetic manipulations of biomolecules and MRI contrast agents. Biomaterials 2005, 26, 7183–7191. [Google Scholar] [CrossRef]
- Bee, A.; Massart, R.; Neveu, S. Synthesis of very fine maghemite particles. J. Magn. Magn. Mater. 1995, 149, 6–9. [Google Scholar] [CrossRef]
- Daoush, W.M. Co-precipitation and magnetic properties of magnetite nanoparticles for potential biomedical applications. J. Nanomed. Res. 2017, 5, 00118. [Google Scholar] [CrossRef]
- Shi, X.; Thomas, T.P.; Myc, L.A.; Kotlyar, A.; Baker, J.R., Jr. Synthesis, characterization, and intracellular uptake of carboxyl-terminated poly(amidoamine) dendrimer-stabilized iron oxide nanoparticles. Phys. Chem. Chem. Phys. PCCP 2007, 9, 5712–5720. [Google Scholar] [CrossRef] [PubMed]
- Molday, R.S.; Mackenzie, D. Immunospecific ferromagnetic iron-dextran reagents for the labeling and magnetic separation of cells. J. Immunol. Methods 1982, 52, 353–367. [Google Scholar] [CrossRef]
- Berret, J.-F.; Schonbeck, N.; Gazeau, F.; El Kharrat, D.; Sandre, O.; Vacher, A.; Airiau, M. Controlled clustering of superparamagnetic nanoparticles using block copolymers: Design of new contrast agents for magnetic resonance imaging. J. Am. Chem. Soc. 2006, 128, 1755–1761. [Google Scholar] [CrossRef]
- Thünemann, A.F.; Schütt, D.; Kaufner, L.; Pison, U.; Möhwald, H. Maghemite nanoparticles protectively coated with poly(ethylene imine) and poly(ethylene oxide)- b lock -poly(glutamic acid). Langmuir 2006, 22, 2351–2357. [Google Scholar] [CrossRef]
- Yoo, M.K.; Kim, I.Y.; Kim, E.M.; Jeong, H.-J.; Lee, C.-M.; Jeong, Y.Y.; Akaike, T.; Cho, C.S. Superparamagnetic iron oxide nanoparticles coated with galactose-carrying polymer for hepatocyte targeting. J. Biomed. Biotechnol. 2007, 2007, 94740. [Google Scholar] [CrossRef] [PubMed]
- Kansara, K.; Patel, P.; Shukla, R.K.; Pandya, A.; Shanker, R.; Kumar, A.; Dhawan, A. Synthesis of biocompatible iron oxide nanoparticles as a drug delivery vehicle. Int. J. Nanomed. 2018, 13, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; He, Q.; Jiang, C. Magnetic iron oxide nanoparticles: synthesis and surface functionalization strategies. Nanoscale Res. Lett. 2008, 3, 397–415. [Google Scholar] [CrossRef] [PubMed]
- Maity, D.; Choo, S.-G.; Yi, J.; Ding, J.; Xue, J.M. Synthesis of magnetite nanoparticles via a solvent-free thermal decomposition route. J. Magn. Magn. Mater. 2009, 321, 1256–1259. [Google Scholar] [CrossRef]
- Yu, W.W.; Falkner, J.C.; Yavuz, C.T.; Colvin, V.L. Synthesis of monodisperse iron oxide nanocrystals by thermal. Chem. Commun. 2004, 0, 2306–2307. [Google Scholar] [CrossRef]
- Park, J.; An, K.; Hwang, Y.; Park, J.-G.; Noh, H.-J.; Kim, J.-Y.; Park, J.-H.; Hwang, N.-M.; Hyeon, T. Ultra-large-scale syntheses of monodisperse nanocrystals. Nat. Mater. 2004, 3, 891–895. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Zeng, H. Size-controlled synthesis of magnetite nanoparticles. J. Am. Chem. Soc. 2002, 124, 8204–8205. [Google Scholar] [CrossRef] [PubMed]
- Giri, S.; Samanta, S.; Maji, S.; Ganguli, S.; Bhaumik, A. Magnetic properties of α-Fe2O3 nanoparticle synthesized by a new hydrothermal method. J. Magn. Magn. Mater. 2005, 285, 296–302. [Google Scholar] [CrossRef]
