Evaluation of Anti-Inflammatory Activities of a Triterpene β-Elemonic Acid in Frankincense In Vivo and In Vitro


Abstract
1. Introduction
2. Results
2.1. Structural Elucidation of the Isolated Compounds
2.2. The Content Determination of β-EA
2.3. Effect s of β-EA on Cell Viability and LPS Induced NO Production
2.4. Effect of β-EA on Inflammatory Factors In Vitro
2.5. Short-Term Toxicity Test
2.6. Effect s of β-EA on Acute Topical Edema
2.7. Effects of β-EA on Acute Systemic Inflammation
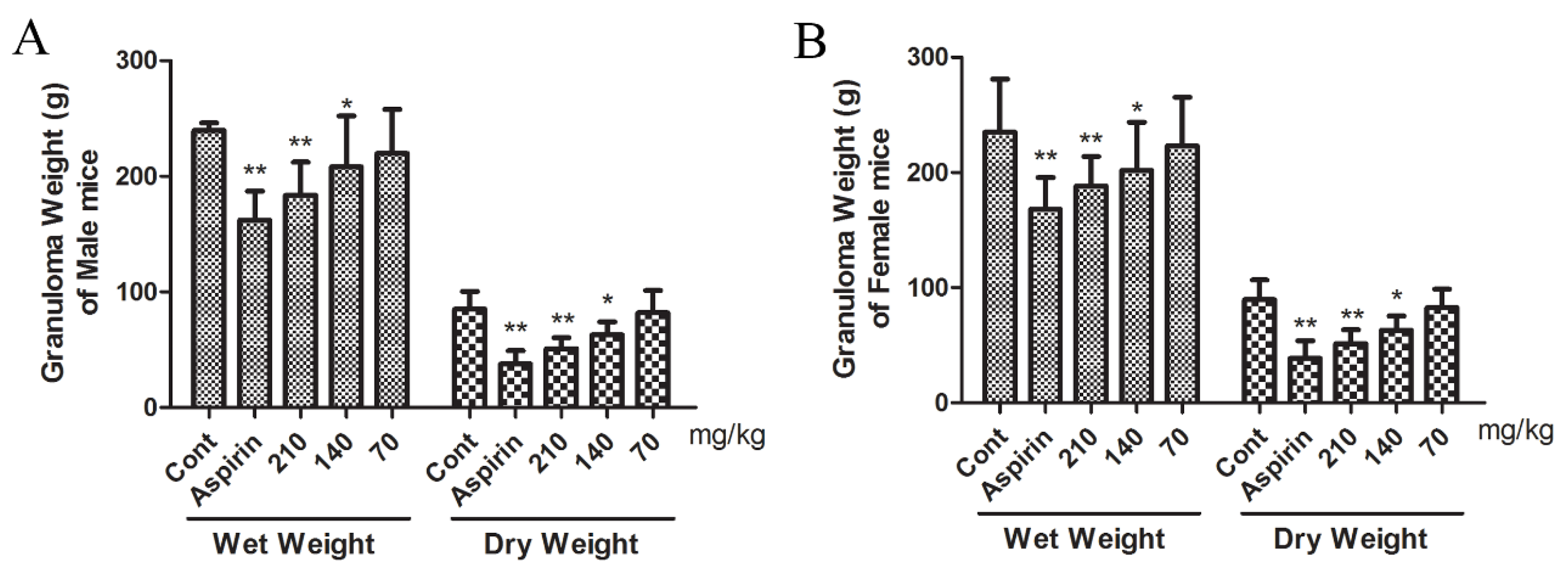
2.8. Effect of β-EA on Cotton Pellet Induced Granuloma Formation
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Isolation and Characterization of Compounds
4.3. Determination of β-EA in Frankincense of Different Origins by HPLC-ELSD
4.4. Cell Viability Assay
4.5. Measurement of Extracellular NO Production
4.6. Expression of Inflammatory Cytokines Using Antibody Array Membrane
4.7. Animals
4.8. Short-Term Toxicity Test
4.9. Xylene-Induced Ear Edema
4.10. Carrageenan-Induced Rat Paw Edema
4.11. Cotton Pellet Induced Granuloma Formation
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fujiwara, N.; Kobayashi, K. Macrophages in inflammation. Curr. Drug Targets Inflamm. Allergy 2005, 4, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Lu, T.-C.; Liao, J.-W.; Huang, T.-H.; Lee, M.-S.; Cheng, H.-Y.; Ho, L.-K.; Kuo, C.-L.; Peng, W.-H. Analgesic and anti-inflammatory activities of ethanol root extract of Mahonia oiwakensis in mice. J. Ethnopharmacol 2009, 125, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Song, X.; Jia, R.; Yin, Z.; Zou, Y.; Li, L.; Yin, L.; He, C.; Liang, X.; Yue, G.; et al. Evaluation of Analgesic and Anti-Inflammatory Activities of Water Extract of Galla Chinensis In Vivo Models. Evid. Based Complement. Alternat. Med. 2018, 2018, 6784032. [Google Scholar] [CrossRef]
- Fung, H.B.; Kirschenbaum, H.L. Selective cyclooxygenase-2 inhibitors for the treatment of arthritis. Clin. Ther. 1999, 21, 1131–1157. [Google Scholar] [CrossRef]
- Derry, S.; Moore, R.A.; Rabbie, R. Topical NSAIDs for chronic musculoskeletal pain in adults. Cochrane Db. Syst. Rev. 2012, 9, CD007400. [Google Scholar]
- Miceli, M.; Bontempo, P.; Nebbioso, A.; Altucci, L. Natural compounds in epigenetics: A current view. Food Chem. Toxicol. 2014, 73, 71–83. [Google Scholar] [CrossRef]
- Choi, O.B.; Park, J.H.; Lee, Y.J.; Lee, C.K.; Won, K.J.; Kim, J.; Lee, H.M.; Kim, B. Olibanum extract inhibits vascular smooth muscle cell migration and proliferation in response to platelet-derived growth factor. Korean J. Physiol. Pharmacol. 2009, 13, 107–113. [Google Scholar] [CrossRef]
- Kimmatkar, N.; Thawani, V.; Hingorani, L.; Khiyani, R. Efficacy and tolerability of Boswellia serrata extract in treatment of osteoarthritis of knee – A randomized double blind placebo controlled trial. Phytomedicine 2003, 10, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Ammon, H.P. Modulation of the immune system by Boswellia serrata extracts and boswellic acids. Phytomedicine 2010, 17, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Bertocchi, M.; Isani, G. Anti-Inflammatory Activity of Boswellia serrata Extracts: An In Vitro Study on Porcine Aortic Endothelial Cells. Oxid. Med. Cell Longev. 2018, 2018, 2504305. [Google Scholar] [CrossRef]
- Cuaz-perolin, C.; Billiet, L.; Bauge, E.; Copin, C.; Scott-Algara, D.; Genze, F.; Buchele, B.; Syrovets, T.; Simmet, T.; Rouis, M. Antiinflammatory and antiatherogenic effects of the NF-kappaB inhibitor acetyl-11-keto-beta-boswellic acid in LPS-challenged ApoE-/- mice. Atherosclerosis Supp. 2008, 9, 44. [Google Scholar] [CrossRef]
- Abdel-Tawab, M.; Werz, O.; Schubert-Zsilavecz, M. Boswellia serrata: An overall assessment of in vitro, preclinical, pharmacokinetic and clinical data. Clin. pharmacokinet. 2011, 50, 349–369. [Google Scholar] [CrossRef] [PubMed]
- Badria, F.A.; Mikhaeil, B.R.; Maatooq, G.T.; Amer, M.M. Immunomodulatory triterpenoids from the oleogum resin of Boswellia carterii Birdwood. Z Naturforsch C. 2003, 58, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Guo, Y.; Luo, Y.-T. Anti-inflammatory and analgesic activities of a novel biflavonoid from shells of Camellia oleifera. Int. J. Mol. Sci. 2012, 13, 12401–12411. [Google Scholar] [CrossRef]
- Banno, N.; Akihisa, T.; Yasukawa, K.; Tokuda, H.; Tabata, K.; Nakamura, Y.; Nishimura, R.; Kimura, Y.; Suzuki, T. Anti-inflammatory activities of the triterpene acids from the resin of Boswellia carteri. Ethnopharmacol 2006, 107, 249–253. [Google Scholar] [CrossRef]
- Frank, M.B.; Yang, Q.; Osban, J.; Azzarello, J.T.; Saban, M.R.; Saban, R.; Ashley, R.A.; Welter, J.C.; Fung, K.-M.; Lin, H.-K. Frankincense oil derived from Boswellia carteri induces tumor cell specific cytotoxicity. BMC Complement. Altern. Med. 2009, 9, 6. [Google Scholar] [CrossRef]