- Ge, S.; Shi, X.; Sun, K.; Li, C.; Baker, J.R., Jr.; Banaszak Holl, M.M.; Orr, B.G. A Facile hydrothermal synthesis of iron oxide nanoparticles with tunable magnetic properties. J. Phys. Chem. C Nanomater. Interf. 2009, 113, 13593–13599. [Google Scholar] [CrossRef] [PubMed]
- Rasouli, E.; Basirun, W.J.; Johan, M.R.; Rezayi, M.; Darroudi, M.; Shameli, K.; Shanavaz, Z.; Akbarzadeh, O.; Izadiyan, Z. Facile and greener hydrothermal honey-based synthesis of Fe3O4/Au core/shell nanoparticles for drug delivery applications. J. Cell. Biochem. 2018, 1–8. [Google Scholar] [CrossRef]
- Wang, S.-B.; Min, Y.-L.; Yu, S.-H. Synthesis and magnetic properties of uniform hematite nanocubes. J. Phys. Chem. C 2007, 111, 3551–3554. [Google Scholar] [CrossRef]
- Takami, S.; Sato, T.; Mousavand, T.; Ohara, S.; Umetsu, M.; Adschiri, T. Hydrothermal synthesis of surface-modified iron oxide nanoparticles. Mater. Lett. 2007, 61, 4769–4772. [Google Scholar] [CrossRef]
- Titirici, M.-M.; Antonietti, M.; Thomas, A. A generalized synthesis of metal oxide hollow spheres using a hydrothermal approach. Chem. Mater. 2006, 18, 3808–3812. [Google Scholar] [CrossRef]
- Wan, L.; Yan, S.; Wang, X.; Li, Z.; Zou, Z. Solvothermal synthesis of monodisperse iron oxides with various morphologies and their applications in removal of Cr(vi). CrystEngComm 2011, 13, 2727. [Google Scholar] [CrossRef]
- Chaianansutcharit, S.; Mekasuwandumrong, O.; Praserthdam, P. Effect of organic solvents on iron oxide nanoparticles by the solvothermal method. Cryst. Growth Des. 2006, 6, 40–45. [Google Scholar] [CrossRef]
- Mishra, D.; Arora, R.; Lahiri, S.; Amritphale, S.S.; Chandra, N. Synthesis and characterization of iron oxide nanoparticles by solvothermal method. Prot. Met. Phys. Chem. Surf. 2014, 50, 628–631. [Google Scholar] [CrossRef]
- Li, S.; Zhang, T.; Tang, R.; Qiu, H.; Wang, C.; Zhou, Z. Solvothermal synthesis and characterization of monodisperse superparamagnetic iron oxide nanoparticles. J. Magn. Magn. Mater. 2015, 379, 226–231. [Google Scholar] [CrossRef]
- Martelli, S.; Mancini, A.; Giorgi, R.; Alexandrescu, R.; Cojocaru, S.; Crunteanu, A.; Voicu, I.; Balu, M.; Morjan, I. Production of iron-oxide nanoparticles by laser-induced pyrolysis of gaseous precursors. Appl. Surf. Sci. 2000, 154, 353–359. [Google Scholar] [CrossRef]
- Malumbres, A.; Martínez, G.; Mallada, R.; Hueso, J.L.; Bomatí-Miguel, O.; Santamaría, J. Continuous production of iron-based nanocrystals by laser pyrolysis. Effect of operating variables on size, composition and magnetic response. Nanotechnology 2013, 24, 325603. [Google Scholar] [CrossRef]
- Costo, R.; Bello, V.; Robic, C.; Port, M.; Marco, J.F.; Puerto Morales, M.; Veintemillas-Verdaguer, S. Ultrasmall iron oxide nanoparticles for biomedical applications: improving the colloidal and magnetic properties. Langmuir 2012, 28, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Martínez, G.; Malumbres, A.; Mallada, R.; Hueso, J.L.; Irusta, S.; Bomatí-Miguel, O.; Santamaría, J. Use of a polyol liquid collection medium to obtain ultrasmall magnetic nanoparticles by laser pyrolysis. Nanotechnology 2012, 23, 425605. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Ren, Z.; Chen, Z.; Zhu, J.; Liang, J.; Liao, R.; Wen, J. Iron oxide nanoparticles as nanocarriers to improve chlorin e6-based sonosensitivity in sonodynamic therapy. Drug Des. Devel. Ther. 2018, 12, 4207–4216. [Google Scholar] [CrossRef] [PubMed]