- Wang, F.; Li, Z.L.; Cui, H.H.; Hua, H.M.; Jing, Y.K.; Liang, S.W. Two new triterpenoids from the resin of Boswellia carterii. J. Asian Nat. Prod. Res. 2011, 13, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.T.; Lu, C.L.; Lin, H.I.; Chen, B.F.; Jow, G.M. beta-Elemonic acid inhibits the cell proliferation of human lung adenocarcinoma A549 cells: The role of MAPK, ROS activation and glutathione depletion. Oncol. Rep. 2016, 35, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Pathak, J.L.; Bakker, A.D.; Luyten, F.P.; Verschueren, P.; Lems, W.F.; Klein-Nulend, J.; Bravenboer, N. Systemic Inflammation Affects Human Osteocyte-Specific Protein and Cytokine Expression. Calcif Tissue Int. 2016, 98, 596–608. [Google Scholar] [CrossRef] [PubMed]
- Morch, R.H.; Dieset, I.; Faerden, A.; Hope, S.; Aas, M.; Nerhus, M.; Gardsjord, E.S.; Joa, I.; Morken, G.; Agartz, I.; et al. Inflammatory evidence for the psychosis continuum model. Psychoneuroendocrinology 2016, 67, 189–197. [Google Scholar] [CrossRef]
- Shanely, R.A.; Nieman, D.C.; Henson, D.A.; Jin, F.; Knab, A.M.; Sha, W. Inflammation and oxidative stress are lower in physically fit and active adults. Scand. J. Med. Sci. Spor. 2013, 23, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Bilancio, A.; Rinaldi, B.; Oliviero, M.A.; Donniacuo, M.; Monti, M.G.; Boscaino, A.; Marino, I.; Friedman, L.; Rossi, F.; Vanhaesebroeck, B.; et al. Inhibition of p110delta PI3K prevents inflammatory response and restenosis after artery injury. Biosci. Rep. 2017, 37, BSR20171112. [Google Scholar] [CrossRef] [PubMed]
- Harada, Y.; Tanaka, K.; Uchida, Y.; Ueno, A.; Oh-Ishi, S.; Yamashita, K.; Ishibashi, M.; Miyazaki, H.; Katori, M. Changes in the levels of prostaglandins and thromboxane and their roles in the accumulation of exudate in rat carrageenin-induced pleurisy--a profile analysis using gas chromatography-mass spectrometry. Prostaglandins 1982, 23, 881–895. [Google Scholar] [CrossRef]
- Siemoneit, U.; Koeberle, A.; Rossi, A.; Dehm, F.; Verhoff, M.; Reckel, S.; Maier, T.J.; Jauch, J.; Northoff, H.; Bernhard, F.; et al. Inhibition of microsomal prostaglandin E2 synthase-1 as a molecular basis for the anti-inflammatory actions of boswellic acids from frankincense. Br. J. Pharmacol. 2011, 162, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Guay, J.; Bateman, K.; Gordon, R.; Mancini, J.; Riendeau, D. Carrageenan-induced paw edema in rat elicits a predominant prostaglandin E2 (PGE2) response in the central nervous system associated with the induction of microsomal PGE2 synthase-1. J. Biol. Chem. 2004, 279, 24866–24872. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Mehmood, M.H.; Ali, A.N.A.; Ahmed, F.S.; Dar, A.; Gilani, A.-H. Studies on anti-inflammatory and analgesic activities of betel nut in rodents. J. Ethnopharmacol. 2011, 135, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Chen, Y.; Zhong, S.; Ji, B.; Wang, J.; Bai, X.; Shi, G. Anti-inflammatory activity of N-butanol extract from Ipomoea stolonifera in vivo and in vitro. PloS One 2014, 9, e95931. [Google Scholar] [CrossRef] [PubMed]
- Saha, K.; Lajis, N.H.; Israf, D.A.; Hamzah, A.S.; Khozirah, S.; Khamis, S.; Syahida, A. Evaluation of antioxidant and nitric oxide inhibitory activities of selected Malaysian medicinal plants. J. Ethnopharmacol. 2004, 92, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Willoughby, D.A.; DiRosa, M. Studies on the mode of action of non-steroid anti-inflammatory drugs. Ann. Rheum. Dis. 1972, 31, 540. [Google Scholar] [CrossRef] [PubMed]
- Di Rosa, M. Biological properties of carrageenan. J. Pharm. Pharmacol. 1972, 24, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Rosa, M.D.; Willoughby, D.A. Screens for anti-inflammatory drugs. J. Pharm. Pharmacol. 1971, 23, 297–298. [Google Scholar] [CrossRef] [PubMed]
- Badole, S.L.; Zanwar, A.A.; Ghule, A.E.; Ghosh, P.; Bodhankar, S.L. Analgesic and anti-inflammatory activity of alcoholic extract of stem bark of Pongamia pinnata (L.) Pierre. Biomed. Aging Pathol. 2012, 2, 19–23. [Google Scholar] [CrossRef]
- Swingle, K.F.; Shideman, F.E. Phases of the inflammatory response to subcutaneous implantation of a cotton pellet and their modification by certain anti-inflammatory agents. J. Pharmacol. Exp. Ther. 1972, 183, 226–234. [Google Scholar] [PubMed]
- Bhattacharya, S.; Pal, S.; Chaudhuri, A.K.N. Pharmacological studies of the antiinflammatory profile of Mikania cordata (Burm) B. L. robinson root extract in rodents. Phytother. Res. 1992, 6, 255–260. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds β-elemonic acid are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dose (mg/kg, i.g.) | Body Weight (g) | Liver (g) | Liver Coefficient | Kidney (g) | Kidney Coefficient | |