- Veintemillas-Verdaguer, S.; Leconte, Y.; Costo, R.; Bomati-Miguel, O.; Bouchet-Fabre, B.; Morales, M.P.; Bonville, P.; Pérez-Rial, S.; Rodriguez, I.; Herlin-Boime, N. Continuous production of inorganic magnetic nanocomposites for biomedical applications by laser pyrolysis. J. Magn. Magn. Mater. 2007, 311, 120–124. [Google Scholar] [CrossRef]
- Seol, S.K.; Kim, D.; Jung, S.; Hwu, Y. Microwave synthesis of gold nanoparticles: Effect of applied microwave power and solution pH. Mater. Chem. Phys. 2011, 131, 331–335. [Google Scholar] [CrossRef]
- Bayazit, M.K.; Yue, J.; Cao, E.; Gavriilidis, A.; Tang, J. Controllable synthesis of gold nanoparticles in aqueous solution by microwave assisted flow chemistry. ACS Sustain. Chem. Eng. 2016, 4, 6435–6442. [Google Scholar] [CrossRef]
- Arshi, N.; Ahmed, F.; Kumar, S.; Anwar, M.S.; Lu, J.; Koo, B.H.; Lee, C.G. Microwave assisted synthesis of gold nanoparticles and their antibacterial activity against Escherichia coli (E. coli). Curr. Appl. Phys. 2011, 11, S360–S363. [Google Scholar] [CrossRef]
- Ngo, V.K.T.; Nguyen, H.P.U.; Huynh, T.P.; Tran, N.N.P.; Lam, Q.V.; Huynh, T.D. Preparation of gold nanoparticles by microwave heating and application of spectroscopy to study conjugate of gold nanoparticles with antibody E. coli O157:H7. Adv. Nat. Sci. Nanosci. Nanotechnol. 2015, 6, 035015. [Google Scholar] [CrossRef]
- Cabello, G.; Davoglio, R.A.; Hartl, F.W.; Marco, J.F.; Pereira, E.C.; Biaggio, S.R.; Varela, H.; Cuesta, A. Microwave-assisted synthesis of Pt-Au nanoparticles with enhanced electrocatalytic activity for the oxidation of formic acid. Electrochim. Acta 2017, 224, 56–63. [Google Scholar] [CrossRef]
- Goel, A.; Meher, M.K.; Gupta, P.; Gulati, K.; Pruthi, V.; Poluri, K.M. Microwave assisted $κ$-carrageenan capped silver nanocomposites for eradication of bacterial biofilms. Carbohydr. Polym. 2019, 206, 854–862. [Google Scholar] [CrossRef] [PubMed]
- Pal, J.; Deb, M.K.; Deshmukh, D.K.; Sen, B.K. Microwave-assisted synthesis of platinum nanoparticles and their catalytic degradation of methyl violet in aqueous solution. Appl. Nanosci. 2014, 4, 61–65. [Google Scholar] [CrossRef]
- Mehta, S.K.; Gupta, S. Time-efficient microwave synthesis of Pd nanoparticles and their electrocatalytic property in oxidation of formic acid and alcohols in alkaline media. J. Appl. Electrochem. 2011, 41, 1407–1417. [Google Scholar] [CrossRef]
- Liu, Y.Q.; Zhang, M.; Wang, F.X.; Pan, G.B. Facile microwave-assisted synthesis of uniform single-crystal copper nanowires with excellent electrical conductivity. RSC Adv. 2012, 2, 11235–11237. [Google Scholar] [CrossRef]
- Baruwati, B.; Varma, R.S. High value products from waste: Grape pomace extract-a three-in-one package for the synthesis of metal nanoparticles. ChemSusChem 2009, 2, 1041–1044. [Google Scholar] [CrossRef] [PubMed]
- Kou, J.; Varma, R.S. Beet juice utilization: Expeditious green synthesis of noble metal nanoparticles (Ag, Au, Pt, and Pd) using microwaves. RSC Adv. 2012, 2, 10283–10290. [Google Scholar] [CrossRef]