|---|---|---|---|---|---|---|
| Control | - | 39.16 ± 3.35 | 1.60 ± 0.21 | 4.08 ± 0.22 | 0.54 ± 0.12 | 13.26 ± 2.60 |
| β-EA | 400 | 39.02 ± 4.57 | 1.56 ± 0.21 | 3.98 ± 0.28 | 0.51 ± 0.13 | 12.95 ± 3.44 |
| β-EA | 200 | 38.60 ± 2.85 | 1.55 ± 0.21 | 4.02 ± 0.32 | 0.50 ± 0.11 | 12.53 ± 2.16 |
| β-EA | 100 | 40.37 ± 3.67 | 1.59 ± 0.20 | 3.94 ± 0.40 | 0.53 ± 0.09 | 13.59 ± 2.71 |
| Parameter | Control | 400 mg/kg | 200 mg/kg | 100 mg/kg |
|---|---|---|---|---|
| ALT | 30.29 ± 4.80 | 27.56 ± 4.83 | 27.70 ± 7.92 | 28.86 ± 5.79 |
| AST | 132.3 ± 15.9 | 132.7 ± 25.7 | 134.50 ±23.5 | 136.6 ± 28.3 |
| ALP | 109.3 ± 41.8 | 119.7 ± 43.6 | 126.9 ± 27.2 | 115.3 ± 22.5 |
| TBIL | 0.24 ± 0.04 | 0.23 ± 0.05 | 0.22 ± 0.04 | 0.22 ± 0.05 |
| TG | 0.73 ± 0.31 | 0.67 ± 0.16 | 0.71 ± 0.16 | 0.77 ± 0.07 |
| TC | 2.27 ± 0.31 | 2.21 ± 0.44 | 2.03 ± 0.29 | 2.11 ± 0.19 |
| GLU | 5.37 ± 0.80 | 5.04 ± 1.10 | 5.31 ± 0.85 | 5.95 ± 1.11 |
| BUN | 6.33 ± 0.31 | 6.17 ± 1.20 | 6.31 ± 1.88 | 7.66 ± 1.99 |
| CRE | 36.04 ± 3.31 | 33.94 ± 4.43 | 37.545.39 | 39.64 ± 5.13 |
| CK | 639.1 ± 219.2 | 708.3 ± 240.9 | 596.2 ± 306.9 | 785.7 ± 270.9 |
| TP | 57.19 ± 2.61 | 54.63 ± 2.69 | 56.19 ± 3.35 | 55.89 ± 3.55 |
| ALB | 23.26 ± 1.46 | 23.23 ± 1.37 | 23.88 ± 1.40 | 23.69 ± 1.90 |
| Dose (mg/kg) | Left Ear (mg) | Right Ear (mg) | Topical Edema (mg) | Inhibition (%) | OD | NO | Inhibition | |
|---|---|---|---|---|---|---|---|---|
| (%) | ||||||||
| Control | - | 15.12 ± 3.12 | 35.87 ± 5.13 | 20.75 ± 4.09 | - | 1.402 ± 0.02 | 39.133 ± 7.68 | - |
| Aspirin | 500 | 15.24 ± 3.20 | 22.78 ± 5.13 | 7.54 ± 1.21** | 63.66 | 0.655 ± 0.06 | 11.451 ± 2.31** | 70.73 |
| β-EA | 300 | 15.13 ± 4.14 | 24.69 ± 4.18 | 9.56 ± 1.62** | 53.92 | 0.715 ± 0.03 | 13.668 ± 2.11** | 65.07 |
| β-EA | 200 | 15.39 ± 3.50 | 28.06 ± 3.16 | 12.67 ± 1.90* | 38.31 | 1.002 ± 0.11 | 21.001 ± 5.08 * | 46.44 |
| β-EA | 100 | 14.98 ± 3.21 | 36.07 ± 6.10 | 21.09 ± 3.23 | −1.6 | 1.145 ± 0.13 | 39.624 ± 8.05 | 16.68 |
| Dose (mg/kg, i.g.) | Swelling Degree (mm) | ||||||
|---|---|---|---|---|---|---|---|
| 0 min | 5 min | 30 min | 1 h | 2 h | 4 h | ||
| Control | - | 2.65 ± 0.12 | 1.65 ± 0.23 | 1.60 ± 0.17 | 1.62 ± 0.16 | 1.58 ± 0.16 | 1.60 ± 0.11 |
| Aspirin | 500 | 2.64 ± 0.20 | 1.56 ± 0.21 | 0.81 ± 0.09 | 0.49 ± 0.03 ** | 0.46 ± 0.19 ** | 0.47 ± 0.01 ** |
| β-EA | 300 | 2.65 ± 0.18 | 1.57 ± 0.13 | 0.86 ± 0.15 * | 0.66 ± 0.14 ** | 0.63 ± 0.15 ** | 0.64 ± 0.04 ** |