- Eshghi, M.; Vaghari, H.; Najian, Y.; Najian, M.J.; Jafarizadeh-Malmiri, H.; Berenjian, A. Microwave-assisted green synthesis of silver nanoparticles using juglans regia leaf extract and evaluation of their physico-chemical and antibacterial properties. Antibiotics 2018, 7, 68. [Google Scholar] [CrossRef]
- El-Naggar, M.E.; Shaheen, T.I.; Fouda, M.M.G.; Hebeish, A.A. Eco-friendly microwave-assisted green and rapid synthesis of well-stabilized gold and core–shell silver–gold nanoparticles. Carbohydr. Polym. 2016, 136, 1128–1136. [Google Scholar] [CrossRef]
- Saloga, P.E.J.; Kästner, C.; Thünemann, A.F. High-speed but not magic: microwave-assisted synthesis of ultra-small silver nanoparticles. Langmuir 2018, 34, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xu, W.; Wu, X.; Li, Q.; Wang, Z.; Zheng, X. Efficient rapid microwave-assisted route to synthesize monodispersive pt nanoparticles and their electrocatalytic activity for methanol oxidation. J. Nanomater. 2014, 2014, 1–5. [Google Scholar] [CrossRef]
- Duan, Z.; Timoshenko, J.; Kunal, P.; House, S.D.; Wan, H.; Jarvis, K.; Bonifacio, C.; Yang, J.C.; Crooks, R.M.; Frenkel, A.I.; et al. Structural characterization of heterogeneous RhAu nanoparticles from a microwave-assisted synthesis. Nanoscale 2018, 22520–22532. [Google Scholar] [CrossRef]
- Mishra, S.K.; Kannan, S. Microwave synthesis of chitosan capped silver-dysprosium bimetallic nanoparticles: a potential nanotheranosis device. Langmuir 2016, 32, 13687–13696. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Wing, C.; Esparza, R.; Vargas-Hernández, C.; Fernández García, M.E.; José-Yacamán, M. Microwave-assisted synthesis of gold nanoparticles self-assembled into self-supported superstructures. Nanoscale 2012, 4, 2281–2287. [Google Scholar] [CrossRef]
- Li, D.; Komarneni, S. Synthesis of Pt nanoparticles and nanorods by microwave-assisted solvothermal technique. Zeitschrift Naturforschung-Sect. B J. Chem. Sci. 2006, 5, 1566–1572. [Google Scholar]
- Pujari, S.P.; Driss, H.; Bannani, F.; Van Lagen, B.; Zuilhof, H. One-pot gram-scale synthesis of hydrogen-terminated silicon nanoparticles. Chem. Mater. 2018, 30, 6503–6512. [Google Scholar] [CrossRef] [PubMed]
- Bhawawet, N.; Essner, J.B.; Atwood, J.L.; Baker, G.A. On the non-innocence of the imidazolium cation in a rapid microwave synthesis of oleylamine-capped gold nanoparticles in an ionic liquid. Chem. Commun. 2018, 54, 7523–7526. [Google Scholar] [CrossRef] [PubMed]
- Barreto, G.P.; Morales, G.; Quintanilla, M.L.L. Microwave assisted synthesis of zno nanoparticles: effect of precursor reagents, temperature, irradiation time, and additives on nano-ZnO morphology development. J. Mater. 2013, 2013, 1–11. [Google Scholar] [CrossRef]
- Hasanpoor, M.; Aliofkhazraei, M.; Delavari, H. Microwave-assisted synthesis of zinc oxide nanoparticles. Procedia Mater. Sci. 2015, 11, 320–325. [Google Scholar] [CrossRef]
- Yusof, N.A.A.; Zain, N.M.; Pauzi, N. Synthesis of ZnO nanoparticles with chitosan as stabilizing agent and their antibacterial properties against Gram-positive and Gram-negative bacteria. Int. J. Biol. Macromol. 2019, 124, 1132–1136. [Google Scholar] [CrossRef] [PubMed]