| β-EA | 200 | 2.64 ± 0.15 | 1.55 ± 0.24 | 1.43 ± 0.08 | 0.79 ± 0.11 * | 0.76 ± 0.14 * | 0.71 ± 0.17 * |
| β-EA | 100 | 2.64 ± 0.09 | 1.56 ± 0.25 | 1.51 ± 0.20 | 1.49 ± 0.10 | 1.48 ± 0.13 | 1.50 ± 0.06 |
| Dose (mg/kg, i.g.) | Left Feet (mg) | Right Feet (mg) | Paw Oedema (mg) | Inhibition (%) | PGE2 (OD/Weights) | |
|---|---|---|---|---|---|---|
| Control | - | 17.12 ± 3.21 | 71.87 ± 15.14 | 54.75 ± 8.89 | - | 1.25 ± 0.18 |
| Aspirin | 500 | 17.24 ± 3.18 | 44.03 ± 13.18 | 26.79 ± 6.17 ** | 48.29 | 0.36 ± 0.07 ** |
| β-EA | 300 | 17.53 ± 5.16 | 47.89 ± 14.11 | 30.36 ± 5.21 ** | 41.31 | 0.65 ± 0.21 ** |
| β-EA | 200 | 17.39 ± 3.19 | 51.90 ± 12.15 | 34.51 ± 4.58 * | 33.31 | 0.86 ± 0.26 * |
| β-EA | 100 | 17.62 ± 4.11 | 58.71 ± 14.82 | 41.09 ± 6.99 | 20.59 | 1.13 ± 0.05 |
| Origin code | Source of Purchase | Specimens Reference Numbers |
|---|---|---|
| A | Gansu Provincial Hospital | RX2016052301 |
| B | Taiyuan Central Hospital | RX2016052302 |
| C | Jincheng General Hospital | RX2016052303 |
| D | Hanchuan Hospital of Traditional Chinese Medicine | RX2016052304 |
| E | Tianjin Lerentang | RX2016052305 |
| F | Tianjin Tongrentang | RX2016052306 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Yu, Y.-l.; Tian, H.; Bai, R.-y.; Bi, Y.-n.; Yuan, X.-m.; Sun, L.-k.; Deng, Y.-r.; Zhou, K. Evaluation of Anti-Inflammatory Activities of a Triterpene β-Elemonic Acid in Frankincense In Vivo and In Vitro. Molecules 2019, 24, 1187. https://doi.org/10.3390/molecules24061187
Zhang Y, Yu Y-l, Tian H, Bai R-y, Bi Y-n, Yuan X-m, Sun L-k, Deng Y-r, Zhou K. Evaluation of Anti-Inflammatory Activities of a Triterpene β-Elemonic Acid in Frankincense In Vivo and In Vitro. Molecules. 2019; 24(6):1187. https://doi.org/10.3390/molecules24061187
Chicago/Turabian StyleZhang, Yue, Ying-li Yu, Hua Tian, Ru-yu Bai, Ya-nan Bi, Xiao-mei Yuan, Li-kang Sun, Yan-ru Deng, and Kun Zhou. 2019. "Evaluation of Anti-Inflammatory Activities of a Triterpene β-Elemonic Acid in Frankincense In Vivo and In Vitro" Molecules 24, no. 6: 1187. https://doi.org/10.3390/molecules24061187
APA StyleZhang, Y., Yu, Y.-l., Tian, H., Bai, R.-y., Bi, Y.-n., Yuan, X.-m., Sun, L.-k., Deng, Y.-r., & Zhou, K. (2019). Evaluation of Anti-Inflammatory Activities of a Triterpene β-Elemonic Acid in Frankincense In Vivo and In Vitro. Molecules, 24(6), 1187. https://doi.org/10.3390/molecules24061187