- Wojnarowicz, J.; Chudoba, T.; Koltsov, I.; Gierlotka, S.; Dworakowska, S.; Lojkowski, W. Size control mechanism of ZnO nanoparticles obtained in microwave solvothermal synthesis. Nanotechnology 2018, 29, 065601. [Google Scholar] [CrossRef] [PubMed]
- Wojnarowicz, J.; Chudoba, T.; Gierlotka, S.; Lojkowski, W. Effect of microwave radiation power on the size of aggregates of ZnO NPs prepared using microwave solvothermal synthesis. Nanomaterials 2018, 8, 343. [Google Scholar] [CrossRef] [PubMed]
- Falk, G.S.; Borlaf, M.; López-Muñoz, M.J.; Fariñas, J.C.; Rodrigues Neto, J.B.; Moreno, R. Microwave-assisted synthesis of TiO2 nanoparticles: photocatalytic activity of powders and thin films. J. Nanopart. Res. 2018, 20, 23. [Google Scholar] [CrossRef]
- Dar, M.I.; Chandiran, A.K.; Grätzel, M.; Nazeeruddin, M.K.; Shivashankar, S.A. Controlled synthesis of TiO2 nanoparticles and nanospheres using a microwave assisted approach for their application in dye-sensitized solar cells. J. Mater. Chem. A 2014, 2, 1662–1667. [Google Scholar] [CrossRef]
- May-Masnou, A.; Soler, L.; Torras, M.; Salles, P.; Llorca, J.; Roig, A. Fast and simple microwave synthesis of TiO2/Au nanoparticles for gas-phase photocatalytic hydrogen generation. Front. Chem. 2018, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Gohain, M.; Som, S.; Kumar, V.; Bezuindenhoudt, B.C.B.; Swart, H.C. Microwave assisted synthesis of ZnO nanoparticles for lighting and dye removal application. Phys. B Condens. Matter 2016, 480, 36–41. [Google Scholar] [CrossRef]
- AmbroŽiČ, G.; Orel, Z.C.; Žigon, M. Microwave-assisted non-aqueous synthesis of ZnO nanoparticles. Mater. Tehnol. 2011, 45, 173–177. [Google Scholar]
- Wada, Y.; Kuramoto, H.; Anand, J.; Kitamura, T.; Sakata, T.; Mori, H.; Yanagida, S. Microwave-assisted size control of CdS nanocrystallites. J. Mater. Chem. 2011, 11, 1936–1940. [Google Scholar] [CrossRef]
- Bharti, D.B.; Bharati, A.V.; Wankhade, A.V. Synthesis, characterization and optical property investigation of CdS nanoparticles. Luminescence 2018, 33, 1445–1449. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, J. Synthesis of Cu–In–S/ZnS quantum dots in aqueous phase by microwave irradiation. J. Nanosci. Nanotechnol. 2016, 16, 8411–8414. [Google Scholar] [CrossRef]
- Shahid, R.; Gorlov, M.; El-Sayed, R.; Toprak, M.S.; Sugunan, A.; Kloo, L.; Muhammed, M. Microwave assisted synthesis of ZnS quantum dots using ionic liquids. Mater. Lett. 2012, 89, 316–319. [Google Scholar] [CrossRef]
- Moghaddam, M.M.; Baghbanzadeh, M.; Keilbach, A.; Kappe, C.O. Microwave-assisted synthesis of CdSe quantum dots: Can the electromagnetic field influence the formation and quality of the resulting nanocrystals? Nanoscale 2012, 4, 7435. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, D.S.M.; de Souza, G.C.S.; Melo, A.; Soares, J.X.; Rodrigues, S.S.M.; Araújo, A.N.; Montenegro, M.C.B.S.M.; Santos, J.L.M. Synthesis of distinctly thiol-capped CdTe quantum dots under microwave heating: Multivariate optimization and characterization. J. Mater. Sci. 2017, 52, 3208–3224. [Google Scholar] [CrossRef]
- Khollam, Y.B.; Dhage, S.R.; Potdar, H.S.; Deshpande, S.B.; Bakare, P.P.; Kulkarni, S.D.; Date, S.K. Microwave hydrothermal preparation of submicron-sized spherical magnetite (Fe3O4) powders. Mater. Lett. 2002, 56, 571–577. [Google Scholar] [CrossRef]
- Hu, L.; Percheron, A.; Chaumont, D.; Brachais, C.-H. Microwave-assisted one-step hydrothermal synthesis of pure iron oxide nanoparticles: Magnetite, maghemite and hematite. J. Sol-Gel Sci. Technol. 2011, 60, 198–205. [Google Scholar] [CrossRef]
- Kooti, M.; Matturi, L. Microwave-assisted fabrication of γ-Fe2O3 Nanoparticles from Tris (acetylacetonato) Iron (III). Int. Nano Lett. 2011, 1, 38–42. [Google Scholar]
- Bano, S.; Nazir, S.; Nazir, A.; Munir, S.; Mahmood, T.; Afzal, M.; Ansari, F.L.; Mazhar, K. Microwave-assisted green synthesis of superparamagnetic nanoparticles using fruit peel extracts: Surface engineering, T2 relaxometry, and photodynamic treatment potential. Int. J. Nanomed. 2016, 11, 3833–3848. [Google Scholar] [CrossRef]
- Sathya, A.; Kalyani, S.; Ranoo, S.; Philip, J. One-step microwave-assisted synthesis of water-dispersible Fe3O4 magnetic nanoclusters for hyperthermia applications. J. Magn. Magn. Mater. 2017, 439, 107–113. [Google Scholar] [CrossRef]
- Aivazoglou, E.; Metaxa, E.; Hristoforou, E. Microwave-assisted synthesis of iron oxide nanoparticles in biocompatible organic environment. AIP Adv. 2018, 8, 48201. [Google Scholar] [CrossRef]
- Liao, X.; Zhu, J.; Zhong, W.; Chen, H.-Y.Y. Synthesis of amorphous Fe2O3 nanoparticles by microwave irradiation. Mater. Lett. 2001, 50, 341–346. [Google Scholar] [CrossRef]
- Hu, X.; Yu, J.C.; Gong, J.; Li, Q.; Li, G. α-Fe2O3 nanorings prepared by a microwave-assisted hydrothermal process and their sensing properties. Adv. Mater. 2007, 19, 2324–2329. [Google Scholar] [CrossRef]
- Ai, Z.; Deng, K.; Wan, Q.; Zhang, L.; Lee, S. Facile microwave-assisted synthesis and magnetic and gas sensing properties of Fe3O4 nanoroses. J. Phys. Chem. C 2010, 114, 6237–6242. [Google Scholar] [CrossRef]
- Wang, W.-W.; Zhu, Y.-J.; Ruan, M.-L. Microwave-assisted synthesis and magnetic property of magnetite and hematite nanoparticles. J. Nanopart. Res. 2007, 9, 419–426. [Google Scholar] [CrossRef]
- Parsons, J.G.; Luna, C.; Botez, C.E.; Elizalde, J.; Gardea-Torresdey, J.L. Microwave-assisted synthesis of iron(III) oxyhydroxides/oxides characterized using transmission electron microscopy, X-ray diffraction, and X-ray absorption spectroscopy. J. Phys. Chem. Solids 2009, 70, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Pascu, O.; Carenza, E.; Gich, M.; Estradé, S.; Peiró, F.; Herranz, G.; Roig, A. Surface reactivity of iron oxide nanoparticles by microwave-assisted synthesis; comparison with the thermal decomposition route. J. Phys. Chem. C 2012, 116, 15108–15116. [Google Scholar] [CrossRef]
- Gonzalez-Moragas, L.; Yu, S.-M.; Murillo-Cremaes, N.; Laromaine, A.; Roig, A. Scale-up synthesis of iron oxide nanoparticles by microwave-assisted thermal decomposition. Chem. Eng. J. 2015, 281, 87–95. [Google Scholar] [CrossRef]
- Carenza, E.; Barceló, V.; Morancho, A.; Montaner, J.; Rosell, A.; Roig, A. Rapid synthesis of water-dispersible superparamagnetic iron oxide nanoparticles by a microwave-assisted route for safe labeling of endothelial progenitor cells. Acta Biomater. 2014, 10, 3775–3785. [Google Scholar] [CrossRef]
- Kozakova, Z.; Kuritka, I.; Babayan, V.; Kazantseva, N.; Pastorek, M. Magnetic iron oxide nanoparticles for high frequency applications. IEEE Trans. Magn. 2013, 49, 995–999. [Google Scholar] [CrossRef]
- Kozakova, Z.; Kuritka, I.; Kazantseva, N.E.; Babayan, V.; Pastorek, M.; Machovsky, M.; Bazant, P.; Saha, P. The formation mechanism of iron oxide nanoparticles within the microwave-assisted solvothermal synthesis and its correlation with the structural and magnetic properties. Dalton Trans. 2015, 44, 21099–21108. [Google Scholar] [CrossRef] [PubMed]
- Kozakova, Z.; Kuritka, I.; Bazant, P.; Pastorek, M.; Babayan, V. Magnetic needle-like iron oxide particles prepared by microwave-assisted thermal decomposition technique. Mater. Lett. 2015, 138, 116–119. [Google Scholar] [CrossRef]
- Kalyani, S.; Sangeetha, J.; Philip, J. Microwave assisted synthesis of ferrite nanoparticles: effect of reaction temperature on particle size and magnetic properties. J. Nanosci. Nanotechnol. 2015, 15, 5768–5774. [Google Scholar] [CrossRef]
- Liang, Y.-J.; Zhang, Y.; Guo, Z.; Xie, J.; Bai, T.; Zou, J.; Gu, N. Ultrafast preparation of monodisperse Fe3O4 nanoparticles by microwave-assisted thermal decomposition. Chem. A Eur. J. 2016, 22, 11807–11815. [Google Scholar] [CrossRef]
- Guru, S.; Mishra, D.; Singh, M.; Amritphale, S.S.; Joshi, S. Effect of SO42−, Cl– and NO3− anions on the formation of iron oxide nanoparticles via microwave synthesis. Prot. Met. Phys. Chem. Surf. 2016, 52, 627–631. [Google Scholar] [CrossRef]
- Guru, S.; Mishra, D.; Amritphale, S.S.; Joshi, S. Influence of glycols in microwave assisted synthesis of ironoxide nanoparticles. Colloid Polym. Sci. 2016, 294, 207–213. [Google Scholar] [CrossRef]
- Sangaiya, P.; Jayaprakash, R. Tuning effect of Sn doping on structural, morphological, optical, electrical and photocatalytic properties of iron oxide nanoparticles. Mater. Sci. Semicond. Process. 2018, 85, 40–51. [Google Scholar] [CrossRef]
- Hammond, O.S.; Eslava, S.; Smith, A.J.; Zhang, J.; Edler, K.J. Microwave-assisted deep eutectic-solvothermal preparation of iron oxide nanoparticles for photoelectrochemical solar water splitting. J. Mater. Chem. A 2017, 5, 16189–16199. [Google Scholar] [CrossRef]
- Bensebaa, F.; Zavaliche, F.; L’Ecuyer, P.; Cochrane, R.W.; Veres, T. Microwave synthesis and characterization of Co–ferrite nanoparticles. J. Colloid Interface Sci. 2004, 277, 104–110. [Google Scholar] [CrossRef]
- Li, C.; Wei, Y.; Liivat, A.; Zhu, Y.; Zhu, J. Microwave-solvothermal synthesis of Fe3O4 magnetic nanoparticles. Mater. Lett. 2013, 107, 23–26. [Google Scholar] [CrossRef]
- Jiang, F.Y.Y.; Wang, C.M.; Fu, Y.; Liu, R.C.C. Synthesis of iron oxide nanocubes via microwave-assisted solvolthermal method. J. Alloys Compd. 2010, 503, L31–L33. [Google Scholar] [CrossRef]
- Blanco-Andujar, C.; Ortega, D.; Southern, P.; Pankhurst, Q.A.; Thanh, N.T.K. High performance multi-core iron oxide nanoparticles for magnetic hyperthermia: microwave synthesis, and the role of core-to-core interactions. Nanoscale 2015, 7, 1768–1775. [Google Scholar] [CrossRef]
- Lastovina, T.A.; Budnyk, A.P.; Soldatov, M.A.; Rusalev, Y.V.; Guda, A.A.; Bogdan, A.S.; Soldatov, A.V. Microwave-assisted synthesis of magnetic iron oxide nanoparticles in oleylamine–oleic acid solutions. Mendeleev Commun. 2017, 27, 487–489. [Google Scholar] [CrossRef]
- Lastovina, T.A.; Budnyk, A.P.; Kubrin, S.P.; Soldatov, A.V. Microwave-assisted synthesis of ultra-small iron oxide nanoparticles for biomedicine. Mendeleev Commun. 2018, 28, 167–169. [Google Scholar] [CrossRef]
- Brollo, M.E.F.; Veintemillas-Verdaguer, S.; Salván, C.M.; Morales, M.D.P. Key parameters on the microwave assisted synthesis of magnetic nanoparticles for MRI contrast agents. Contrast Media Mol. Imaging 2017, 2017, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Burdinski, D.; Bohlender, C. Synthesis and Use of Iron Oleate. EP2588416A1, 21 June 2011. PCT/IB2011/052708. [Google Scholar]
- Williams, M.J.; Sánchez, E.; Aluri, E.R.; Douglas, F.J.; MacLaren, D.A.; Collins, O.M.; Cussen, E.J.; Budge, J.D.; Sanders, L.C.; Michaelis, M.; et al. Microwave-assisted synthesis of highly crystalline, multifunctional iron oxide nanocomposites for imaging applications. RSC Adv. 2016, 6, 83520–83528. [Google Scholar] [CrossRef]
- Fernández-Barahona, I.; Ruiz-Cabello, J.; Herranz, F.; Pellico, J. Synthesis of 68Ga core-doped iron oxide nanoparticles for dual positron emission tomography/(T1) magnetic resonance imaging. J. Vis. Exp. 2018. [Google Scholar] [CrossRef] [PubMed]
- Pellico, J.; Lechuga-Vieco, A.V.; Almarza, E.; Hidalgo, A.; Mesa-Nuñez, C.; Fernández-Barahona, I.; Quintana, J.A.; Bueren, J.; Enríquez, J.A.; Ruiz-Cabello, J.; et al. In vivo imaging of lung inflammation with neutrophil-specific 68Ga nano-radiotracer. Sci. Rep. 2017, 7, 13242. [Google Scholar] [CrossRef]
- Pellico, J.; Fernández-Barahona, I.; Benito, M.; Gaitán-Simón, Á.; Gutiérrez, L.; Ruiz-Cabello, J.; Herranz, F. Unambiguous detection of atherosclerosis using bioorthogonal nanomaterials. Nanomed. Nanotechnol. Biol. Med. 2019, 17, 26–35. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Synthesis Method | Advantages | Disadvantages | Time Range |
|---|---|---|---|
| Coprecipitation |
|
| Hours–days |
| Thermal Decomposition |
|
| Hours–days |
| Hydrothermal and Solvothermal Synthesis |
|
| Hours–days |
| Pyrolysis |
|
| Hours |
| Microwave Synthesis |
| Microwave reactor required | Seconds–hours |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández-Barahona, I.; Muñoz-Hernando, M.; Herranz, F. Microwave-Driven Synthesis of Iron-Oxide Nanoparticles for Molecular Imaging. Molecules 2019, 24, 1224. https://doi.org/10.3390/molecules24071224
Fernández-Barahona I, Muñoz-Hernando M, Herranz F. Microwave-Driven Synthesis of Iron-Oxide Nanoparticles for Molecular Imaging. Molecules. 2019; 24(7):1224. https://doi.org/10.3390/molecules24071224
Chicago/Turabian StyleFernández-Barahona, Irene, Maria Muñoz-Hernando, and Fernando Herranz. 2019. "Microwave-Driven Synthesis of Iron-Oxide Nanoparticles for Molecular Imaging" Molecules 24, no. 7: 1224. https://doi.org/10.3390/molecules24071224
APA StyleFernández-Barahona, I., Muñoz-Hernando, M., & Herranz, F. (2019). Microwave-Driven Synthesis of Iron-Oxide Nanoparticles for Molecular Imaging. Molecules, 24(7), 1224. https://doi.org/10.3390/molecules24071224